

Abstract
This protocol describes the labeling of epidermal growth factor receptor (EGFR) on COS7 fibroblast cells, and subsequent correlative fluorescence microscopy and environmental scanning electron microscopy (ESEM) of whole cells in hydrated state. Fluorescent quantum dots (QDs) were coupled to EGFR via a two-step labeling protocol, providing an efficient and specific protein labeling, while avoiding label-induced clustering of the receptor. Fluorescence microscopy provided overview images of the cellular locations of the EGFR. The scanning transmission electron microscopy (STEM) detector was used to detect the QD labels with nanoscale resolution. The resulting correlative images provide data of the cellular EGFR distribution, and the stoichiometry at the single molecular level in the natural context of the hydrated intact cell. ESEM-STEM images revealed the receptor to be present as monomer, as homodimer, and in small clusters. Labeling with two different QDs, i.e., one emitting at 655 nm and at 800 revealed similar characteristic results.
Keywords: Bioengineering, Issue 103, quantum dot, specific protein label, fluorescence microscopy, environmental scanning electron microscopy, ESEM, scanning transmission electron microscopy, STEM
Introduction
A new approach was introduced recently, to image whole cells with labeled proteins in liquid state using STEM1-3, or correlative fluorescence microscopy and STEM4-7. This methodology is capable of studying the spatial distribution of membrane proteins and protein complexes including their stoichiometry at the single molecule level in the intact plasma membranes of whole cells. Here, we describe a protocol involving a two-step specific labeling of receptor proteins with fluorescent quantum dots (QDs)8, and correlative light microscopy and ESEM-STEM. As example, we studied the EGFR, a membrane protein that belongs to the protein kinase superfamily. Upon ligand binding, the receptor activates and then forms a dimer with another activated EGFR; the subsequent signal cascade drives cell proliferation9. Mutations leading to abnormal EGFR expression or activity play a central role in many types of cancer10. Studying the spatial arrangement of this membrane receptor in whole cells provides important context information about its function and possible changes thereof11-14. But it remains challenging to probe the distribution of EGFR monomers, dimers, and clusters directly on a (sub-)cellular level with biochemical- or conventional microscopy methods15. Our method is capable of visualizing EGFRs with a spatial resolution of a few nanometers such that the locations of proteins in a complex can be determined. Most importantly, the obtained information about the stoichiometric distribution relates to the native situation of the protein in the intact cell.
In order to achieve a short distance between the label and the receptor, EGFR was directly labeled via its ligand EGF, using a biotin-streptavidin bond to a QD. This label can be detected both with fluorescence- and electron microscopy. We have used this label for correlative imaging of COS7 cells in liquid enclosed in a microfluidic chamber previously1,16,17. However, on account of a multiple of streptavidin molecules per QD, this label may induce receptor clustering, leading to a low labeling efficiency or rather expensive experiments, and it is not possible to conclude on the stoichiometry of the protein under investigation. Label induced receptor clustering can be avoided altogether by employing the two-step labeling procedure presented here, resulting in a probe comprising biotinylated EGF to which only one streptavidin-quantum dot (STR-QD) is coupled. The cells are first incubated with biotinylated EGF, and then fixed. The fixation step determines the time point at which protein processes are stopped (depending on the speed of fixation). STR-QD is applied thereafter as second step of the labeling protocol. Clustering does not take place since dynamic membrane motion of the proteins is hindered once the proteins are fixed. Moreover, the STR-QD solution, which is the most expensive component of the experiment, can be applied in an optimized low concentration, and this second step of labeling is not time-crucial, i.e., the QD can be coupled in several minutes.
To study the stoichiometry of EGFR as distributed in whole cells, cellular samples were prepared on silicon microchips18. After applying the two-step QD labeling, and the recording of fluorescence microscopy images, the cells were rinsed with pure water, and imaged in hydrated state using ESEM-STEM19. The resulting correlative images provide information about the cellular EGFR distribution, and the stoichiometry in their natural context of hydrated cells. A similar method was used to study HER2 proteins in breast cancer cells in a recent study7.
Protocol
1. Preparation of Labeling and Fixation Reagents
Prepare labeling buffer (BSA-GEL-PBS) using autoclaved phosphate buffered saline, pH 7.4 (PBS). Supplement with 1% BSA (bovine albumin fraction V, biotin-free) and 0.2% gelatin (GEL, from cold water fish skin) and vortex/shake well. Note: Can be stored for ~5 days at 4 °C.
Prepare washing buffer (BSA-PBS) from autoclaved phosphate buffered saline, pH 7.4. Supplement with 1% BSA and vortex/shake well. Note: Can be stored for ~5 days at 4 °C.
Prepare 50 mM borate buffer (BB) from 200 mM boric acid stock solution, pH adjusted to 8.3 with 200 mM sodium tetraborate. Note: Can be stored for ~12 months at 4 °C.
Prepare 0.1 M cacodylate buffer (CB) from 0.2 M sodium cacodylate stock solution, pH adjusted to 7.4, supplemented with 0.1 M sucrose. Note: Opened stock solution can be stored for ~5 days at 4 °C.
Prepare 0.1 M glycine in PBS (GLY–PBS). Note: Can be stored for ~2 months at 4 °C.
Prepare 3% paraformaldehyde in CB (3% PFA) from freshly made or opened vial of 16% PFA, stock solution, EM grade. Note: Opened stock solution can be stored for ~5 days at 4 °C.
Prepare 2% glutaraldehyde in CB (2% GA) from freshly made (or opened vial of) 25% GA stock solution, EM grade. Note: Opened stock solution can be stored for ~5 days at 4 °C.
- Prepare 300 nM EGF-biotin labeling solution
- Dilute 6 µM EGF-biotin stock solution in BSA-GEL-PBS, use a volume of 100-200 µl per microchip. Use within a few hr.
- Prepare 10 nM STR-QD labeling solution
- Centrifuge the streptavidin-QD stock solution for 4 min at 8,000 x g, take supernatant and dilute 1:10 in BB. Different types of QDs can be used, for example, QD655, or QD800. Dilute to final concentration in BSA-GEL-PBS, use 75-100 µl per microchip. Use within a few hr.
2. Preparation of Silicon Nitride Membrane Microchips
Clean a microscope glass slide, as used for light microscopy, with a clean room tissue and high performance liquid chromatography (HPLC) grade ethanol.
In a laminar airflow workbench with a low dust level, open a 100 mm diameter Petri dish, and lay the cleaned microscope glass into the dish; leave the dish open.
With clean tweezers, preferentially coated with carbon fiber or other coating, take 4-10 microchips with thin (50 nm) silicon nitride membranes, one after the other, out of their storage box and onto the glass slide. Grab the microchips gently at their sides and avoid touching the fragile upper side, consisting of the thin silicon nitride membrane. Take great care to always keep the membrane side up and do not touch it. Note: The microchips should be rectangular and have flat edges perpendicular to their silicon nitride covered face, so that they can be gripped easily.
Close the lid of the petri dish and bring it to a fume hood. Get a new clean room tissue and deposit it beside the petri dish. Prepare two clean 200 ml glass beakers by filling one with 50-70 ml acetone, and the other with a similar volume of ethanol, both of HPLC grade. Transfer the microchips from the glass slide first onto the clean room tissue.
From the tissue, transfer microchips one by one into the first beaker with acetone for removing the protective resist layer. Wait 2 min, while gently moving the beaker a few times to ensure that the microchips are well rinsed.
Transfer the microchips directly into the beaker with ethanol, without letting them dry out. Wait 2 min. Then transfer the beaker to the laminar airflow workbench. Get a new clean room tissue.
Transfer the chips onto the tissue and let them dry for a few min. Transfer the microchips onto the glass slide in the Petri dish, close the dish, and bring it to the plasma cleaner.
Deposit the glass slide with the microchips into the plasma cleaner. Run a 5 min cleaning program to render the surface of the silicon nitride membrane hydrophilic. Note: Suitable settings applied for our plasma cleaner are: 70 mTorr, gas flow of 11.5 sccm for O2 and 35.0 sccm for Ar, 50 W forward radio frequency (RF) target, 5 W RF range and max. reflected RF.
Put the glass slide with the microchip back into the petri dish, and close its lid. Take care to keep the microchips sterile from now on.
Examine the window areas of the microchips under a light microscope for possible membrane ruptures or remaining dirt particles.
Bring the petri dish to the laminar airflow workbench.
Place the following items in the laminar airflow workbench: tubes/bottles containing sterilized 0.01% poly-L-lysine solution (PLL, mol. wt 150,000–300,000, sterile-filtered, and cell culture tested), HPLC grade water, Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum, DMEM without serum, a new clean room tissue, a sterile 24-well plate (a 12-well plate can also be used), a sterile 96-well plate, sterile pipet tips, a pipette, and flat, rounded tip, tweezers.
Take the 24-well plate, and fill one well with PLL, and two wells with HPLC grade water, use an approx. volume of 1 ml per well. Take the 96-well plate and fill for every microchip two wells with serum supplemented DMEM and one well with serum-free DMEM, use an approx. volume of 200 µl per well.
From now on use tweezers with sterile tips to handle the microchips. Achieve this, for instance, by briefly flaming and subsequently cooling off the tips of the tweezers.
First transfer the microchips from the glass slide onto the clean room tissue. From the tissue, transfer up to six microchips into the PLL-filled well, incubate the microchips for 5 min. Note: For an experiment with more microchips, fill additional wells in the 24-well plate.
Subsequently rinse the microchips by placing them, one-by-one, into the first water-filled well, and from there into the second water-filled well. Don't leave the microchips in the water longer than about a minute to avoid removal of PLL.
Finally place them individually into the DMEM-filled wells of the 96-well plate. Store the 96-well plate in the CO2 incubator until the cell suspension is prepared.
3. Preparation of Cells on Microchips
Perform a subculture of COS7 cells to prepare a cell suspension with a concentration of approx. 5 × 105 cells/ml. Make sure the cells are mono-dispersed.
Take the 96-well plate with the microchips and add one droplet of cell suspension to each microchip. Wait 5 min.
Start to examine the window areas of the microchips with an inverted microscope to choose the right moment for transferring it into a new well with serum-free DMEM. Note: A good density of cells is achieved with approximately one cell per 2,500 µm2 area, corresponding to 24 COS7 cells on a window size of 150 x 400 µm. Higher cell densities have the risk to hinder maximal flattening of the cells. Be aware that after a cell has settled down onto the SIN membrane, it needs 3-5 min to develop enough adhesion to resist floating off during transfer into the new well.
Transfer microchip to the next well when sufficient cells have adhered onto the window. Keep the microchips always with the cells facing up and avoid any touching of this side.
After the transfer, examine each microchip window again. If too many cells were washed off and the number of remaining cells is insufficient, re-triturate the cell suspension (to break up newly formed cell clumps) and repeat steps 3.1-3.5.
When all transferred microchips have enough adhering cells on their window areas, transfer the chips into the last well with serum-free DMEM. Place the 96-well plate back into the CO2 incubator and let the cells recover and flatten out for several hr, best O/N.
4. EGFR Labeling, Fixation, and Fluorescence Microscopy
For the following steps, use a 96-well and a 24-well plate. Pre-fill wells with the respective solutions, and use one row or column per microchip. The 24- well plate is used under the fume hood for the fixation steps with CB, PFA and GA. Always transfer microchips with the cells facing up and avoid any touching of this side.
Rinse microchips by placing the microchips in a well with fresh BSA-GEL-PBS, then place 96 well plate in incubator at 37 °C for 5 min.
Incubate with 300 nM EGF-biotin solution for 3 min at 37 °C. Because the cells will start endocytosis of EGF-labeled EGFRs, proceed as fast as possible to step 4.4.
Rinse 3 times with PBS, and 1 time with CB (now the 96 well plate is again at RT).
Incubate in 3% PFA for 10 min.
Rinse chips 1 time with CB, and 3 times with PBS.
Incubate in GLY-PBS for 2 min, rinse 2 times with 1 time PBS.
Incubate in 10 nM STR-QD for 10 min.
Rinse 4 times with BSA-PBS.
Submerge a microchip in a glass-bottom 35 mm dish pre-filled with BSA-PBS at RT.
- Acquire differential interference contrast (DIC) images and fluorescence images of QD-labeled cells.
- Use a 40X air objective for overview images of cells labeled with QD655. Use an oil immersion objective (for example, 63X) to allow the most efficient collection of the emitted fluorescence light; this is especially needed when imaging cells labeled with QD800.
- Use a filter cube that exposes the QDs to fluorescence excitation light between 340 and 380 nm, and allows the collection of emitted light above 420 nm suitable for most QDs (Filter cube A in this case).
- Minimize the light intensity to avoid too intense light that might damage the sample. Set the exposure time to at least 300 msec, so that the signals of all QDs are captured, noting that the fluorescence of QDs has a blinking behavior.
- Adjust the light intensity, and the amplification to provide bright images without visible image noise, while minimizing the light intensity. Note: Alternatively, shorter exposure time can be used if image series are taken from the same region. These series can be processed to yield a maximum projection image, leading to averaging and thus noise reduction of the resulting image.
Place the imaged microchip back (cells facing up) into a well of a 96-well plate filled with BSA-PBS.
Rinse microchips 1 times in CB.
Incubate in 2% GA for 10 min.
Rinse 1 time in CB, and 3 times with BSA-PBS.
Store microchips until ESEM imaging, in BSA-PBS pre-filled wells of a new 96-well plate. For every microchip fill a row of four wells with HPLC-grade water. Note: The samples can be kept at 4 °C for several weeks, however, the best results are achieved when the complete imaging series is performed within two days. QDs will slowly start to detach from the sample, it is therefore not recommended to wait more than 10 days before the ESEM imaging is done. Note: Fluorescence images are stored for future analysis. The positions of the cells in the images can easily be correlated with electron microscopy images via localizing the corners of the SiN window and measuring the positions relative to the corners. Note: It is advisable to print high quality fluorescence images and use them for orientation purposes when recording the ESEM images.
5. Wet ESEM-STEM of Whole Cells
- Prepare the ESEM for wet STEM
- Mount the gaseous secondary electron detector (GSED), and the Peltier stage, containing the STEM detector (located under the sample).
- Start the flow of cooling water through the stage, and set the temperature to 3 °C.
- Set the stage x-y coordinates to zero, and adjust the working distance to approximately 6 mm.
During the ESEM imaging, keep the 96-well plate with the microchips cool, for instance on frozen cooling elements, in a Styrofoam box.
For loading a sample into the precooled ESEM-STEM stage, take the 96-well plate out of the cooling container and place it close to the opened ESEM stage. Put a new clean room tissue beside it.
Quickly rinse a microchip (with the cells facing up) by dipping it four times, each time for about a second, into a well filled with HPLC-grade water.
Blot the backside of the microchip briefly on the clean room tissue and place the microchip into the precooled stage. Make sure that the cells remain wet, this can be recognized by a reflective surface (drying results in a dull surface).
Wet the sample surface with 3 µl of cooled HPLC-grade water (make sure not to touch the surface with the pipet tip), and fix the sample in the stage.
Place three additional 3 µl water droplets on the stage close to the sample.
Close the specimen chamber
Purge the specimen chamber by cycling the pressure five times between 800 and 1,500 Pa. End the cycling with 800 Pa.
Switch the electron beam (30 kV) on and examine the sample under the lowest magnification with both detectors, i.e., the GSED, and the STEM detector to search for the location of the microchip window. Note: If the water layer is too thick, the window may not be visible. In this case, start lowering the pressure to 760 Pa and wait a few min. The window first becomes visible with the GSED.
Position the SiN membrane window in the center of the image by moving the specimen stage and reduce the pressure to 750-720 Pa. Note: When the water layer is thin enough, the window also becomes visible with the STEM detector.
From now on use the dark field segment of the STEM detector to record images. First, acquire one or two images showing all cells on the complete window area.
Compare these ESEM images with the light microscopy images, locate the corners of the SiN window.
Correlate the coordinate frames of both microscopy modalities by comparing the magnification, rotation, and possible mirroring. Locate the same cells in both images.
Select very small dirt particles or a cell region with small features to adjust imaging settings, such as the eucentric height, the fine focus and the correction of lens stigmatism. Work at a magnification of at least 50.000X, use an electron probe current around 0.6 nA, a pixel-dwell time of 30 µs, and an image size of 1024 x 884 pixels. Note: Other settings may also be used as long as an increase of the electron dose is avoided. Note: Even when working very clean, the chip surfaces will in practice always contain some dirt particles, because the protocol is not executed in a clean room environment.
To record STEM images from the QD-labeled cells, refer to the fluorescence images and choose a cell of interest. Note: It is best to start with a cell showing strong QD signals.
Record a STEM overview image of this cell.
Go to a peripheral region where the cell border is visible and zoom in to reach 50.000X magnification.
Record images showing individual QDs, use pixel dwell times of 20-30 µsec. In order to avoid radiation damage, image every region only once with high magnification. Note: Usually, at this stage both, the focus and the correction of the stigmatism, need some fine adjustment.
After the recording of a sufficient number of high magnification images, zoom-out, and acquire another STEM overview image, depicting the regions just recorded with high-magnification as dark rectangles. Note: These overview images will later serve to correlate ESEM and fluorescence images and to determine the precise location of the high magnification images within the cellular context.
Proceed to the next cell of interest, start with an overview image, record a series of high magnification images, and end with an overview image.
At the end of an ESEM imaging session, increase the pressure to 850-900 Pa before starting to vent the chamber. Note: This increase of pressure improves the chance of recovering the microchip with a still wetted surface; however, this cannot always be achieved. A sample that has never been dried out can be restored in BSA-PBS and, if desired, reinvestigated in the ESEM at a later point in time.
Representative Results
Figure 1 and 2 show representative images of QD655 labeled, membrane-bound EGFR visualized in intact, fully hydrated COS-7 cells. The DIC image in Figure 1 a gives an impression of the membrane topography of the cells, and the corresponding fluorescence image in Figure 1b depicts the distribution of EGFR after 3 min of EGF-Biotin incubation. Figure 1A and B are each stitched together from two images recorded with a 40X air objective. The EGF activated EGFR is distributed over the entire cellular surface. In most cells a slight enhancement of fluorescence (Figure 1B) can be seen at the cell edges, indicating a locally increased occurrence of EGFR20. Control experiments conducted using a similar protocol verified specific EGFR labeling (data not shown). These controls included: 1) a control labeling without previous incubation of the cells with EGF-biotin, i.e., only incubation with STR-QDs, and 2) an incubation with non-biotinylated EGF and STR-QDs.
The three cells marked with rectangles were further investigated with ESEM-STEM. The ESEM-STEM images depicted in Figure 1C-E show low magnification overviews of these cells. These transmission images reflect whole cells in hydrated state with a thin layer of water residing over the cell placed on a silicon microchip with silicon nitride (SiN) viewing window. The vacuum chamber in the microscope contained water vapor, and the balance between sample temperature and pressure was adjusted to maintain a thin layer of liquid over the cells. The individual cells can be easily recognized on account of their shape and location on the SiN membrane window. The low-magnification ESEM-STEM images reveal fine structures, such as filopodia, extending from the cell edge towards neighboring cells, and some structures inside the thinner cell regions. Thick central cellular regions including the nucleus appear white because transmission through the sample is not possible for electrons of the used energy (30 keV).
High-resolution ESEM-STEM images were recorded in the thinner, peripheral regions. The spatial resolution for ESEM-STEM of nanoparticles in the thin regions of cells in a thin liquid layer was determined in a previous study6 to amount to 3 nm. Four high-resolution images shown in Figure 2A-D were recorded at the locations of the small rectangles in Figure 1C-E. The used magnification was sufficient to discern individual QDs, appearing as bright, bullet-shaped rods, each bound to an individual EGFR. The QD655 has typical dimensions of 6 x 14 nm2.
Figure 2A (corresponding to the rectangular area indicated on the cell in Figure 1C) shows QD labels on a membrane fold diagonally crossing the image. This membrane structure has a higher EGFR density than the surrounding membrane regions. At several locations, two labels were at close proximity. Two examples with distances of 20 and 24 nm are indicated with arrowheads. These pairs of labels are interpreted as belonging to EGFR dimers. Figure 2B gives an example from the edge of a cell (Figure 1D, left rectangle), EGFR monomers and dimers can be seen as well. Figure 2C (Figure 1C, right rectangle) was recorded of a region with a lower fluorescence signal, i.e., lower EGFR density. Nevertheless, EGFR was also found here in dimers as well. In addition, two clusters of 10 or 11 EGFRs were present (see ellipses). Figure 2D shows another example of a membrane fold (Figure 1E) with smaller clusters including 5-6 EGFRs, in addition to several monomers and dimers.
As an example of the kind of information that can be obtained from this data, the pair correlation function21 g(x) was determined for all label positions in Figure 2. Note that this analysis is not part of this protocol and the procedure was described elsewhere6,7. g(x) is a measure of the chance of a particle to be found within a certain radial distance x from a reference particle. g(x) = 1 represents a random distribution, and a larger value is evidence for clustering. The positions of a total of 210 labels were automatically detected in the four images of Figure 2A-D, and g(x) was calculated using a bin size of 5 nm and a smoothing filter with a bandwidth of 10 nm. The g(x) curve (Figure 3) shows a preferred center-to-center QD distance of 25 nm. EGFRs are thus not randomly oriented but a significant fraction of them resides at this preferred distance. From an approximate molecular model6 (Figure 3 inset) we estimate that the center-to-center distance between the QD and the EGF binding pocket amounts to ~14 nm, and center-to-center distance between the two QDs attached to a EGFR dimer to be ~27 nm (this value is likely to vary by a few nanometers due to the flexibility of the linker). The preferred label distance is thus consistent with the expected label distance for the EGFR dimer within the precision of the method. The g(x) curve is larger than unity for distance of up to 300 nm, indicating the presence of clusters, consistent with the data. This analysis shows that the stoichiometry of the EGFR can be studied with our method.
Figure 4 shows a similar result as Figure 2, except that the labeling was performed with EGF-QD800, and a 63X oil immersion objective was used for the fluorescence image. This data confirms that these typical results are also found when the EGFR labeling is done with QDs, which emit in the near red spectrum. Figure 4A is the fluorescence image of a representative cell, Figure 4B shows the same cell in ESEM-STEM overview mode and Figure 4C is a 150,000 x magnification image, recorded at the upper membrane border of the cell (see rectangle in Figure 4B). Similar to Figure 2A and D, the images capture QD-labeled EGFR on a membrane fold and shows monomeric, dimeric, and clustered receptors. Note that the electron dense QD core appears somewhat smaller and rounder than the cores of QDs emitting at 655 nm, consistent with their smaller core size22 of ~5 nm.
 Figure 1. Correlative DIC, fluorescence and ESEM-STEM of EGF-QD655 labeled EGFR on fully hydrated COS-7 cells. (A) DIC image of the cells grown on the SiN membrane window area, giving a topographic impression of the plasma membrane. (B) Fluorescence image showing cells on the centrally located SiN membrane window (marked by the dashed rectangle). (C-E) Three ESEM-STEM low magnification images from the cells marked with rectangles in A and B. These cell overview images serve to determine the precise location of subsequently recorded high-resolution images. The location of four high-resolution images (shown in Figure 2A-D) are marked with white rectangles. Please click here to view a larger version of this figure.
Figure 1. Correlative DIC, fluorescence and ESEM-STEM of EGF-QD655 labeled EGFR on fully hydrated COS-7 cells. (A) DIC image of the cells grown on the SiN membrane window area, giving a topographic impression of the plasma membrane. (B) Fluorescence image showing cells on the centrally located SiN membrane window (marked by the dashed rectangle). (C-E) Three ESEM-STEM low magnification images from the cells marked with rectangles in A and B. These cell overview images serve to determine the precise location of subsequently recorded high-resolution images. The location of four high-resolution images (shown in Figure 2A-D) are marked with white rectangles. Please click here to view a larger version of this figure.
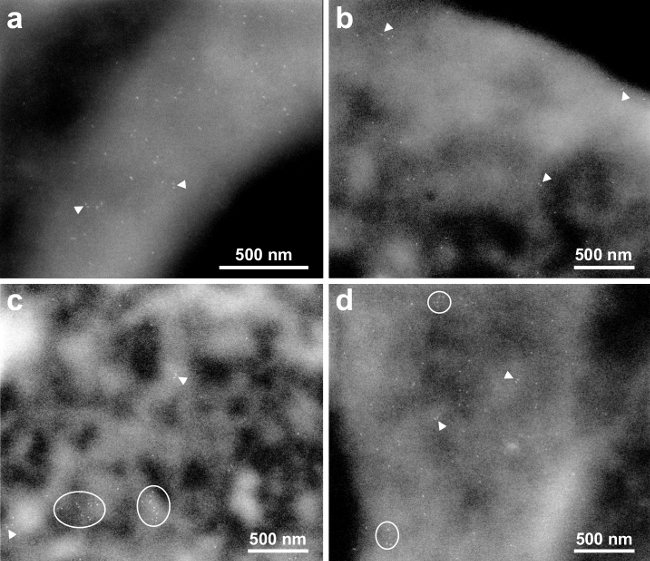 Figure 2. High-resolution ESEM-STEM images depicting membrane bound EGFRs. (A) Micrograph at 75,000X magnification of the area marked in Figure 1B. Many monomers are visible. Several dimers are indicated with the arrowheads. (B) and (C) images acquired at 50,000X magnification of the left, respectively right, membrane areas marked in Figure 1C. Clusters of EGFRs are outlined. (D) image recorded with 50,000X magnification at the location marked in Figure 1D. Please click here to view a larger version of this figure.
Figure 2. High-resolution ESEM-STEM images depicting membrane bound EGFRs. (A) Micrograph at 75,000X magnification of the area marked in Figure 1B. Many monomers are visible. Several dimers are indicated with the arrowheads. (B) and (C) images acquired at 50,000X magnification of the left, respectively right, membrane areas marked in Figure 1C. Clusters of EGFRs are outlined. (D) image recorded with 50,000X magnification at the location marked in Figure 1D. Please click here to view a larger version of this figure.
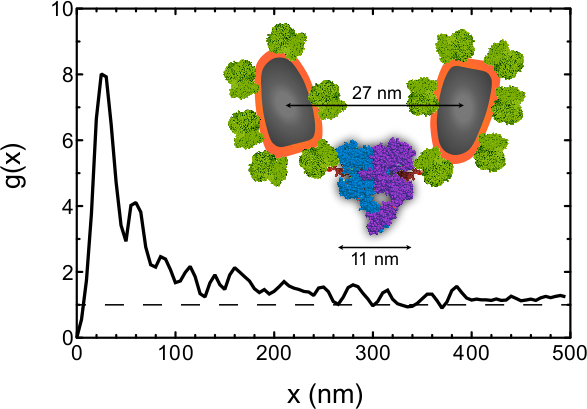 Figure 3. Pair correlation function g(x) as function of the radial distance x determined for the inter-particle distances of all 210 labels detected in Figure 2A-D. The peak at 25 nm indicates that a center-to-center QD distance has a much higher chance of occurring than random, whereby a g(x) = 1 indicates a random chance. The dashed line is a guide to the eye representing g(x) of a random distribution. The inset shows an approximate molecular model of the EGFR dimer with bound EGF and streptavidin coated QDs attached via biotin. The models of streptavidin, EGF and the EGFR were obtained from CPK models of the 1stp (streptavidin), 1EGF (EGF), 1NQL, 2JWA, 1M17, 1IVO and 2GS6 (EGFR) structures in the RCSB Protein Protein Databank, created by Jmol Version 12.2.15. The biotin model is as drawn in RCSB Ligand Explorer Version 1.0. Please click here to view a larger version of this figure.
Figure 3. Pair correlation function g(x) as function of the radial distance x determined for the inter-particle distances of all 210 labels detected in Figure 2A-D. The peak at 25 nm indicates that a center-to-center QD distance has a much higher chance of occurring than random, whereby a g(x) = 1 indicates a random chance. The dashed line is a guide to the eye representing g(x) of a random distribution. The inset shows an approximate molecular model of the EGFR dimer with bound EGF and streptavidin coated QDs attached via biotin. The models of streptavidin, EGF and the EGFR were obtained from CPK models of the 1stp (streptavidin), 1EGF (EGF), 1NQL, 2JWA, 1M17, 1IVO and 2GS6 (EGFR) structures in the RCSB Protein Protein Databank, created by Jmol Version 12.2.15. The biotin model is as drawn in RCSB Ligand Explorer Version 1.0. Please click here to view a larger version of this figure.
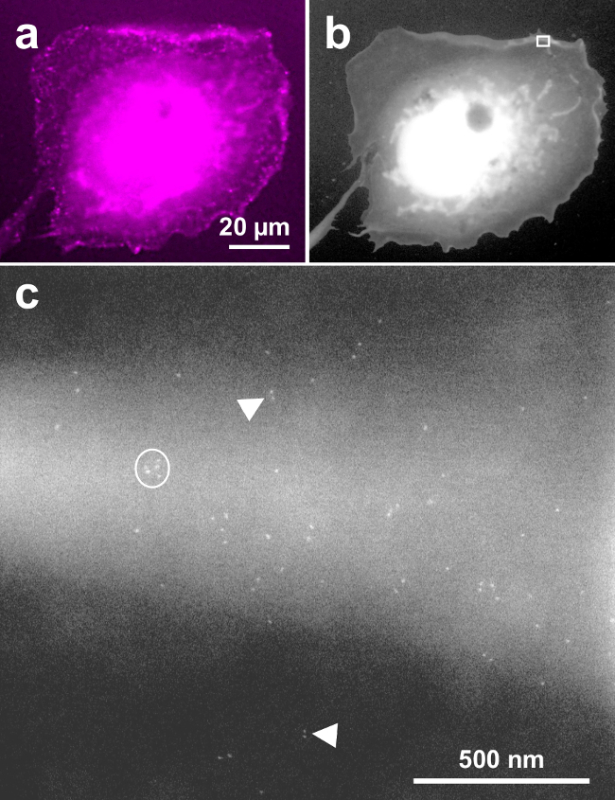 Figure 4. Exemplary COS-7 cell labeled with EGF-QD800 and imaged with correlative fluorescence and ESEM-STEM. (A) Fluorescence image, (B) low magnification overview ESEM-STEM image. (C) High-resolution image recorded at the location of the rectangle in b at 150,000X magnification. Monomeric, dimeric, and clustered EGFRs are detected similar to labeling with EGF-QD655. Please click here to view a larger version of this figure.
Figure 4. Exemplary COS-7 cell labeled with EGF-QD800 and imaged with correlative fluorescence and ESEM-STEM. (A) Fluorescence image, (B) low magnification overview ESEM-STEM image. (C) High-resolution image recorded at the location of the rectangle in b at 150,000X magnification. Monomeric, dimeric, and clustered EGFRs are detected similar to labeling with EGF-QD655. Please click here to view a larger version of this figure.
Discussion
Studying the spatial distribution and stoichiometry of the membrane proteins, such as the EGFR, in whole cells provides important context information about its function and possible changes thereof11-14. It is challenging to study the distribution of monomers, dimers, and clusters directly on a subcellular level using commonly used biochemical methods, for which information is lost about the localization of the protein in the cell or differences between cells15. The labeling and microscopy methods described here allow the visualization of protein complexes on a length scale of a few nanometers so that its stoichiometry can be studied within the context of individual, intact cells4-7. This is not possible with (super resolution) light microscopy23 because its resolution is insufficient to distinguish molecules engaged in a protein complex. Förster Resonance Energy Transfer (FRET) is an ensemble averaging technique24, and does not necessarily detect the dimers and higher order clusters as a consequence of the short range of the energy transfer25. Proximity ligation assays26 do not actually measure distances and are thus not capable of distinguishing between a cellular region with a high protein density, inevitably leading to a number of detected proximities by random chance, from a cellular region with a similar protein density but containing protein complexes exhibiting distinct distances between the labels. The required high resolution to visualize the constituents of protein complexes is achieved by transmission electron microscopy (TEM). However, conventional TEM of whole cells is limited by the requirement of thin samples/sections slicing through the plasma membrane27, or plasma membrane ripping or breaking off28,29 resulting in randomly formed small pieces of membrane. The plasma membrane is thus not imaged as a whole, leading to a lack of possibly crucial cellular context information.
Other experimental challenges for the study of protein stoichiometry in cells arise from the applied protein labeling. Commonly used immunolabeling bears the difficulty that one-to-one stoichiometry between receptor and label can only be achieved with a monovalent probe; this is not the case when primary and secondary antibodies are required. To obtain information about the protein stoichiometry, it is required that a probe binds uniquely to one epitope on the receptor and has one binding site for a fluorescent probe or a high atomic number nanoparticle on the other side. Secondly, the precision achievable with antibody-labeling is restricted on account of their large size30 to 30 nm, so that a discrimination between neighboring single proteins, homodimers, or larger clusters is not feasible, at least not for receptors of the EGFR family. Much smaller specific labels are known in literature and can be applied even for intracellular labeling31 but those are not commonly used. The length of the whole label (EGF-biotin-streptavidin-QD) is of a similar dimension as the EGFR, and sufficiently small to be able to detect the dimer. Moreover, the described two-step labeling procedure avoids labeling induced receptor clustering.
Our method is not very difficult, not much more than fluorescence microscopy of labeled proteins. However, an experiment needs to be carried out with great care, since the number of steps adds up and an error in one of the steps may lead to a complete failure in the experiment. Handling the microchips is not more tedious than the handling of TEM grids but some training empty chips is recommended. ESEM-STEM of whole hydrated cells is probably the most difficult aspect, and requires at least a skilled operator and several days of practice in order to reach a resolution around 3 nm as needed to visualize the QDs. Radiation damage of the specimen under investigation presents a risk. The pressure and the temperature should be carefully watched to maintain a water layer. Moreover, the thickness of the water layer may vary between cells. It is advisable to monitor the presence of the water layer using the gaseous electron detector above the specimen, as described elsewhere6. Cellular regions should only be imaged once or twice to avoid damage.
A key limitation of the method is that high-resolution information about the ultrastructure is absent. Other techniques, for example, cryo TEM, are needed to study the protein structure, and the cellular ultrastructure15. Secondly, studying the dynamic interplay of a multiple of proteins in live cell in time-lapse microscope is not feasible on account of radiation damage, and requires state-of-the-art light microscopy23. Currently, our method is not capable of determining the absolute level of dimers since the labeling efficiency is unknown but we expect to add such data in future. It is nevertheless possible to tell if dimers are present or not. From the literature is it known, that even a labeling efficiency around 15% is enough to detect dimers with sufficient statistical significance32.
It is readily possible to study other membrane-bound receptors via their ligand, via biotinylated Fab fragments of specific antibodies, or via other small linkers7 using the described two-step labeling protocol. Since we have already demonstrated that two different colors (sizes) of QDs can be used, and gold nanoparticle labeling was used in prior work6, it seems feasible to label multiple protein species in the same experiment. The key challenge for the study of the stoichiometry of multiple protein species is ensuring a similar labeling efficiency for both species or at least a normalization of the obtained relative stoichiometry on the respective labeling efficiencies. Yet, the two different QDs used in this study do not seem to differ much in labeling efficiency (see Figures 2 and 4). The described method involving two-step labeling with QDs, and correlative fluorescence microscopy and ESEM-STEM presents a viable method to study the complex interplay of membrane proteins in the intact plasma membranes of whole cells.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank E. Arzt for his support through INM, M. Koch for help with the ESEM, and DENS Solutions for providing the microchips. Research in part supported by the Leibniz Competition 2013.
References
- Dukes MJ, Peckys DB, de Jonge N. Correlative fluorescence microscopy and scanning transmission electron microscopy of quantum-dot-labeled proteins in whole cells in liquid. ACS Nano. 2010;4:4110–4116. doi: 10.1021/nn1010232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama H, et al. Atmospheric scanning electron microscope observes cells and tissues in open medium through silicon nitride film. J. Struct. Biol. 2010;169:438–449. doi: 10.1016/j.jsb.2010.01.005. [DOI] [PubMed] [Google Scholar]
- Kinoshita T, et al. Immuno-electron microscopy of primary cell cultures from genetically modified animals in liquid by atmospheric scanning electron microscopy. Microsc. Microanal. 2014;20:469–483. doi: 10.1017/S1431927614000178. [DOI] [PubMed] [Google Scholar]
- Jonge N, Peckys DB, Kremers GJ, Piston DW. Electron microscopy of whole cells in liquid with nanometer resolution. Proc. Natl. Acad. Sci. 2009;106:2159–2164. doi: 10.1073/pnas.0809567106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peckys DB, de Jonge N. Liquid Scanning Transmission Electron Microscopy: Imaging Protein Complexes in their Native Environment in Whole Eukaryotic Cells. Microsc. Microanal. 2014;20:189–198. doi: 10.1017/S1431927614000099. [DOI] [PubMed] [Google Scholar]
- Peckys DB, Baudoin JP, Eder M, Werner U, de Jonge N. Epidermal growth factor receptor subunit locations determined in hydrated cells with environmental scanning electron microscopy. Sci. Rep. 2013;3:2621–2626. doi: 10.1038/srep02626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peckys DB, Korf U, de Jonge N. Local variations of HER2 dimerization in breast cancer cells discovered by correlative fluorescence- and liquid electron microscopy. Science Advances. 2015;under revision doi: 10.1126/sciadv.1500165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giepmans BN, Deerinck TJ, Smarr BL, Jones YZ, Ellisman MH. Correlated light and electron microscopic imaging of multiple endogenous proteins using Quantum dots. Nat. Meth. 2005;2:743–749. doi: 10.1038/nmeth791. [DOI] [PubMed] [Google Scholar]
- Herbst RS. Review of epidermal growth factor receptor biology. Int. J.Radiat. Oncol. Biol. Phys. 2004;59:S21–S26. doi: 10.1016/j.ijrobp.2003.11.041. [DOI] [PubMed] [Google Scholar]
- Normanno N, et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366:2–16. doi: 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]
- Hofman EG, et al. EGF induces coalescence of different lipid rafts. J. Cell Sci. 2008;121:2519–2528. doi: 10.1242/jcs.028753. [DOI] [PubMed] [Google Scholar]
- Irwin ME, Mueller KL, Bohin N, Ge Y, Boerner JL. Lipid raft localization of EGFR alters the response of cancer cells to the EGFR tyrosine kinase inhibitor gefitinib. J. Cell Physiol. 2011;226:2316–2328. doi: 10.1002/jcp.22570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillemeier BF, Pfeiffer JR, Surviladze Z, Wilson BS, Davis MM. Plasma membrane-associated proteins are clustered into islands attached to the cytoskeleton. Proc. Natl. Acad. Sci. 2006;103:18992–18997. doi: 10.1073/pnas.0609009103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayawala K, Vlachos DG, Edwards JS. Heterogeneities in EGF receptor density at the cell surface can lead to concave up scatchard plot of EGF binding. FEBS Lett. 2005;579:3043–3047. doi: 10.1016/j.febslet.2005.04.059. [DOI] [PubMed] [Google Scholar]
- Valley CC, Lidke KA, Lidke DS. The spatiotemporal organization of ErbB receptors: insights from microscopy. Cold Spring Harb. Perspect. Biol. 2014;6 doi: 10.1101/cshperspect.a020735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peckys DB, Bandmann V, de Jonge N. Correlative fluorescence- and scanning transmission electron microscopy of quantum dot labeled proteins on whole cells in liquid. Meth. Cell Biol. 2014;124:305–322. doi: 10.1016/B978-0-12-801075-4.00014-8. [DOI] [PubMed] [Google Scholar]
- Peckys DB, Dukes MJ, de Jonge N. Correlative fluorescence and electron microscopy of quantum dot labeled proteins on whole cells in liquid. Methods Mol. Biol. 2014;1117:527–540. doi: 10.1007/978-1-62703-776-1_23. [DOI] [PubMed] [Google Scholar]
- Ring EA, Peckys DB, Dukes MJ, Baudoin JP, de Jonge N. Silicon nitride windows for electron microscopy of whole cells. J. Microsc. 2011;243:273–283. doi: 10.1111/j.1365-2818.2011.03501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogner A, Thollet G, Basset D, Jouneau PH, Gauthier C. Wet STEM: A new development in environmental SEM for imaging nano-objects included in a liquid phase. Ultramicroscopy. 2005;104:290–301. doi: 10.1016/j.ultramic.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Chung I, Akita R, Vandlen R, Toomre D, Schlessinger J, Mellman I. Spatial control of EGF receptor activation by reversible dimerization on living cells. Nature. 2010;464:783–787. doi: 10.1038/nature08827. [DOI] [PubMed] [Google Scholar]
- Stoyan D, Stoyan H. Estimating pair correlation functions of planar cluster processes. Biom. J. 1996;38:259–271. [Google Scholar]
- Gao J, et al. In vivo tumor-targeted fluorescence imaging using near-infrared non-cadmium quantum dots. Bioconjug. Chem. 2010;21:604–609. doi: 10.1021/bc900323v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Manley S. Putting super-resolution fluorescence microscopy to work. Nat. Meth. 2009;6:21–23. doi: 10.1038/nmeth.f.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piston DW, Kremers GJ. Fluorescent protein FRET: the good, the bad and the ugly. Trends Biochem. Sci. 2007;32:407–414. doi: 10.1016/j.tibs.2007.08.003. [DOI] [PubMed] [Google Scholar]
- Warren CM, Landgraf R. Signaling through ERBB receptors: Multiple layers of diversity and control. Cell. Signal. 2006;18:923–933. doi: 10.1016/j.cellsig.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Leuchowius KJ, et al. Parallel visualization of multiple protein complexes in individual cells in tumor tissue. Mol. Cell. Proteomics. 2013;12:1563–1571. doi: 10.1074/mcp.O112.023374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoenger A, McIntosh JR. Probing the macromolecular organization of cells by electron tomography. Curr. Opin. Cell Biol. 2009;21:89–96. doi: 10.1016/j.ceb.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Leiderman K, Pfeiffer JR, Wilson BS, Oliver JM, Steinberg SL. Characterizing the topography of membrane receptors and signaling molecules from spatial patterns obtained using nanometer-scale electron-dense probes and electron microscopy. Micron. 2006;37:14–34. doi: 10.1016/j.micron.2005.03.014. [DOI] [PubMed] [Google Scholar]
- Pinto da Silva P, Kan FW. Label-fracture: a method for high resolution labeling of cell surfaces. J. Cell Biol. 1984;99:1156–1161. doi: 10.1083/jcb.99.3.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergersen LH, Storm-Mathisen J, Gundersen V. Immunogold quantification of amino acids and proteins in complex subcellular compartments. Nat. Protoc. 2008;3:144–152. doi: 10.1038/nprot.2007.525. [DOI] [PubMed] [Google Scholar]
- Orlov I, et al. Live cell immunogold labelling of RNA polymerase. II. Sci. Rep. 2015;5:8324. doi: 10.1038/srep08324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala GJ, Kaschek D, Blumenthal B, Reth M, Timmer J, Schamel WW. Pre-clustering of the B cell antigen receptor demonstrated by mathematically extended electron microscopy. Front. Immunol. 2013;4:427. doi: 10.3389/fimmu.2013.00427. [DOI] [PMC free article] [PubMed] [Google Scholar]