

Abstract
The large majority of in vitro nanotoxicological studies have used immortalized cell lines for their practicality. However, results from nanoparticle toxicity testing in immortalized cell lines or primary cells have shown discrepancies, highlighting the need to extend the use of primary cells for in vitro assays. This protocol describes the isolation of mouse liver macrophages, named Kupffer cells, and their use to study nanoparticle toxicity. Kupffer cells are the most abundant macrophage population in the body and constitute part of the reticulo-endothelial system (RES), responsible for the capture of circulating nanoparticles. The Kupffer cell isolation method reported here is based on a 2-step perfusion method followed by purification on density gradient. The method, based on collagenase digestion and density centrifugation, is adapted from the original protocol developed by Smedsrød et al. designed for rat liver cell isolation and provides high yield (up to 14 x 106 cells per mouse) and high purity (>95%) of Kupffer cells. This isolation method does not require sophisticated or expensive equipment and therefore represents an ideal compromise between complexity and cell yield. The use of heavier mice (35-45 g) improves the yield of the isolation method but also facilitates remarkably the procedure of portal vein cannulation. The toxicity of functionalized carbon nanotubes f-CNTs was measured in this model by the modified LDH assay. This method assesses cell viability by measuring the lack of structural integrity of Kupffer cell membrane after incubation with f-CNTs. Toxicity induced by f-CNTs can be measured consistently using this assay, highlighting that isolated Kupffer cells are useful for nanoparticle toxicity testing. The overall understanding of nanotoxicology could benefit from such models, making the nanoparticle selection for clinical translation more efficient.
Keywords: Immunology, Issue 102, Kupffer cell, isolation, nanoparticle, nanotoxicity, primary cells, carbon nanotubes, modified LDH assay.
Introduction
The field of nanotoxicology research aims to characterize the biological effect of nanoparticles. Toxicological studies based on in vivo investigations remain the most accurate methods. However, their use is limited by their cost, labor and time requirements1. As alternatives, in vitro assays have been used because of their simplicity and the possibility of developing high-throughput in vitro testing platforms2 - so extending the number of conditions tested. Most nanotoxicological studies are performed using in vitro assays with immortalized cell lines. However, there are concerns regarding the extrapolation of these experimental findings to in vivo toxicological effects3. Indeed, the properties of immortalized cell lines can be significantly different from tissues they were derived from, e.g. genetic transformation4, deterioration of key morphological features5, loss of cellular polarity6 and functional alterations such as the regulation of inflammatory mediators7.
Kupffer cells are the most abundant macrophage population in the body and are directly in contact with blood by lining the wall of liver sinusoids. As part of the reticulo-endothelial system (RES), these macrophages are responsible for the capture of circulating nanoparticles and therefore, constitute a highly suitable model to study nanoparticle toxicity. In vivo8and in vitro9 studies of inflammatory responses associated with Kupffer cells exposed to nanoparticles have been published elsewhere. Kupffer cells have also been involved in the pathogenesis of liver diseases such as alcoholic liver disease10, liver fibrosis11 or viral hepatitis12. It was reported that isolated Kupffer cells provide useful insight to describe cellular mechanisms involved in liver disruption13,14.
Several methods have been reported to isolate and purify Kupffer cells. Cell isolation can result from mechanical or enzymatic dissociation15. Collagenase digestion shows the advantage of conserving the functional integrity of Kupffer cells, while leading to high Kupffer cell yield16. Many experimental approaches, varying in complexity and costs, have been used to separate Kupffer cells from other liver cell populations. For example, Kupffer cell purity can be achieved by immunoaffinity17, flow electrophoresis18, selective adhesion16 or by centrifugal techniques16, which select cells according to their size and density. A combination of these methods can be chosen to increase the purity of the population16. There is no consensus about the ideal method for Kupffer cell isolation, as it mostly depends on the application and available equipment. However, in the case of nanoparticle toxicity testing, the simplicity and high yield of the technique associated with the functional preservation of Kupffer cells appeared to be most suitable for this application.
The Kupffer cell isolation method reported here is based on a 2-step perfusion method followed by purification on density gradient. The method was modified from the original protocol developed by Smedsrød et al.16 designed for rat liver cell isolation. Most studies reported and described the isolation of Kupffer cells from rat livers. Herein, we describe a method to isolate Kupffer cells from mouse liver, at a high yield and purity. The use of mice reduces the experiment cost and allows the processing of several livers to obtain large amounts of Kupffer cells for nanoparticle toxicity testing.
In the following protocol, Kupffer cells were incubated with functionalized carbon nanotubes (f-CNTs). The unique physico-chemical properties of CNTs –i.e. high length to diameter ratio and large surface area, have made CNTs interesting candidates as vectors for therapy and diagnosis purposes. However, concerns have been raised regarding the toxicity of CNTs19, and the development of new in vitro tests aims at increasing the understanding of CNT biological effect. Toxicity in Kupffer cell is associated with a lack of structural integrity of the cell membrane. This is measured by the loss of the cytoplasmic enzyme LDH from the cell into the supernatant. The principle of this method, therefore, is to remove any released LDH and measure what is left in the cells20. This is done in preference to measuring the released LDH in the supernatant because the presence of CNTs in the supernatant interferes with the assay21.
We propose the use of this simple and cost effective Kupffer cell isolation method to isolate high number of functional Kupffer cells. This allows the screening of toxicity of a range of nanoparticles, in a relevant primary macrophage model.
Protocol
All animal experiments were executed in compliance with all relevant guidelines, regulations and regulatory agencies. The protocol being demonstrated was performed under the guidance and approval of the UK Home office regulation
1. Perfusion and Cell Collection (Figure 1)
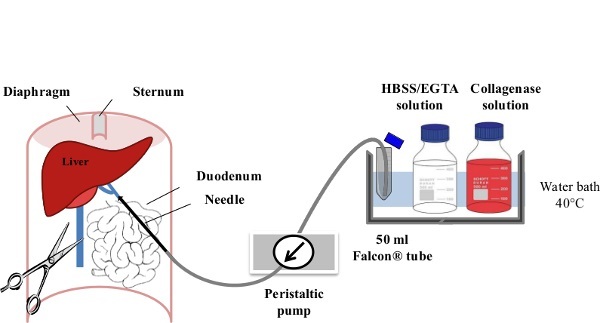 Figure 1: Liver Perfusion. After anaesthesia of the mouse, the digestive tract is laterally moved to the left of the abdomen in order to make the portal vein (PV) accessible. The PV is cannulated using a slow flow rate (1-3 ml/min) of EGTA/HBSS Solution and the inferior veina cava (IVC) is immediately ruptured to avoid any excess pressure within the liver. Within the first minute of perfusion, the flow rate is gradually increased to 7 ml/min. The Collagenase Solution is then perfused at 10 ml/min until its full digestion is achieved.
Figure 1: Liver Perfusion. After anaesthesia of the mouse, the digestive tract is laterally moved to the left of the abdomen in order to make the portal vein (PV) accessible. The PV is cannulated using a slow flow rate (1-3 ml/min) of EGTA/HBSS Solution and the inferior veina cava (IVC) is immediately ruptured to avoid any excess pressure within the liver. Within the first minute of perfusion, the flow rate is gradually increased to 7 ml/min. The Collagenase Solution is then perfused at 10 ml/min until its full digestion is achieved.
Prepare freshly all reagents described in the material table.
Warm the EGTA (Ethylene Glycol Tetra-acetic Acid)/HBSS (Hank's Balanced Salt Solution) Solution (50 ml per mouse) and the Collagenase Solution (100 ml per mouse) for 30 min at 40 °C.
Rinse the pump flexible tubing first with 70% ethanol. Pour 40 ml of EGTA/HBSS Solution into a centrifuge tube immersed in the water bath and rinse the pump flexible tubing with pre-warmed EGTA/HBSS Solution.
Perform terminal anesthesia using a barbiturate to reliably produce unconsciousness before respiratory depression and death. Inject phenobarbitone at 1 mg/kg, i.p. into a female or male CD1 mouse (35-45 g). Confirm the anesthesia by toe pinching.
Shave abdominal hairs and sterilize the abdominal surface using 70% ethanol solution.
Cut through the abdominal cavity and expose the portal vein and inferior vena cava by moving the intestine laterally to the left of the abdomen.
Start the pump at a speed of 1-3 ml/min with EGTA/HBSS Solution and cannulate the portal vein using a 23 g butterfly needle (wings cut). Clamp the section of the portal vein cannulated with the 23 G needle and then flip the serrefine forceps at the surface of the opened mouse abdomen. The liver should quickly pale within the first 30 sec of EGTA/HBSS Solution perfusion.
Rapidly incise the lower part of the inferior vena cava to avoid excess pressure building in the liver, and then increase the flow rate gradually to 7 ml/min, over the first minute of perfusion. The animal dies due to bleeding secondary to vena caval venipuncture.
When less than 5 ml of EGTA/HBSS Solution is remaining into the centrifuge tube, fill it up with Collagenase Solution (40-50 ml). Increase the flow rate gradually to 10 ml/min, over 30 sec.
Make the liver swell by applying pressure to the inferior vena cava, using tweezers, for 5-10 sec intervals. This can be done periodically (5-10 times during digestion). This step will improve the liver cell dissociation and reduce the perfusion time with collagenase.
When less than 10 ml of Collagenase solution remains within the centrifuge tube, pour 40-50 ml of pre-warmed Collagenase solution into the centrifuge tube.
After 10-15 min of perfusion and about 70-80 ml of Collagenase Solution perfused, apply a small pressure on the surface of the liver with a forceps. A print of the pressure indicates that liver cells are dissociated.
Remove the liver from the abdominal cavity as one piece and place it in a centrifuge tube containing 20-30 ml of Kupffer Cells Isolation Medium. Keep liver cells on ice or at 4 °C for a maximum period of 3 hr to avoid affecting liver cell viability.
Repeat the perfusion procedure on several animals if needed, while keeping the perfused liver on ice. Pool one to three livers for purification and proceed with stage 3.
2. Preparation of the Density Gradient for Centrifugation (Figure 2)
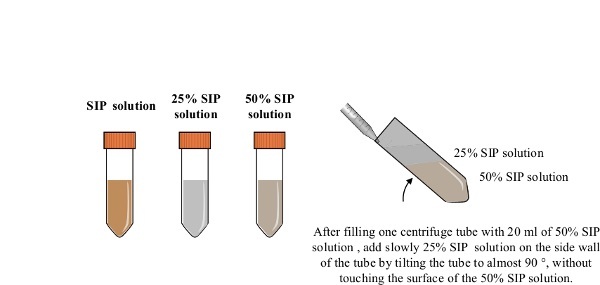 Figure 2: Preparation of the density gradient. All procedures are carried out under sterile condition. The SIP is prepared by mixing 15.3 ml of coated silica particle solution with 1.7 ml of 10 x PBS. A 5 ml of SIP are mixed with 15 ml of PBS to make 20 ml of 25% SIP solution. 10 ml of SIP are mixed with 10 ml of PBS to make 20 ml of 50% SIP solution. A centrifuge tube containing the 50% SIP solution (20 ml) is tilted and the 25% SIP solution (20 ml) is slowly added using a serological 25 ml pipette.
Figure 2: Preparation of the density gradient. All procedures are carried out under sterile condition. The SIP is prepared by mixing 15.3 ml of coated silica particle solution with 1.7 ml of 10 x PBS. A 5 ml of SIP are mixed with 15 ml of PBS to make 20 ml of 25% SIP solution. 10 ml of SIP are mixed with 10 ml of PBS to make 20 ml of 50% SIP solution. A centrifuge tube containing the 50% SIP solution (20 ml) is tilted and the 25% SIP solution (20 ml) is slowly added using a serological 25 ml pipette.
Proceed with the following steps (section 2., 3. & 5.) under sterile condition and keep cells on ice or at 4 °C. Prepare the SIP (Solution of Isotonic coated silica Particles) by mixing 15.3 ml of coated silica particle solution with 1.7 ml of 10 x PBS.
To make 20 ml of 25% SIP solution, mix 5 ml of SIP with 15 ml of PBS. To make 20 ml of 50% SIP solution, mix 10 ml of SIP with 10 ml of PBS.
Fill one centrifuge tube with 20 ml of the 50% SIP solution. Tilt the centrifuge tube at an angle close to 90° and add slowly the 25% SIP solution (20 ml) on the side wall of the tube using a 25 ml serological pipette, without touching the surface of the 50% SIP layer. When adding the 25% SIP solution, progressively reduce the angle of the tube. Keep the density gradient on ice until use.
3. Kupffer Cell Purification (Figure 3)
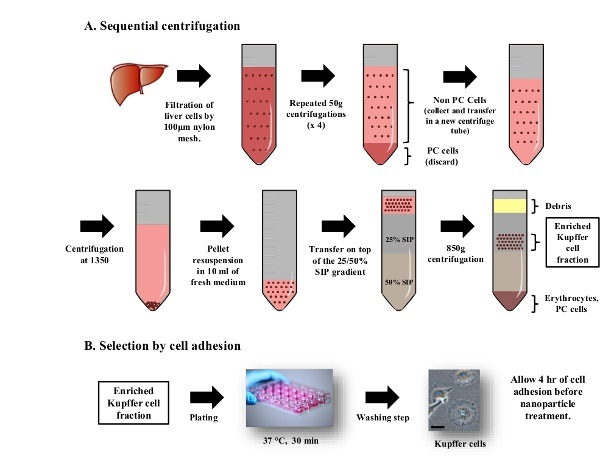 Figure 3: Purification of Kupffer Cells by density gradient centrifugation and cell adhesion. All procedures are carried out under sterile condition. (A) After rupture of the Glisson’s capsule, liver cells are filtered through a 100 µm strainer. The suspension is centrifuged at x 50 g to discard hepatocytes (pellets) and collect the non-parenchymal cell fraction (supernatant). This step is repeated 3 times. Non-parenchymal cells are added on top of a discontinuous isotonic gradient and centrifuged at 800 x g for 15 min. Kupffer cells collected from the 25% SIP cushion are purified further by cell adhesion selection. (B) Cells are plated in 24-well plate and incubated at 37 °C and 5% CO2 for 30 min. Non-adherent cells are washed once with 500 µl of HBSS. Kupffer cells display their adherent morphology 4 hr after plating, when f-CNTs can be added to the cells. The scale bar represents 25 µm. Please click here to view a larger version of this figure.
Figure 3: Purification of Kupffer Cells by density gradient centrifugation and cell adhesion. All procedures are carried out under sterile condition. (A) After rupture of the Glisson’s capsule, liver cells are filtered through a 100 µm strainer. The suspension is centrifuged at x 50 g to discard hepatocytes (pellets) and collect the non-parenchymal cell fraction (supernatant). This step is repeated 3 times. Non-parenchymal cells are added on top of a discontinuous isotonic gradient and centrifuged at 800 x g for 15 min. Kupffer cells collected from the 25% SIP cushion are purified further by cell adhesion selection. (B) Cells are plated in 24-well plate and incubated at 37 °C and 5% CO2 for 30 min. Non-adherent cells are washed once with 500 µl of HBSS. Kupffer cells display their adherent morphology 4 hr after plating, when f-CNTs can be added to the cells. The scale bar represents 25 µm. Please click here to view a larger version of this figure.
Put one perfused liver in a Petri dish with 15 ml of Kupffer Cell Isolation Medium. Rupture the Glisson’s capsule (membrane of the liver) using scissors and release all liver cells into the Kupffer cell isolation medium. Filter the solution though a 100 µm cell strainer to be collected in a centrifuge tube. Repeat the perfusion procedure on additional perfused livers and pool the cells in the same centrifuge tube (15 ml from one mouse, up to 45 ml from three mice).
Centrifuge the cell suspension at 50 x g for 2 min at 4 °C. Parenchymal cells (hepatocytes) will be in the pellet and non-parenchymal cells (including Kupffer cells) will be in the supernatant.
Collect the supernatant in a clean centrifuge tube and centrifuge at 50 x g for 2 min each. Repeat this step three more times.
Centrifuge at 1,350 x g for 15 min to pellet non-parenchymal cells. Discard the supernatant and resuspend the pellet in 10 ml of Kupffer Cell Isolation Medium.
Add the non-parenchymal cell solution on the discontinuous isotonic gradient 25/50% as described in 2.3. Centrifuge at 850 x g for 15 min without acceleration or break.
Localize the enriched Kupffer cell fraction appearing turbid within the 25% SIP fraction close to the 25/50% SIP interface (Figure 3). Using a 10 ml serological pipette, aspirate about 12 ml of the enriched Kupffer cell fraction. Transfer cells into a centrifuge tube containing 35-40 ml of Kupffer Cell Isolation Medium. Mix gently and centrifuge at 1,350 x g for 15 min at 4 °C to pellet cells.
Discard the supernatant and resuspend cells in 5-10 ml of pre-warmed Kupffer Cell Culture Medium. Count cells (haemocytometer) and measure the viability using trypan blue staining.
Plate the purified non-parenchymal cells in 24-well plates at a density of 5 x 105 cells/well. Incubate the cells at 37 °C and 5% CO2 (Figure 3). After 30 min, gently remove the medium, wash with pre-warmed Hank's Balanced Salt Solution (HBSS) once then replace it with 500 µl of fresh and pre-warmed Kupffer Cell Culture Medium (per well).
Leave Kupffer cells for at least 4 hr at 37 °C and 5% CO2 before treatment with nanoparticles, to allow them to acquire their adherent morphology.
4. Characterization
- Kupffer Cells Purity
- Prepare fresh PBS/BSA solution used to maintain Kupffer cell viability. Wash cells once with 500 µl of PBS/BSA solution. Remove the PBS/BSA solution and detach Kupffer cells in 500 µl of fresh PBS/BSA solution using gentle scraping. When cells are plated in 24-well plates, shorten the extremities of scraper blades with scissors to make the detachment easier to proceed. Transfer cells into flow cytometry tubes.
- Count Kupffer cells using a haemocytometer and centrifuge 1 x 105 cells at 1,350 x g. Resuspend Kupffer cells in 30 µl of F4/80 antibody (neat). Mix well and incubate at room temperature for 30 min. Keep the unstained cells (not incubated with the antibody) on ice.
- Add 2 ml of PBS/BSA solution in stained cells, centrifuge at 1,350 g for 5 min and discard the resulting supernatant. Resuspend stained cells in 200 µl of PBS/BSA.
- For flow cytometry analysis, gate 10,000 unstained Kupffer cells according to their size (forward scatter) and granularity (side scatter). Analyze fluorescence of gated cells in FL-1 channel.
- Repeat the procedure for stained Kupffer cells keeping the same settings used for the flow cytometry analysis of unstained Kupffer cells. Select and quantify the fluorescence of stained cells to assess the purity of Kupffer cell.
- Kupffer Cell Phagocytic Activity
- Replace media with 0.5% (v/v) fluorescent beads in Kupffer Cell Culture Medium and incubate for 4 hr at 37 °C and 5% CO2.
- In order to remove non-internalized beads, centrifuge Kupffer cells at 1,350 x g, discard the supernatant and resuspend cells in 500 µl PBS/BSA Solution. Repeat this step 2 more times.
- Scrape Kupffer cells in 500 µl of PBS/BSA Solution and transfer cells into a flow cytometry tube.
- Centrifuge Kupffer cells at 1,350 x g, discard the supernatant and resuspend cells in 200 µl of PBS/BSA Solution.
- For flow cytometry analysis, gate 10,000 unstained Kupffer cells according to their size (forward scatter) and granularity (side scatter). Analyze fluorescence of gated cells in FL-2 channel.
5. Incubation of Chemically Functionalized Carbon Nanotubes (f -CNTs)
Prepare a dispersion of f-CNTs (1 mg/ml) in water by sonication for 15 min before use. f-CNTs are prepared in-house22.
Prepare 2x concentrated f-CNTsdispersion in Kupffer Cell Culture Medium.Sonicate f-CNTs dispersed in medium for 2 min before proceeding with step 5.3).
For each well, replace 250 µl of old media with 250 µl of 2x f-CNTs dispersion. Untreated wells (250 µl of conditioned medium + 250 µl of fresh Kupffer Cell Culture Medium) and wells treated with 10% DMSO are used as negative and positive control, respectively.
Incubate Kupffer cells at 37 °C and 5% CO2 for 24 or 72 hr.
6. Toxicity Assessment of f -CNTs in Kupffer Cells by the Modified Lactate Dehydrogenase (LDH) Assay
Discard the supernatant.
Add 200 µl (per well of 24-well plate) of Lysis Buffer and incubate at 37 °C for 30 min-1 hr.
After carrying out vigorous pipetting, collect lysed Kupffer cells into microcentrifuge tubes and centrifuge at 40,000 x g for 10 min to pellet f-CNTs that were taken up by cells and the cell debris.
Collect the supernatants (ƒ-CNT free) into microcentrifuge tubes and store at -20 °C or proceed to step 6.5).
Transfer 50 µl into a 96-well plate and add an equal volume of the substrate mix solution (LDH kit) to each well. Cover the plate and incubate at room temperature for 15 min before adding 50 µl of the stop solution (LDH kit). Add triplicates of blank wells containing 50 µl of lysis buffer, 50 µl of substrate mix solution and 50 µl of stop solution.
Read the absorbance at 490 nm in a microplate reader and use the following formula to calculate the (%) cell viability.
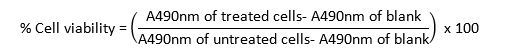
Representative Results
The number of non-purified parenchymal cells was consistent and ranged between 8 and 14 x 106 cells per mouse (17 isolations were performed). Each mouse isolation was sufficient to plate 16 to 28 wells. Cell viability by trypan blue staining showed cell viability ~95%. Kupffer cells showed a round shape within 30 min of incubation at 37 °C, related to their incomplete adherent morphology (Figure 4A). At 4 hr of incubation and afterwards, cells were spread and cell clusters started to form.
Kupffer cells were then characterized to confirm their purity and phagocytic activity. Cells were stained with F4/80 antibody to demonstrate the Kupffer cell purity. Flow cytometry analysis showed Kupffer cell purity above 95% (Figure 4B). Cells were incubated with 1 µm fluorescent beads, 12 hr after plating, to confirm their ability to take up large particles. Flow cytometry analysis showed that more than 85% of Kupffer cells are phagocytic, i.e. can take up the 1 µm beads (Figure 4C). After 72 hr of culture, 40% of Kupffer cells were phagocytic, indicating that Kupffer cells partially lost their phagocytic activity.
Microscopy imaging of Kupffer cells incubated with f-CNTs for 24 and 72 hr revealed similar cell morphology in comparison to naïve cells (Figure 5A). Kupffer cells incubated with 10% DMSO (positive control) displayed necrotic morphology (Figure 5A). Following the analysis of the modified LDH assay, Kupffer cells treated with 10% DMSO (positive control) for 24 and 72 hr showed high toxicity (Figure 5B). In comparison, f-CNTs induced slight but significant reduction in cell viability only when cells were exposed at 50 µg/ml for 72 hr (Figure 5B).
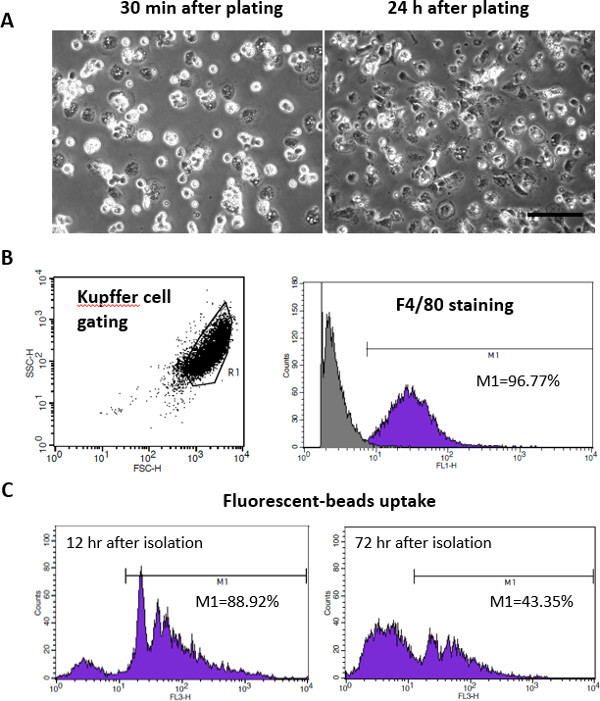 Figure 4: Purity and Uptake Abilities of Kupffer cells. (A) Microscopic images of Kupffer cells plated after 30 min or 24 hr at 37 °C with an atmosphere of 5% CO2. The scale bar presents 50 µm. (B) Flow cytometry analysis of Kupffer cells was carried out following FSC/SSC cell gating. Kupffer cell purity was determined using F4/80 antibody staining and showed to be above 95%. (C) Kupffer cells were incubated for 4 hr in presence of 1 µm fluorescent beads to assess their phagocytic activity. The functional properties of Kupffer were maintained better in cells freshly isolated (12 hr after plating) compared to cells tested after 3 days.
Figure 4: Purity and Uptake Abilities of Kupffer cells. (A) Microscopic images of Kupffer cells plated after 30 min or 24 hr at 37 °C with an atmosphere of 5% CO2. The scale bar presents 50 µm. (B) Flow cytometry analysis of Kupffer cells was carried out following FSC/SSC cell gating. Kupffer cell purity was determined using F4/80 antibody staining and showed to be above 95%. (C) Kupffer cells were incubated for 4 hr in presence of 1 µm fluorescent beads to assess their phagocytic activity. The functional properties of Kupffer were maintained better in cells freshly isolated (12 hr after plating) compared to cells tested after 3 days.
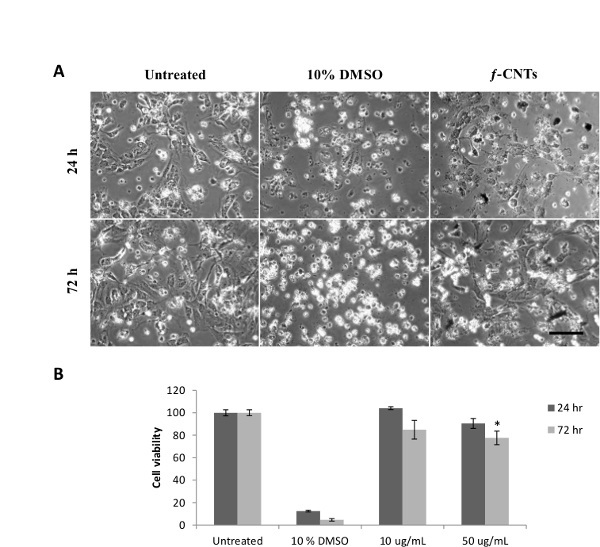 Figure 5: Uptake and toxicity of f-CNTs in Kupffer cells. (A) Microscopy images of Kupffer cells. Images show Kupffer cells treated with 10% DMSO and 50 µg/ml of f-CNTs at 37 °C for 24 and 72 hr. The scale bar presents 50 µm. (B) Modified LDH assay. Low cell viability was seen in 10% DMSO treated cells (positive controls) while f-CNTs significantly affected cell viability only after exposition to 50 µg/ml for 72 hr. *P <0.05 relative to the naïve condition (excluding 10% DMSO condition) using analysis of variance (one-way ANOVA) with post hoc analysis by the Tukey test. The scale bar corresponds to 50 µm. Please click here to view a larger version of this figure.
Figure 5: Uptake and toxicity of f-CNTs in Kupffer cells. (A) Microscopy images of Kupffer cells. Images show Kupffer cells treated with 10% DMSO and 50 µg/ml of f-CNTs at 37 °C for 24 and 72 hr. The scale bar presents 50 µm. (B) Modified LDH assay. Low cell viability was seen in 10% DMSO treated cells (positive controls) while f-CNTs significantly affected cell viability only after exposition to 50 µg/ml for 72 hr. *P <0.05 relative to the naïve condition (excluding 10% DMSO condition) using analysis of variance (one-way ANOVA) with post hoc analysis by the Tukey test. The scale bar corresponds to 50 µm. Please click here to view a larger version of this figure.
Discussion
The following steps are critical to achieve high yield and high viability of Kupffer cells. Aseptic conditions should be used to limit the risk of bacterial and fungal contamination. All instruments need to be sterilized before use. Reagents should be freshly prepared before carrying out the isolation procedure.
The choice of collagenase IV, with low tryptic activity, is crucial. Different lots from the same supplier have different enzymatic activity and they may need to be compared initially to select the most appropriate batch for Kupffer cell isolation. This can be judged by assessing cell yield and cell viability. It is also recommended to contact the supplier to provide batches that have been used for similar applications. Once decided on the batch to be used, reserve it for future use.
The cannulation procedure requires training. Plan the use of several animals to become confident with the portal vein cannulation procedure. With practice, successful cannulation will be easily achieved and the procedure can be handled by one person. Note that the use of heavy animals increases the diameter of the portal vein and therefore facilitates the procedure. When the back wall of the portal vein is perforated, it is technically difficult to access the vein again, especially if you are new with this procedure and therefore, it is preferable to start with a new animal.
The HBSS and Collagenase Solutions should have their pH adjusted to 7.4 and be pre-warmed at 40 °C to achieve an output temperature from the peristaltic pump of 37 °C. The perfusion with EGTA/HBSS Solution (Ca2+ and Mg2+ free) containing EGTA is needed for the disruption of Ca2+-dependent adhesion molecules, named desmosome, weakening the cell-cell interaction. Collagenase type IV is used to detach liver cells from the extracellular matrix and so lead to liver cell dissociation. The perfusion time for the two solutions should not exceed 15 min for CD1 mice but other strains, such as C57Bl/6, require slightly longer liver digestion time.
To achieve a successful isolation, it is necessary to obtain high cell yield and viability. If the experimenter has no experience in liver cell isolation, it is advised to combine hepatocytes pellets from the first 3 centrifugations at 50 x g and count cells by trypan blue exclusion assay. The hepatocyte yield should be >20 x 106 cells and the hepatocyte viability >50%. Low yields result essentially from poor cell dissociation leading indirectly to lower Kupffer cell yield. It should be confirmed that the liver is properly digested at the end of the perfusion step. When suitable hepatocyte yield is achieved in combination with low hepatocyte viability, this can result from an excessive collagenase digestion but is more likely explained by an inappropriate liver perfusion procedure or the use of a contaminated reagent/material.
After plating, Kupffer cells are sensitive and should be washed gently. At least 4 hr are needed for Kupffer cells to acquire their adherent morphology. The treatment is generally carried out 4-24 hr after plating, as Kupffer cells show their maximum phagocytic activity within 1 day after plating.
In our protocol, we describe the dissociation of CD1 mouse liver cells followed by the purification of Kupffer cells by density gradient centrifugation and selection by adhesion. The characterization of Kupffer cells, by flow cytometry, demonstrates that high yield and purity of Kupffer cells can be achieved using this method.
This Kupffer cell isolation method represents a valuable compromise between complexity and cell yield. However, it is reported that certain Kupffer cell methods can be easier to conduct, such as the protocol described by Wu et al., where the liver is digested ex vivo23. However, the method described by Wu et al. leads to a limited amount of cells and lower cell purity. Higher yield and purity can be obtained but require expensive and/or sophisticated equipment and can be time consuming.
The original protocol developed by Smedsrød et al.16 used a comparable protocol based on 2-step perfusion method (HBSS + collagenase digestion) followed by centrifugation on Percoll gradient and selection of the lower Percoll cushion for further purification by surface adhesion. By modifying the centrifugation procedure and collecting the upper Percoll cushion, we increased the enriched Kupffer cell fraction from 5.25 x 106 to 8.2 x 106 cells per gram of liver. This improvement could be attributed to the use of EGTA during the early perfusion step in order to facilitate the dissociation of liver cells. Moreover, the use of flow cytometry analysis allowed the reliable quantitative characterization of Kupffer cell purity and phagocytic activity. With a maximum yield of 14 x 106 purified non-parenchymal cells per mouse liver. The present isolation method achieve higher amount of Kupffer cells per mouse liver to what has been reported in literature24. This improvement can be explained by the use of heavier mouse (35-45 g) compared to other methods24. Beside this increase in cell yield, the use of larger animals facilitates remarkably the procedure of portal vein cannulation.
High-throughput in vitro testing can be carried out using this primary phagocytic model, therefore mimicking the in vivo toxicological impact of nanoparticles. The overall understanding of nanotoxicology could benefit from such models, making the nanoparticle selection for clinical translation more efficient. In addition, it is in line with implementing the concept of 3 R (reduction, refinement and replacement) in animal biomedical research.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
This work was supported by EU FP7-ITN Marie-Curie Network program RADDEL. MB and KAJ would like to acknowledge Joe Varguese and Rui Serra Maia for their help and suggestions along the optimization of the Kupffer cell isolation.
References
- Hoffmann D, et al. Performance of novel kidney biomarkers in preclinical toxicity studies. Toxicol Sci. 2010;116(1):8–22. doi: 10.1093/toxsci/kfq029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nel A, et al. Nanomaterial Toxicity Testing in the 21st Century: Use of a Predictive Toxicological Approach and High-Throughput Screening. Accounts of Chemical Research. 2013;46(3):607–621. doi: 10.1021/ar300022h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bregoli L, et al. Toxicity of antimony trioxide nanoparticles on human hematopoietic progenitor cells and comparison to cell lines. Toxicology. 2009;262(2):121–129. doi: 10.1016/j.tox.2009.05.017. [DOI] [PubMed] [Google Scholar]
- Yamasaki K, et al. Genomic Aberrations and Cellular Heterogeneity in SV40-Immortalized Human Corneal Epithelial Cells. Investigative Ophthalmology, & Visual Science. 2009;50(2):604–613. doi: 10.1167/iovs.08-2239. [DOI] [PubMed] [Google Scholar]
- Motoyoshi Y, et al. Megalin contributes to the early injury of proximal tubule cells during nonselective proteinuria. Kidney International. 1038;74(10):1262–1269. doi: 10.1038/ki.2008.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prozialeck WC, Edwards JR, Lamar PC, Smith CS. Epithelial barrier characteristics and expression of cell adhesion molecules in proximal tubule-derived cell lines commonly used for in vitro toxicity studies. Toxicology in Vitro. 2006;20(6):942–953. doi: 10.1016/j.tiv.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Chamberlain LM, Godek ML, Gonzalez-Juarrero M, Grainger DW. Phenotypic non-equivalence of murine (monocyte-) macrophage cells in biomaterial and inflammatory models. J Biomed Mater Res A. 2009;88(4):858–871. doi: 10.1002/jbm.a.31930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kermanizadeh A, et al. The role of Kupffer cells in the hepatic response to silver nanoparticles. Nanotoxicology. 2013. [DOI] [PubMed]
- Fischer HC, Hauck TS, Gomez-Aristizabal A, Chan WCW. Exploring Primary Liver Macrophages for Studying Quantum Dot Interactions with Biological Systems. Advanced Materials. 2010;22(23):2520–2524. doi: 10.1002/adma.200904231. [DOI] [PubMed] [Google Scholar]
- Wan JH, et al. M2 Kupffer Cells Promote M1 Kupffer Cell Apoptosis: A Protective Mechanism Against Alcoholic and Nonalcoholic Fatty Liver Disease. Hepatology. 2014;59(1):130–142. doi: 10.1002/hep.26607. [DOI] [PubMed] [Google Scholar]
- Matsuoka M, Tsukamoto H. Stimulation of hepatic lipocyte collagen production by Kupffer cell-derived transforming growth factor β: Implication for a pathogenetic role in alcoholic liver fibrogenesis. Hepatology. 1990;11(4):599–605. doi: 10.1002/hep.1840110412. [DOI] [PubMed] [Google Scholar]
- Polakos NK, et al. Kupffer Cell-Dependent Hepatitis Occurs during Influenza Infection. The American Journal of Pathology. 2006;168(4):1169–1178. doi: 10.2353/ajpath.2006.050875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner A, Keyhani A, Reiner R, Holdstock G, Wright R. Proteolytic enzymes released by liver macrophages may promote hepatic injury in a rat model of hepatic damage. Gastroenterology. 1981;80(4):647–654. [PubMed] [Google Scholar]
- Knittel T, et al. Expression patterns of matrix metalloproteinases and their inhibitors in parenchymal and non-parenchymal cells of rat liver: regulation by TNF-α and TGF-β1. Journal of Hepatology. 1999;30(1):48–60. doi: 10.1016/s0168-8278(99)80007-5. [DOI] [PubMed] [Google Scholar]
- Branster MV, Morton RK. Isolation of Intact Liver Cells. Nature. 1038;180(4597):1283–1284. doi: 10.1038/1801283a0. [DOI] [PubMed] [Google Scholar]
- Smedsrød B, Pertoft H. Preparation of pure hepatocytes and reticuloendothelial cells in high yield from a single rat liver by means of Percoll centrifugation and selective adherence. Journal of Leukocyte Biology. 1985;38(2):213–230. doi: 10.1002/jlb.38.2.213. [DOI] [PubMed] [Google Scholar]
- Morin O, Patry P, Lafleur L. Heterogeneity of endothelial cells of adult rat liver as resolved by sedimentation velocity and flow cytometry. J Cell Physiol. 1984;119(3):327–334. doi: 10.1002/jcp.1041190311. [DOI] [PubMed] [Google Scholar]
- Miller SB, et al. Purification of cells from livers of carcinogen-treated rats by free-flow electrophoresis. Cancer Res. 1983;43(9):4176–4179. [PubMed] [Google Scholar]
- Firme CP, Bandaru PR. Toxicity issues in the application of carbon nanotubes to biological systems. Nanomedicine. 2010;6(2):245–256. doi: 10.1016/j.nano.2009.07.003. [DOI] [PubMed] [Google Scholar]
- Ali-Boucetta H, Al-Jamal KT, Kostarelos K. Cytotoxic assessment of carbon nanotube interaction with cell cultures. Methods in Molecular Biology. 2011;726:299–312. doi: 10.1007/978-1-61779-052-2_19. [DOI] [PubMed] [Google Scholar]
- Wang G, et al. Understanding and correcting for carbon nanotube interferences with a commercial LDH cytotoxicity assay. Toxicology. 2012;299(2-3):99–111. doi: 10.1016/j.tox.2012.05.012. [DOI] [PubMed] [Google Scholar]
- Wang JT-W, et al. Magnetically Decorated Multiwalled Carbon Nanotubes as Dual MRI and SPECT Contrast Agents. Advanced Functional Materials. 2014;24(13):1880–1894. doi: 10.1002/adfm.201302892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li PZ, Li JZ, Li M, Gong JP, He K. An efficient method to isolate and culture mouse Kupffer cells. Immunol Lett. 2014;158(1-2):52–56. doi: 10.1016/j.imlet.2013.12.002. [DOI] [PubMed] [Google Scholar]
- Froh M, Konno A, Thurman RG. Isolation of liver Kupffer cells. Curr Protoc Toxicol. 2003;Chapter 14:Unit14 14. doi: 10.1002/0471140856.tx1404s14. [DOI] [PubMed] [Google Scholar]