

Abstract
Macromolecular delivery strategies typically utilize the endocytic pathway as a route of cellular entry. However, endosomal entrapment severely limits the efficiency with which macromolecules penetrate the cytosolic space of cells. Recently, we have circumvented this problem by identifying the reagent dfTAT, a disulfide bond dimer of the peptide TAT labeled with the fluorophore tetramethylrhodamine. We have generated a fluorescently labeled dimer of the prototypical cell-penetrating peptide (CPP) TAT, dfTAT, which penetrates live cells and reaches the cytosolic space of cells with a particularly high efficiency. Cytosolic delivery of dfTAT is achieved in multiple cell lines, including primary cells. Moreover, delivery does not noticeably impact cell viability, proliferation or gene expression. dfTAT can deliver small molecules, peptides, antibodies, biologically active enzymes and a transcription factor. In this report, we describe the protocols involved in dfTAT synthesis and cellular delivery. The manuscript describes how to control the amount of protein delivered to the cytosolic space of cells by varying the amount of protein administered extracellularly. Finally, the current limitations of this new technology and steps involved in validating delivery are discussed. The described protocols should be extremely useful for cell-based assays as well as for the ex vivo manipulation and reprogramming of cells.
Keywords: Bioengineering, Issue 103, Cell-penetrating peptide, cytosolic delivery, protein, fluorescence microscopy, cell culture.
Introduction
The delivery of proteins, peptides or cell-impermeable small molecules into live cells is often desirable in many biological or biotechnological applications (cellular imaging, functional assays, cellular reprogramming, etc.)1-4. Many delivery approaches have already been reported, including microinjection, electroporation, or use of carrier agents (e.g., cell-penetrating peptides such as TAT, lipids)5-7. Each technique typically has specific pros and cons that might make these approaches adequate for certain applications but not for others. Common issues involve poor delivery efficiencies and/or lack of control of how much material is delivered8,9, toxicity or deleterious physiological impact10,11, lack of temporal control, delivery in few cells but not in a whole population (e.g., microinjection)12, and complex chemical conjugation or formulation schemes11.
Recently, we have developed a novel delivery strategy that circumvents these limitations. This strategy relies on a peptide named dfTAT (dimer fluorescent TAT)13. dfTAT is derived from the well-know cell–penetrating peptide (CPP) TAT. dfTAT contains two disulfide bonded copies of TAT labeled with the fluorophore tetramethylrhodamine. Despite their similarities, TAT and dfTAT differ significantly in activity. TAT is typically internalized into cells by endocytosis. This CPP remains however mostly trapped inside endosomes (this usually lead to a punctate distribution of the peptide inside cells when examined by fluorescence microscopy). Like TAT, dfTAT is efficiently endocytosed by cells. However, dfTAT does not stay trapped inside endosomes. Instead, it mediates endosomal leakage in a manner that is extremely efficient. The endosomolytic activity of dfTAT can then be exploited to deliver macromolecules by a simple incubation assay.
The current understanding of the delivery process is as follows. dfTAT induces macropinocytosis. As a result, cells incubated with dfTAT take up soluble proteins, peptides, or small molecules (molecules of interest, MOI) present in media by fluid-phase endocytosis (see Figure 1). Interactions between dfTAT and MOI are not necessary as long as both entities traffic together within the endocytic pathway. As dfTAT reaches a certain threshold within the lumen of endocytic organelles, it expresses its endosomal leakage activity (the molecular details remain to be fully characterized). The content of the lumen of leaky organelles, and therefore the MOI, is then released into the cells. This approach is therefore very convenient as no conjugation or formulation schemes with MOI are required. Moreover, because dfTAT is not directly modifying a MOI, it should also not interfere with MOIs function once intracellular delivery is achieved. In addition, the concentration of dfTAT used delivery is independent of that of MOI in media. For instance, dfTAT concentration can be kept constant between different experiments to guarantee reproducible efficiencies in endosomal leakage. In contrast, concentration of MOI in media can be gradually changed to achieve desired levels of MOI delivered in cytosol.
The high endosomal leakage efficiency achieved with dfTAT is remarkably innocuous to many of the cells tested to date. This is a surprise because endocytic organelles are important component of the cells and one would expect that the dramatic leakage mediated by dfTAT would be accompanied by deleterious cellular responses. Yet, treated cells proliferate at the same rate as untreated cells and do not display any significant changes in their transcriptome. Moreover, delivery can be repeated within minutes with reproducible delivery efficiencies, indicating that cells can either tolerate or recover from the delivery process without losing their capacity for endocytosis or endosomal leakage. Subtle cellular responses might take place during dfTAT delivery and the molecular details of what these responses might be remain to be explored. Yet, by combining high efficiency, convenience of protocols, and lack of toxicity, this delivery approach should prove immediately useful in many cell-based applications. The protocols presented herein are aimed at making this technology accessible to the research community.
Protocol
1. SPPS: CK(TMR)TATG (fTAT) Synthesis
Note: dfTAT is produced in two steps: synthesis of the monomer fTAT by solid phase peptide synthesis followed by disulfide bond dimerization to form dfTAT.
Swell 500 mg of rink amide MBHA resin in dimethylformamide (DMF) in a standard 50 ml SPPS vessel for 1 hr.
Perform Fmoc deprotection by incubating the Fmoc protected resin in a 20% piperidine solution (20% piperidine in DMF, 10 ml (0.30 mmol). Carry out deprotection steps twice (1 x 5 min and 1 x 15 min) with a washing step using DMF (the washing step includes rinsing the resin multiple times with a total volume of approximately 150 ml of DMF and removing the solvent by vacuum filtration) in between reactions.
- Synthesize CK(TMR)RKKRRQRRRG (fTAT) on the rink amide MBHA resin. Use the following Fmoc protected amino acids: Fmoc-Lys(Mtt)-OH (only on N-terminus), Fmoc-Lys(Boc)-OH, Fmoc-Gly-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Gln(Trt)-OH, and Fmoc-Cys(Trt)-OH. Carry out the reaction using N2 (20 PSI) to agitate the reaction. Perform all reactions at RT.
- Carry out each amino acid coupling reaction for 4 hr. For each coupling, use the Fmoc protected amino acid (1.2 mmol), N,N,N ′ ,N ′ -Tetramethyl- O- (1 H -benzotriazol-1-yl)uronium hexafluorophosphate (HBTU) (0.44 g, 1.1 mmol), and diisopropylethylamine (DIEA) (0.51 ml, 3.0 mmol) dissolved in DMF. Following the 4 hr coupling, wash the resin with DMF and perform the Fmoc deprotection step as described in step 1.2.
- Repeat until the linear peptide chain of fTAT (Linear peptide chain: CKRKKRRQRRRG no TMR) was synthesized.
- Then wash resin thoroughly using first DMF and following that with dichloromethane (DCM). Rinse the resin multiple times with a total volume of approximately 150 ml of DMF or DCM and remove the solvent by vacuum filtration.
- Use MALDI-TOF to confirm that the peptide obtained has the correct molecular weight.
- Make a matrix mix by adding 1 mg of matrix to a solution composed of 50 µl of 0.01% TFA in water solution and 50 µl of acetonitrile.
- To confirm linear peptide chain mass, use α-Cyano-4-hydroxycinnamic acid. Run an analytical HPLC of the crude peptide and collect 50 µl from the top of the pure peak.
- To prepare the sample to plate on the MALDI plate: Add 8 µl of the peptide to 2 µl of the matrix solution and place on the MALDI plate to air dry or on a 37 °C heat plate. Note: Arginine is known to be an amino acid difficult to couple in SPPS. Successful coupling is established by performing a Kaiser test14 (e.g., ninhydrin test). This test allows for the detection of free amines that might remain uncoupled on the resin. In case a blue color is detected (indicative of the presence of free amines), coupling steps are repeated.
- For CK(-NH-TMR)TATG (fTAT), cleave the Mtt protecting group at the -amino group of Lys on CK(-NH-Mtt)TATG with a solution composed of 1% trifluoroacetic acid (TFA) and 2% triisopropylsilane (TIS) in DCM.
- As the removal of MTT protecting group would result in the appearance of a yellow color, incubate the resin with 20 ml of the above solution for 5 min, repeat this until no yellow color is observed. Wash the resin with DCM and DMF in between. Additionally since TIS is a scavenger that would scavenge the MTT, once no yellow color appears in the solution in the presence of MTT.
- Add an additional solution with 1% TFA in DCM (no TIS) to the resin to insure no more MTT is removed and the solution remains clear.
Dissolve the three components: Carboxytetramethylrhodamine (TMR), HBTU, and DIEA (4, 3.9 and 10 equivalence in respect to the amount of resin (in mg)) in DMF and add this mixture to the resin and carry out the reaction O/N using dry N2 to provide agitation.
- Following Fmoc-deprotection and amino acid conjugation, wash the resin with DCM and allow it to dry. For complete cleavage of the peptide from the resin, add a solution containing 92.5% TFA, 2.5% H2O, 2.5%, TIS, and 2.5% ethanedithiol (EDT) (total volume = 4 ml) to the peptidyl-resin for 3 hr at RT to achieve global deprotection and cleavage from the resin.
- Precipitate the crude peptide products using cold anhydrous ethyl ether (Et2O) by draining the solution from step 1.6 into 40 ml of Et2O, spin this down at 4 °C and 4,000 x g for 20 - 25 min. Repeat this step to allow washing of the precipitate with cold anhydrous Et2O.
- Resuspend the precipitates in water (5 ml or until precipitate is completely dissolved) and lyophilize. Then resuspend the products obtained in 0.1% aqueous TFA/acetonitrile.
Perform HPLC analysis with an analytical C18 column (5 µm, 4 x 150 mm) to analyze each peptide. Use a flow rate of 1 ml/min, and detection at 214 nm and 550 nm.
Perform semi-preparative HPLC on a C18 10 x 250 mm column for peptide purification. Use a flow rate of 4 ml/min, and detection at 214 nm and 550 nm. For all runs, use linear gradients of 0.1% aqueous TFA (solvent A) and 90% acetonitrile, 9.9% water, and 0.1% TFA (solvent B).
Confirm the correct identity of the peptides by MALDI-TOF according to manufacturer’s protocol: fTAT, expected mass: 2,041.17, observed mass: 2040.66. DEAC-K9, expected mass: 1,412.97, observed mass: 1,415.59. Use an α- cyano-4-hydroxycinnamic acid matrix for the MALDI-TOF.
2. Oxidation Reaction: dfTAT Generation
Aerate phosphate buffered saline (PBS) pH 7.4 for 1 hr (at least 5 ml for the reaction below).
Dissolve fTAT (0.3 mg, 1.5 x 10-4 mmol) in aerated PBS pH 7.4 (5 ml). Make sure that the pH is between 7.0 - 7.5 after the addition of the peptide. If not add sodium hydroxide (1 M, in small increments 1 - 5 µl) to bring the pH back up to 7.4.
Note: Oxygen dissolved in PBS acts to oxidize the thiol groups on fTAT and form a disulfide bond.
Nutate the reaction O/N to allow it to react until completion (100% yield based on HPLC analysis). Purify the product using reverse-phase HPLC and analyze by mass spectrometry (MALDI-TOF) as in step 1.9. Expected mass: 4,080.34, observed mass: 4,084.21.
Lyophilize pure dfTAT (0.71 x10-4 mmol) and resuspend in 200 µl water (concentration pure dfTAT = 356.3 µM).
3. Measuring dfTAT Concentration
Resuspend an aliquot of the purified dfTAT (typically 1 µl depending on the amount of peptide purified) in a 149 µl of 50 mM TCEP solution.
Note: In this step dfTAT is reduced to its monomer counterpart fTAT to eliminate the absorbance quenching that occurs due to the close proximity of the TMR fluorophore in dfTAT (the extinction coefficient ε of TMR in dfTAT is reduced in comparison to the ε of TMR in reduced fTAT).
Allow the sample to react for approximately 20 min (analytical HPLC can be used to confirm formation of fTAT).
Add all the solution to the quartz cuvette and measure the absorbance at 556 nm.
Using Beer’s Law ( A= εcl : ε= 91,500 M-1 cm-1) to calculate the concentration of fTAT, determine the concentration of dfTAT, and divide [fTAT] by two.
4. Cellular Delivery Experiments
Seed the cells (HeLa, HDF, etc.) in a dish at a confluency of 80 - 90% (e.g., 8-well or 24-well dish). Grow cells in an appropriate medium (e.g., DMEM supplemented with 10% FBS and Pen/Strep) until 80 - 90% confluency in a 37 °C humidified atmosphere containing 5% CO2.
Wash the cells three times (3X) with PBS (by adding 200 µl of PBS and then removing it three times).
Make a 5 µM working concentration of dfTAT by diluting a stock of dfTAT (in water) in nrL-15 media (for an 8 well dish the total volume should be 200 µl). A concentration of 5 µM dfTAT leads to efficient delivery (high level of cytosolic delivery in more than 90% of cells present in a dish) in most cell types tested to date (Figure 3). However, lower or higher concentrations might be more adequate for cell types with a high or low propensity for penetration. NOTE ABOUT MEDIA: nrL-15 is used for the delivery of dfTAT since it does not contain cysteine which could reduce the disulfide bond in dfTAT. However, our data indicate that both regular L-15 (with cysteine) and DMEM can be used as the delivery media. L-15 contains the reducing amino acid cysteine but cysteine presumably oxidizes in the media to form cystine (DMEM is formulated with cystine). dfTAT therefore remains intact in these media and delivery works at the same efficiency as that obtained with nrL15.
Incubate cells with dfTAT (5 µM) with or without cargo (e.g., EGFP (10 µM)) and keep at 37 °C for 1 hr (incubation time can be reduced but dfTAT typically requires approximately 30 to 45 min to induce endosomal leakage).
Wash cells with heparin (1 mg/ml) in L-15 (3 washes are recommended) to remove dfTAT bound to the plasma membrane of cells.
Incubate cells with cell-impermeable nuclear stain, e.g., Sytox Blue, Sytox Green (2 µM in nrL-15), to determine whether the plasma membrane of cells is compromised (dead cells will be stained while live cells will not) according to manufacturer’s protocol.
Image cells using a fluorescence microscope (100X oil immersion or 20X objective). Image dfTAT using a RFP filter (Ex = 560 ± 20 nm/Em = 630 ± 35 nm). Note: Successful delivery leads to a diffuse fluorescence of dfTAT throughout the cell (assessing the delivery of protein or peptide of interest will depend on application). Staining of nucleoli by fTAT (the reduced product of dfTAT upon cytosolic entry) can be used as an indication that the fluorescence detected is intracellular. fTAT will degrade within few hours. At this point, the fluorescence of the degradation fragments will appear as punctate. This should not be confused for the punctate distribution that is also seen if dfTAT remains unsuccessfully trapped inside endosomes (this can happen if dfTAT is present at too low of a concentration).
5. Controling Concentration of MOI Delivered
Identify the “optimal delivery” concentration required to achieve efficient cytosolic release of dfTAT in the cell type used. Perform fluorescence microscopy on cells incubated with increasing concentrations of dfTAT. The optimal dfTAT concentration is defined as the minimal concentration that leads to clear cytosolic “diffuse” TMR fluorescence in approximately 100% of cells in a culture.
Vary the concentration of MOI used in co-incubation protocol while keeping the concentration of dfTAT constant (e.g., using dfTAT’s optimal delivery concentration). Changing the incubation time to less than 1 hr could also be an option to vary the amount of MOI delivered.
Representative Results
To assess the difference between fTAT and dfTAT, HeLa cells are incubated for 1 hr with each peptide to determine the difference in their cellular localization. The internalization of the two CPP’s was assessed using fluorescence microscopy. Figure 2 shows that fTAT (20 µM) localizes in a punctate distribution. This distribution is consistent with the peptide remaining entrapped inside endosomes. In contrast, the fluorescence signal of dfTAT (5 µM) displays a homogenous distribution throughout the cytosol and nucleus. Figure 3 shows that the cytosolic distribution of dfTAT is observed in a number of different cell lines including COLO 316, NIH 3T3, HaCaT, the difficult to transfect Neuro-2a, MCH58, the primary cell line HDF, DRG-F11 and NCL-H1299. The 20X images shows that a very high percentage (>80%) in a dish display a cytosolic distribution of dfTAT with no cellular toxicity (no Sytox blue nuclear staining).
To determine whether dfTAT-mediated endosomal leakage delivers large proteins into the cytosol of cells. EGFP (26 kDa) is chosen as a model protein. This is because its fluorescence can be used to detect its delivery into the cells by observing the localization of the green fluorescence (if correctly folded). To do this assay, EGFP and dTAT were incubated for 1 hr with cells, as shown in Figure 4, EGFP displayed a cytosolic and nuclear green fluorescent distribution similar to what is observed for dfTAT in more than 90% of cells without observable toxicity.
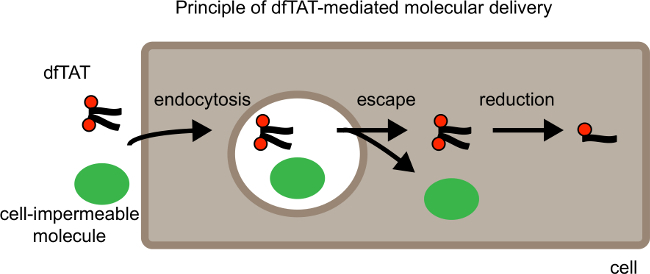 Figure 1. Schematic Showing Principle of dfTAT-mediated Molecular Delivery. From left to Right. Schematic shows dfTAT cellular delivery along with a cell impermeable cargo. First dfTAT induces endocytosis which results in the uptake of dfTAT along with the cargo into endocytic vesicles. In the second step dfTAT escapes from the endocytic vesicles which results in the release of dfTAT and the cell impermeable molecule into the cytosol of cells. The cytosol of mammalian cells is a reductive environment, therefore in the cytosol dfTAT is reduced to its monomer counterpart fTAT. Please click here to view a larger version of this figure.
Figure 1. Schematic Showing Principle of dfTAT-mediated Molecular Delivery. From left to Right. Schematic shows dfTAT cellular delivery along with a cell impermeable cargo. First dfTAT induces endocytosis which results in the uptake of dfTAT along with the cargo into endocytic vesicles. In the second step dfTAT escapes from the endocytic vesicles which results in the release of dfTAT and the cell impermeable molecule into the cytosol of cells. The cytosol of mammalian cells is a reductive environment, therefore in the cytosol dfTAT is reduced to its monomer counterpart fTAT. Please click here to view a larger version of this figure.
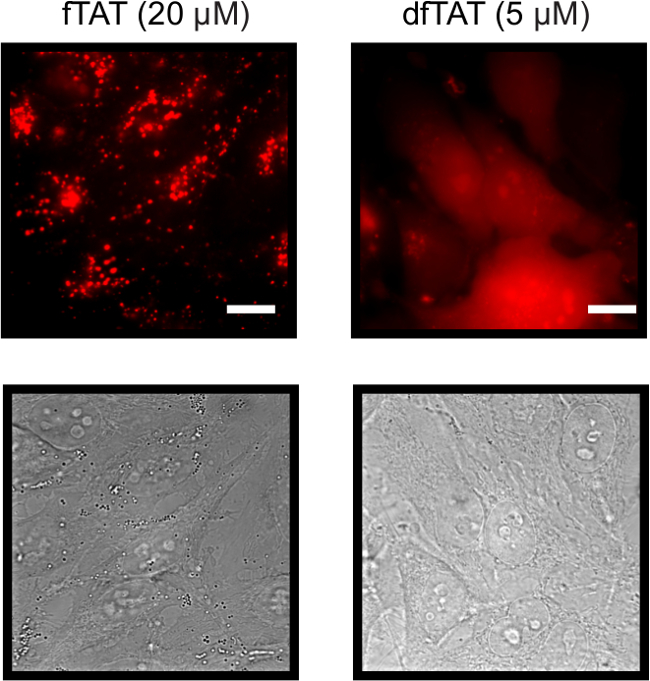 Figure 2. Fluorescence and Bright Field Images of Both HeLa cells Incubated with 20 µM fTAT (left panel) and 5 µM dfTAT (right panel) using a 100X Objective. fTAT monochrome images shows cells that exhibit a fluorescence punctate distribution while dfTAT images shows cells displaying a homogenous cytosolic and nuclear fluorescence distribution. Scale bar: 10 µm. Please click here to view a larger version of this figure.
Figure 2. Fluorescence and Bright Field Images of Both HeLa cells Incubated with 20 µM fTAT (left panel) and 5 µM dfTAT (right panel) using a 100X Objective. fTAT monochrome images shows cells that exhibit a fluorescence punctate distribution while dfTAT images shows cells displaying a homogenous cytosolic and nuclear fluorescence distribution. Scale bar: 10 µm. Please click here to view a larger version of this figure.
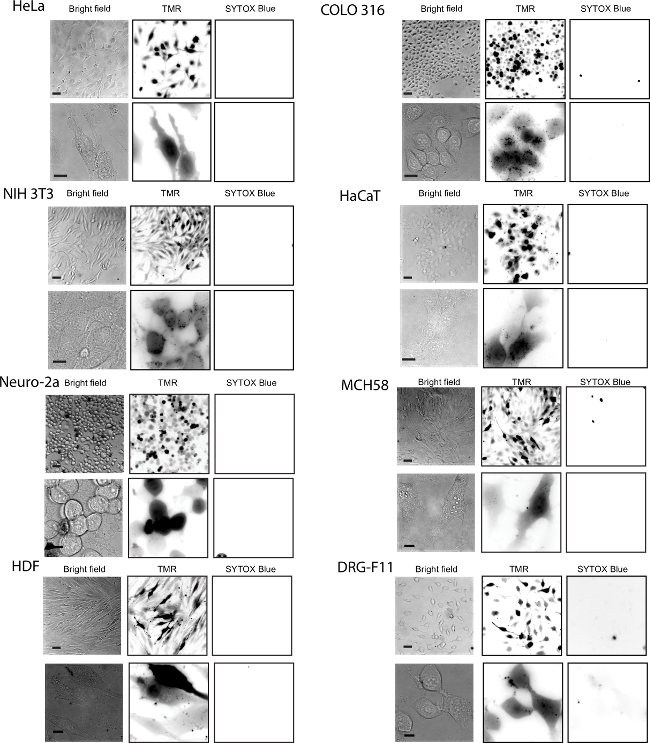 Figure 3. Efficient Delivery of dfTAT into Live Cells was Achieved in Multiple Cell Lines. The cell lines tested were the following: HeLa, NIH 3T3, COLO 316 and HaCaT, Neuro-2a, MCH58, HDF, DRG-F11 and NCL- H1299. Cells were incubated with 5 µM dfTAT for 1 hr followed by a washing step according to protocol and imaged. The fluorescence signal detected was in the cytosol and nucleus of cells (top panel: 20X objective, bottom panel: 100X objective). The cell-impermeable nuclear stain SYTOX Blue was used to assess cell viability after dfTAT treatment. Scale bars, 20X objective: 50 µm; 100X objective: 10 µm. Please click here to view a larger version of this figure.
Figure 3. Efficient Delivery of dfTAT into Live Cells was Achieved in Multiple Cell Lines. The cell lines tested were the following: HeLa, NIH 3T3, COLO 316 and HaCaT, Neuro-2a, MCH58, HDF, DRG-F11 and NCL- H1299. Cells were incubated with 5 µM dfTAT for 1 hr followed by a washing step according to protocol and imaged. The fluorescence signal detected was in the cytosol and nucleus of cells (top panel: 20X objective, bottom panel: 100X objective). The cell-impermeable nuclear stain SYTOX Blue was used to assess cell viability after dfTAT treatment. Scale bars, 20X objective: 50 µm; 100X objective: 10 µm. Please click here to view a larger version of this figure.
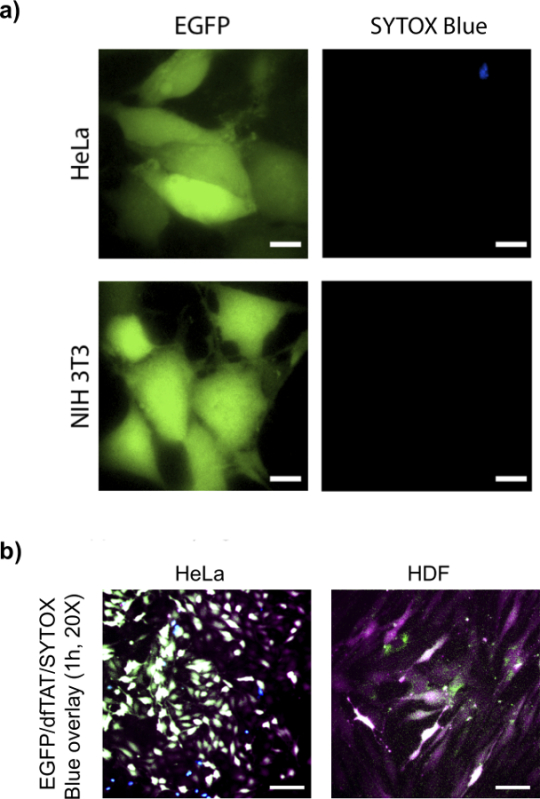 Figure 4. dfTAT Delivers Intact EGFP into Different Cell Lines. (A) HeLa (top panel) and NIH 3T3 (bottom panel) cells were incubated with EGFP (10 µM) and dfTAT (5 µM) for 1 hr and following step were performed according to protocol. Images show a homogenous cytosolic fluorescence distribution of EGFP in both cell lines. Scale bars, 10 µm. (B) HeLa and primary HDF cells were incubated with EGFP (10 µM) and dfTAT (5 µM) according to protocol. 20X images show a homogenous cytosolic fluorescence distribution of EGFP and dfTAT in HeLa and primary HDF cells. Overlay (pseudocolor: white) indicate presence of dfTAT (pseudocolor: purple), EGFP (pseudocolor: green) in the cytosolic space of both HeLa and HDF cells. Sytox blue (shown in blue) was used to asses for cell viability. Scale bars: 50 µm. Please click here to view a larger version of this figure.
Figure 4. dfTAT Delivers Intact EGFP into Different Cell Lines. (A) HeLa (top panel) and NIH 3T3 (bottom panel) cells were incubated with EGFP (10 µM) and dfTAT (5 µM) for 1 hr and following step were performed according to protocol. Images show a homogenous cytosolic fluorescence distribution of EGFP in both cell lines. Scale bars, 10 µm. (B) HeLa and primary HDF cells were incubated with EGFP (10 µM) and dfTAT (5 µM) according to protocol. 20X images show a homogenous cytosolic fluorescence distribution of EGFP and dfTAT in HeLa and primary HDF cells. Overlay (pseudocolor: white) indicate presence of dfTAT (pseudocolor: purple), EGFP (pseudocolor: green) in the cytosolic space of both HeLa and HDF cells. Sytox blue (shown in blue) was used to asses for cell viability. Scale bars: 50 µm. Please click here to view a larger version of this figure.
Discussion
The cells used for dfTAT delivery should not be overly confluent (>90% confluency) since this might affect the delivery efficiency. The cells should also be healthy: dead cells in the culture can release apoptotic fragments with which dfTAT can interact (e.g., DNA from degraded nuclei). This in turn can interfere with delivery efficiency and the quality of imaging. Cells should be washed thoroughly to remove FBS before adding dfTAT. BSA present in FBS can bind to dfTAT and this can lower the delivery efficiency of dfTAT. Washes should however be performed with care as excessive force might cause adherent cells to detach from the culture dish or cause a stress that results in lower endocytic uptake.When delivering a protein/ peptide using dfTAT, the protein/peptide stock solution sample should be sufficiently concentrated to avoid excessive dilution of the nrL-15 media during incubation: e.g., adding on 5 - 10 µl of sample to 200 µl nrL-15 is recommended.
Fluorescently-labeled cell penetrating peptides can display a light-inducible membrane-disrupting activity.15 This is the case for dfTAT when high intensity light doses are used. It can manifest itself by rupture of intracellular organelles (e.g., endosomes, mitochondria), cell surface blebbing, and cell death. To minimize these effects, care should be taken to keep light exposure to a minimum (standard conditions required for imaging by confocal or epifluorescence are typically tolerated).
When delivering MOI such as proteins, one is confronted to the problem that every macromolecule is unique and that, consequently, every delivery experiment might require fine-tuning (more so than in the case of DNA transfection where the molecules delivered are always a negatively charged polymer made of A, T, G, and C). Troubleshooting is therefore important and several aspects should be considered. First, proteins with very low pIs can bind electrostatically to dfTAT and inactivate it. In contrast, proteins with very high pIs might compete with dfTAT for binding with negatively charged glycosaminoglycans on the cell surface. This might then reduce endocytosis and diminish uptake below endosomolytic thresholds. In both cases, a possible solution to this problem is increasing dfTAT concentration.
As one would expect, due to the presence of cellular proteases (such as cathepsins16) along the endocytic pathway, degradation of the MOI during delivery is a concern. While dfTAT can deliver intact proteins, the amount of protein that is fully or partially degraded during the delivery process has not been established. This is again an issue that is MOI-dependent and that should be monitored depending on the application pursued.
dfTAT is a highly efficient delivery agent. The molecular basis for dfTAT’s activity remains however unclear. In particular, the structural or chemical features that are required to achieve endosomal escape have not been identified. It is therefore currently not possible to predict how much the structure of dfTAT can be modified without altering delivery efficiencies. We have already established that the disulfide bond present in dfTAT can be replaced by a non-reducible linker without deleterious effects. Additional structure-activity relationships are currently being established.
Our data suggest that it is possible to decrease the incubation time of the peptide to less than 1 hr (as low as 5 min has been performed). However, the release of dfTAT inside cells is not observed in a large population of cells until approximately 15-30 min. This suggests that short incubation time may be sufficient for dfTAT endocytosis or cellular uptake. However time is required for endosomal maturation necessary for dfTAT escape from the endocytic pathway.
Finally, we have established that multiple MOI can be delivered at once (using the protocols presented herein, with the only exception that more than one MOI is used during co-incubation). In addition, dfTAT-mediated delivery can be repeated (delivery steps repeated 20 min apart have been tested). This did not impact cell viability. We therefore believe that the delivery protocols presented herein are extremely convenient, efficient, and innocuous to cells.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This article was supported by Award Number R01GM087227 from the National Institute of General Medical Sciences, the Norman Ackerman Advanced Research Program, and the Robert A. Welch foundation (Grant A-1769).
References
- Didenko VV, Ngo H, Baskin DS. Polyethyleneimine as a transmembrane carrier of fluorescently labeled proteins and antibodies. Anal Biochem. 2005;344:168–173. doi: 10.1016/j.ab.2005.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi T, et al. Direct and Rapid Cytosolic Delivery Using Cell-Penetrating Peptides Mediated by Pyrenebutyrate. ACS Chem Biol. 2006;1:299–303. doi: 10.1021/cb600127m. [DOI] [PubMed] [Google Scholar]
- Morris MC, Depollier J, Mery J, Heitz F, Divita G. A peptide carrier for the delivery of biologically active proteins into mammalian cells. Nat Biotech. 2001;19:1173–1176. doi: 10.1038/nbt1201-1173. [DOI] [PubMed] [Google Scholar]
- Zhang H, et al. Reprogramming of somatic cells via TAT-mediated protein transduction of recombinant factors. Biomaterials. 2012;33:5047–5055. doi: 10.1016/j.biomaterials.2012.03.061. [DOI] [PubMed] [Google Scholar]
- Torchilin V. Intracellular delivery of protein and peptide therapeutics. Drug Discov Today: Technol. 2005;5:e95–e103. doi: 10.1016/j.ddtec.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Chakravarty P, Qian W, El-Sayed MA, Prausnitz MR. Delivery of molecules into cells using carbon nanoparticles activated by femtosecond laser pulses. Nature nanotechnol. 2010;5:607–611. doi: 10.1038/nnano.2010.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczmarczyk SJ, Sitaraman K, Young HA, Hughes SH, Chatterjee DK. Protein delivery using engineered virus-like particles. Proc Natl Acad Sci U S A. 2011;108:16998–17003. doi: 10.1073/pnas.1101874108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan C, Lu B, Chen H, Bishop C. Reprogramming human fibroblasts using HIV-1 TAT recombinant proteins OCT4, SOX2, KLF4 and c-MYC. Mol Biol Rep. 2010;37:2117–2124. doi: 10.1007/s11033-009-9680-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fittipaldi A, et al. Cell Membrane Lipid Rafts Mediate Caveolar Endocytosis of HIV-1 Tat Fusion Proteins. J Biol Chem. 2003;278:34141–34149. doi: 10.1074/jbc.M303045200. [DOI] [PubMed] [Google Scholar]
- Lee YJ, Erazo-Oliveras A, Pellois JP. Delivery of macromolecules into live cells by simple co-incubation with a peptide. Chembiochem. 2010;11:325–330. doi: 10.1002/cbic.200900527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angeles-Boza AM, Erazo-Oliveras A, Lee YJ, Pellois JP. Generation of endosomolytic reagents by branching of cell-penetrating peptides: tools for the delivery of bioactive compounds to live cells in cis or trans. Bioconjugate chem. 2010;21:2164–2167. doi: 10.1021/bc100130r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walev I, et al. Delivery of proteins into living cells by reversible membrane permeabilization with streptolysin-O. Proc Natl Acad Sci U S A. 2001;98:3185–3190. doi: 10.1073/pnas.051429498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erazo-Oliveras A, et al. Protein delivery into live cells by incubation with an endosomolytic agent. Nat methods. 2014;11:861–867. doi: 10.1038/nmeth.2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser E, Colescott RL, Bossinger CD, Cook PI. Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Anal Biochem. 1970;34:595–598. doi: 10.1016/0003-2697(70)90146-6. [DOI] [PubMed] [Google Scholar]
- Muthukrishnan N, Johnson GA, Lim J, Simanek EE, Pellois JP. TAT-mediated photochemical internalization results in cell killing by causing the release of calcium into the cytosol of cells. Biochim biophys acta. 2012;1820:1734–1743. doi: 10.1016/j.bbagen.2012.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lautwein A, et al. Human B lymphoblastoid cells contain distinct patterns of cathepsin activity in endocytic compartments and regulate MHC class II transport in a cathepsin S-independent manner. J Leukoc Biol. 2004;75:844–855. doi: 10.1189/jlb.0803367. [DOI] [PubMed] [Google Scholar]