

Abstract
Meta-omic technologies such as metagenomics, metatranscriptomics and metaproteomics can aid in the understanding of microbial community structure and metabolism. Although powerful, metagenomics alone can only elucidate functional potential. On the other hand, metaproteomics enables the description of the expressed in situ metabolism and function of a community. Here we describe a protocol for cell lysis, protein and DNA isolation, as well as peptide digestion and extraction from marine microbial cells collected on a cartridge filter unit (such as the Sterivex filter unit) and preserved in an RNA stabilization solution (like RNAlater). In mass spectrometry-based proteomics studies, the identification of peptides and proteins is performed by comparing peptide tandem mass spectra to a database of translated nucleotide sequences. Including the metagenome of a sample in the search database increases the number of peptides and proteins that can be identified from the mass spectra. Hence, in this protocol DNA is isolated from the same filter, which can be used subsequently for metagenomic analysis.
Keywords: Environmental Sciences, Issue 103, Environmental microbiology, microbial ecology, metaproteomics, proteomics, metagenomics, marine, metabolism, biogeochemistry
Introduction
Microorganisms are ubiquitous and play essential roles in Earth’s biogeochemical cycles 1. Currently, there are numerous molecular approaches available for characterizing microbial community structure and function. Most common is the analysis of 16S rRNA gene sequences PCR-amplified from environmental DNA 2–4. A disadvantage of 16S rRNA gene analysis is that it only provides information on phylogenetic identity and community structure, with little information on metabolic function. In contrast, approaches such as metagenomics, metatranscriptomics and metaproteomics provide information on community structure and metabolism. Metagenomics, or the analysis of the gene content of an assemblage of organisms, provides information about the structure and functional potential of the community 5–8. Although powerful, this functional potential may not correspond to the metabolic activities of the organisms. An organism’s genotype is represented by its genes, each of which can be transcribed to RNA and further translated to protein, resulting in a phenotype. Thus, to aid in the understanding of microbial functional activity in an environment, post-genomic analysis should be performed 9. Metatranscriptomics, or the analysis of RNA transcripts is useful because it reveals which genes are transcribed in any given environment. However, mRNA levels do not always match their corresponding protein levels due to translational regulation, RNA half-life, and the fact that multiple protein copies can be generated for every mRNA 10.
For these reasons metaproteomics is now recognized as an important tool for environmental microbiology. Common metaproteomic analyses use a shotgun proteomic approach where the near full complement of proteins in a complex sample are purified and analyzed simultaneously, usually through enzymatic digestion into peptides and analysis on a mass spectrometer. Subsequent tandem mass spectrometry (MS/MS) “peptide fingerprinting” is used to determine the peptide sequence and potential protein of origin by protein database searching (for a review see 11). Proteomic work has come a long way in the past 25 years thanks to the increase in genomic data availability and the increase in the sensitivity and accuracy of mass spectrometers allowing for high-throughput protein identification and quantification 11,12. Since proteins are the final product of gene expression, metaproteomic data can help determine which organisms are active in any given environment and what proteins they are expressing. This is advantageous when trying to determine how a particular set of environmental variables will affect the phenotype of an organism or community. Early on, MS/MS-based metaproteomic studies in the ocean were used to identify specific proteins in targeted microbial lineages, with the first study focusing on the light driven proton pump proteorhodopsin in SAR11 marine bacteria 13. More recently, comparative metaproteomic analyses have elucidated differential protein expression patterns between complex communities. Examples include the identification of temporal shifts in metabolism in the coastal Northwest Atlantic Ocean 14 or the Antarctic Peninsula 5. Other studies have described variations in protein expression patterns across spatial scales, for instance, along a geographical transect from a low-nutrient ocean gyre to a highly productive coastal upwelling system 15. For further reviews of metaproteomics we recommend Schneider et al. (2010) 9 and Williams et al. (2014) 16. Targeted proteomics has also been employed in recent years to quantify expression of specific metabolic pathways in the environment 17,18.
There are three main phases in metaproteomic analysis. The first phase is sample preparation, which includes sample collection, cell lysis and concentration of protein. Sample collection in marine microbiology often entails the filtration of seawater through a pre-filter to remove larger eukaryotic cells, particles and particle-associated bacteria, followed by filtration for the capture of free living microbial cells, commonly with the use of a 0.22 µm cartridge filter unit 19,20. These filters are incased in a plastic cylinder and a cell lysis and protein extraction protocol that can be performed within the filter unit would be a valuable tool. Once biomass is obtained, the cells must be lysed to allow for protein extraction. Several methods can be employed, including guanidine-HCl lysis 21 and sodium dodecyl sulfate (SDS)-based lysis methods. Although detergents like SDS are very efficient at disrupting membranes and solubilizing many protein types, concentrations as low as 0.1% can interfere with downstream protein digestion and MS analysis 22. Of major concern is the negative effects of SDS on trypsin digestion efficiency, resolving power of reverse phase liquid chromatography and ion suppression or accumulation inside the ion source 23.
The second phase is fractionation and analysis, where proteins are subjected to enzymatic digestion followed by LC MS/MS analysis, resulting in a m/z fragmentation pattern that can be used to ascertain the primary amino acid sequence of the initial tryptic peptide. Various digestion methods can be performed depending on the types of detergents used, as well as the downstream mass spectrometry workflow. In our protocol, 1-D PAGE electrophoresis followed by removal of SDS from the gel is utilized in order to remove any detergent contamination. The analysis of proteins that are difficult to solubilize, such as membrane proteins, requires the use of high concentrations of SDS or other detergents. This leads to compatibility issues with SDS-gel electrophoresis. If the objective of a study requires the solubilization of these hard to solubilize proteins, the tube-gel system can be used 22,24. The tube-gel method incorporates proteins within the gel matrix without the use of electrophoresis. Subsequently any detergents used for solubilization are removed before protein digestion.
The third phase is the bioinformatic analysis. In this phase the MS/MS peptide data are searched against a database of translated nucleotide sequences to determine which peptides and proteins are present in the sample. The identification of peptides is dependent on the database it is searched against. Marine metaproteomic data are commonly searched against databases comprised of reference genomes, metagenomic data such as the Global Ocean Sampling dataset 25, as well as single cell amplified genomes from uncultivated lineages 26,27. Protein identification can also be increased by the inclusion of metagenomic sequences from the same sample as the metaproteomic data was derived 5.
Here we provide a protocol for the generation of peptides suitable for MS/MS-based analysis from microbial biomass collected by filtration and stored in an RNA stabilization solution. The protocol described here allows for DNA and protein to be isolated from the same sample so that all steps leading up to the protein and DNA precipitations are identical. From a practical perspective, less filtration is required since only one filter is required for both protein and DNA extraction. We would also like to acknowledge that this protocol was created through the combination, adaptation and modification of two previously published protocols. The cell lysis steps are adapted from Saito et al. (2011) 28 and the in-gel trypsin digest component is adapted from Shevchenko et al. (2007) 29.
Protocol
1. Prepare Reagents
Prepare SDS-extraction solution: 0.1 M Tris-HCl pH 7.5, 5 % glycerol, 10 mM EDTA and 1 % SDS. Filter-sterilize using a 0.22 µm filter and store at 4 °C.
- Prepare stock reagents needed for the polyacrylamide gel.
- Prepare 1.5 M Tris-HCL pH 8.8. Filter-sterilize using a 0.22 µm filter and store at room temperature.
- 0.5 M Tris-HCL pH 6.8. Filter-sterilize using a 0.22 µm filter and store at room temperature.
- Prepare 10 % SDS. Filter sterilize using a 0.22 µm filter and store at room temperature.
- Prepare solutions needed for in-gel trypsin digest and peptide extraction.
- Prepare 100 mM ammonium bicarbonate. Filter-sterilize using a 0.22 µm filter and store at 4 °C.
- Prepare 1 M DTT stocks. Suspended in 100 mM ammonium bicarbonate and store at -80 °C.
- Prepare 550 mM iodoacetamide stocks. Suspended in 100 mM ammonium bicarbonate and store at -80 °C.
- Prepare 100 ng/µl trypsin stocks. Aliquot 60 µl into 1.5 ml microcentrifuge tubes and store at -80 °C.
- Prepare extraction solution: 1% formic acid, 2% acetonitrile.
- Prepare resuspension solution: 5% acetonitrile and 0.1% formic acid.
2. Perform Cell Lysis in Cartridge Filter Unit with Cells Preserved in RNA Stabilization Solution
Expel the RNA stabilization solution from the cartridge filter unit (1.5 ml) and into a 2 ml microcentrifuge tube using a 60 ml syringe attached at the luer-lock end of the filter.
Centrifuge the 2 ml microcentrifuge tube for 10 min at 17,000 x g to pellet any cellular debris. This is done so that the filter in step 2.3 does not clog.
Transfer supernatant to a 10 K ultracentrifugal filter unit to capture any proteins originating from cells that have lysed from the freeze/thaw of the sample. Do not discard the pellet. It will be used in step 2.5. Perform centrifugation of the ultracentrifugal filter unit at 3,270 x g for 30 min or until the volume has reduced to about 600 µl.
Melt the tip of a p10 tip and block the non luer-lock side of the cartridge filter unit with the open end of the tip. This will ensure that no extraction buffer and biomass escapes during steps 2.5-2.7.
Suspend the pellet in 1 ml SDS-extraction solution and pipette it into the original cartridge filter unit. The easiest way to pipette into the cartridge filter unit is to stack a p200 tip onto a p1000 tip and use this double tip for pipetting.
Add 1 ml of SDS extraction solution to the cartridge filter unit so the total volume is about 2 ml and incubate for 10 min at room temperature while rotating in a hybridization oven. Place 3 crumpled lab wipes at the bottom of a 50 ml conical tube. Parafilm both ends of the cartridge filter unit closed and put the cartridge filter unit into the 50 ml tube. Close the lid and put the tube into the hybridization oven.
After 10 min place the Sterivex filter on a foam floater and secure it in place with a 5 ml syringe on the luer-lock end. Float and incubate in a 95 °C water-bath for 15 min.
Let the cartridge filter unit cool and rotate at room temperature for 1 hour (as in step 2.6).
Expel the SDS extraction solution/cell lysate out of the filter with a 60 ml syringe into the same ultracentrifugal filter unit as before (step 2.3). Add 1 ml of fresh SDS extraction solution to the cartridge filter unit and mix for 30 sec by hand, then expel into the ultracentrifugal filter unit using the 60 ml syringe. This is to rinse the cartridge filter unit to ensure all proteins have been removed.
Perform centrifugation on the ultracentrifugal filter unit for 45 min at 3,270 x g or until the volume in the filter unit is less than 600 µl.
Discard flow-through and top up the ultracentrifugal filter unit with fresh SDS-extraction solution.
Centrifuge the filter unit for another 45 min at 3,270 x g.
Repeat steps 2.11 and 2.12 twice more. Ensure the final volume in the ultracentrifugal filter unit is at most 600 µl at the end of the final spin.
At this point, split the concentrate in two. One fraction will be used for DNA precipitation and the other for protein precipitation. Note: The fractionation amount depends on the amount of biomass that was filtered and the intended uses of the products. In our case, we split the concentrate with 10% towards DNA precipitation and 90% towards protein precipitation.
3. Protein Precipitation
Add 4 volumes methanol:acetone (50:50) to one volume of concentrate and vortex for 10 sec. Incubate overnight at -20 °C.
Spin down at 17,000 x g for 30 min. Decant the supernatant and let the pellet (may be invisible) dry in a speedvac for 1 hr (or until dry). Note: Do not over dry the pellet as this may make it difficult to resuspend.
Suspend the pellet in 25 µl of SDS-extraction solution. Let sit for one hour then resuspend by pipetting up and down.
Quantify protein using a protein assay kit and the manufacturers instructions.
4. DNA Precipitation
Add SDS-extraction solution to the concentrate fraction to be used for DNA precipitation until the 500 µl mark. This step is simply to increase the volume of the solution, making it easier to work with.
Add 0.583 volumes of a protein precipitation reagent (such as the MPC protein precipitation reagent) and vortex for 10 sec. You should see a white precipitate form. Note: We have also used phenol:chloroform DNA extraction methods, but it is more difficult due to the low volumes. We obtained better DNA yields using the MPC Protein Precipitation Reagent.
Centrifuge at 17,000 x g and 4 °C for 10 min.
Transfer supernatant to another 1.5 ml microcentrifuge tube and add 0.95 volumes of isopropanol. Invert 30-40 times.
Centrifuge for 10 min at 4 °C at maximum speed.
Carefully decant and discard the supernatant.
Rinse twice with 750 µl of 70% ethanol.
Remove as much ethanol as possible by pipetting, then let air-dry. Note: Do not over dry the DNA as this may make it difficult to resuspend.
Resuspend in 25 µl of low TE buffer, pH 8 (10 mM Tris-HCl, 0.1 mM EDTA).
Quantify the DNA using a dsDNA assay kit and the manufacturers instructions. Perform agarose gel (1%) electrophoresis on 3 µl of the DNA to check its quality.
5. SDS-PAGE Gel of Proteins
Prepare sample buffer (950 µl Laemmli sample buffer and 50 µl β-mercaptoethanol).
Add the corresponding volume for 15 µg of protein, or a max of 20 µl to equal volume of sample buffer and boil for 4 min. Samples can now be stored at -80 °C until SDS-PAGE can be performed.
Prepare 10% acrylamide resolving gel with a 5% acrylamide stacking gel.
Load the gel with samples and 4 µl of a protein ladder. Run the gel at constant 120 V until the 250 kDa ladder marker has just reached the resolving gel.
Stain the gel using a coomassie based stain according to the manufacturers instructions.
6. In-gel Trypsin Digest and Peptide Extraction
Note: All steps from here until 6.10 are performed in a biological safety cabinet to minimize contamination.
Cut off excess gel under the 10 kDa ladder mark for each lane. Each lane will be analyzed on the MS/MS separately. Cut each lane into 1 mm x 1 mm squares to increase surface area and place all squares from the lane into a low-binding micro-centrifuge tube. Repeat this for all lanes. (1% acetic acid can be used to prevent the gel from drying out and becoming difficult to cut). Note: Minimizing contamination is critical at this stage, so gel slicing is performed in a biological safety cabinet on the glass SDS-PAGE gel mold used to make the gel.
- De-stain
- Dispense 50 µl of 100 mM ammonium bicarbonate into each low-binding tube, close cap and incubate at 37 °C for 10 min.
- Dispense 50 µl of acetonitrile, close cap and incubate at 37 °C for 5 min.
- Aspirate and discard 150 µl.
- Repeat steps 6.2.1 and 6.2.2.
- Aspirate and discard 95 µl.
- Dehydration
- Dispense 50 µl acetonitrile, close cap and wait 5 min. Aspirate and discard 45 µl and wait 10 min.
- Reduction
- Dispense 50 µl DTT (10 mM, diluted in 100 mM ammonium bicarbonate), close cap and wait 30 min at 37 °C.
- Alkylation
- Dispense 50 µl iodoacetamide (55 mM, diluted in 100 mM ammonium bicarbonate), close cap and wait 20 min at 37 °C.
- Dispense 100 µl of acetonitrile, close cap and incubate for 5 min at 37 °C. Aspirate and discard 195 µl.
- Wash
- Dispense 50 µl ammonium bicarbonate, close cap and incubate for 10 min at 37 °C.
- Dispense 50 µl of acetonitrile, close cap and incubate at 37 °C for 5 min. Aspirate and discard 120 µl.
- Dehydration
- Dispense 50 µl acetonitrile, close cap and wait 5 min. Aspirate and discard 45 µl.
- Dispense 50 µl of acetonitrile, wait 5 min.
- Aspirate and discard 75 µl and wait 5 min.
- Digestion
- Dispense 25-30 µl of 6 ng/µl Trypsin (until all gel fragments are covered). Close cap and wait 30 min at room temperature.
- Incubate overnight (4.5 hr minimum) at 37 °C.
- Let sit at room temperature for 30 min.
- Extraction and Peptide Transfer
- Dispense 30 µl of extraction solution and cover with lid for 30 min on ice.
- Aspirate 30 µl and dispense aspirated volume in a new labeled low-binding tube.
- Dispense 12 µl of extraction solution and 12 µl of acetonitrile into the original tube (with gel). Close cap and incubate for 30 min on ice.
- Aspirate 15 µl and deposit into the new tube.
- Repeat steps (6.9.3 and 6.9.4).
Place new tubes in a SpeedVac until dry.
Resuspend in 50 µl of resuspension solution. Vortex on medium for 10 min to resuspend.
Pipette peptide solution into either nano/LC vials or 96-well plates suitable for nano/LC MS/MS work.
Representative Results
As a demonstration, we performed the protocol on two seawater samples collected from the surface and the chlorophyll maximum of the coastal ocean in Northern Canada. While at sea, 6-7 L of seawater was passed through a 3 µm GF/D prefilter, then microbial cells were collected onto a 0.22 µm cartridge filter unit following the protocol of Walsh et al. 20. Cells were immediately stored in an RNA stabilization solution until further processing. Upon returning to the lab, we performed the protocol as it is presented here. The concentrated cell lysate was divided; protein was precipitated from 90% of the volume, while DNA was precipitated from the remaining 10% of the volume. We recovered 24-26 µg of protein and 250-308 ng of high quality DNA from these samples (Figure 1). After the in-gel trypsin digestion and peptide extraction, we subjected the peptides to MS/MS analysis using a nano-LC coupled to the Orbitrap Elite mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). From the peptides, we generated over 23,000 MS/MS spectra per sample. Peptides and proteins were then identified by searching these spectra against a custom in-house sequence database using the PEAKS bioinformatics tool (BSI, Waterloo, ON, Canada). The database was comprised of predicted proteins from marine reference genomes and metagenomes. The search resulted in the identification of around 1,000 peptides and 700-800 proteins for each sample. Naturally, these results are dependent on microbial cell abundance, MS instrumentation, and protein search database and algorithms. Nonetheless, these results demonstrate that this protocol has the potential to produce adequate tryptic peptides suitable for identify hundreds of proteins in the environment. Moreover, since metagenomic libraries can be constructed from as little as 100 ng of DNA 30, this protocol also has potential to provide adequate quantities of DNA to generate matched metagenomic-metaproteomic datasets.
Taxonomic and functional composition of the metaproteomes was analyzed using a combination of BLASTp and the MEGAN (Metagenome analyzer) software package 31,32(Figure 2). Proteins assigned to Alpha-proteobacteria were the most highly represented in the dataset, the vast majority of which were assigned to the SAR11 clade. The Rhododobacterales clade of Alpha-proteobacteria was also highly represented and identified most often in surface waters. Proteins assigned to Bacteroidetes were evenly distributed between the surface and chlorophyll maximum, but Flavobacteria proteins were identified to a greater degree at the chlorophyll maximum. Gamma-proteobacterial proteins were evenly distributed throughout the water column while Beta-proteobacterial proteins were found predominately in the surface. From a functional perspective, a wide range of metabolic pathways were identified. Vertical structuring of these metabolic pathways was apparent. For example, proteins associated with amino acid metabolism, carbohydrate metabolism and prokaryotic carbon fixation pathways were identified primarily at the surface, and nitrogen metabolism was found exclusively at the surface. Photosynthetic carbon fixation proteins were observed primarily at the chlorophyll maximum while proteins involved in photosynthesis were identified evenly between the surface and chlorophyll maximum. These results demonstrate that a wide variety of proteins from a diversity of microbial taxa can be detected using the protocol presented here.
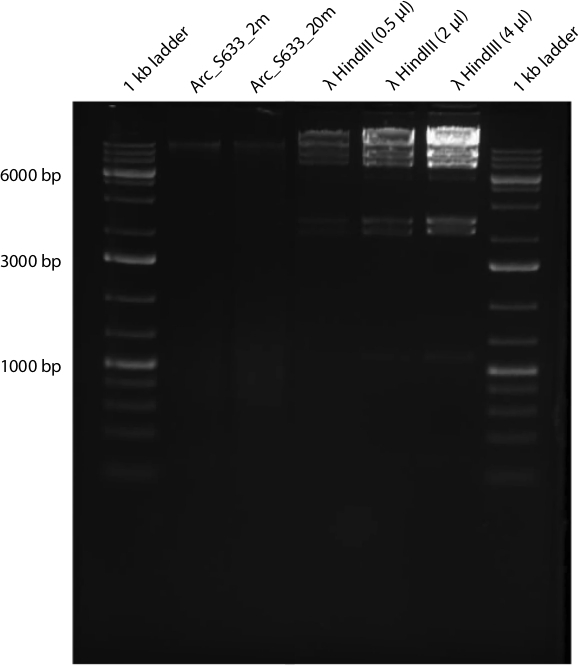 Figure 1: Genomic DNA from 2 depths at Arctic station S633. The first lane contains 4 µl of a 1 kb DNA ladder, lane 2 contains 3 µl of genomic DNA extraction from S633_2 m, lane 3 contains 3 µl of genomic DNA extraction from S633_20 m and lanes 4-6 contain 0.5 µl (85 ng), 2 µl (333 ng) and 4 µl (667 ng) of HindIII digested lambda DNA.
Figure 1: Genomic DNA from 2 depths at Arctic station S633. The first lane contains 4 µl of a 1 kb DNA ladder, lane 2 contains 3 µl of genomic DNA extraction from S633_2 m, lane 3 contains 3 µl of genomic DNA extraction from S633_20 m and lanes 4-6 contain 0.5 µl (85 ng), 2 µl (333 ng) and 4 µl (667 ng) of HindIII digested lambda DNA.
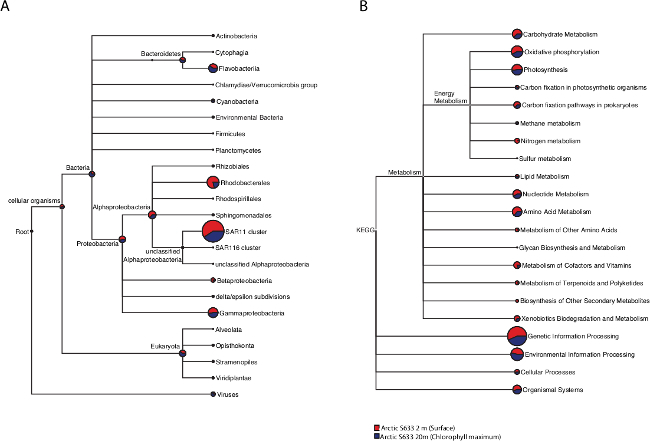 Figure 2: Taxonomic and functional analysis of 2 depths at Arctic station S633. Taxonomic diversity comparison of the Arctic station 633 surface and chlorophyll maximum waters (A) created using MEGAN. Functional diversity comparison of the Arctic station 633 surface and chlorophyll maximum waters (B) created using MEGAN to query against the KEGG database. Please click here to view a larger version of this figure.
Figure 2: Taxonomic and functional analysis of 2 depths at Arctic station S633. Taxonomic diversity comparison of the Arctic station 633 surface and chlorophyll maximum waters (A) created using MEGAN. Functional diversity comparison of the Arctic station 633 surface and chlorophyll maximum waters (B) created using MEGAN to query against the KEGG database. Please click here to view a larger version of this figure.
Discussion
Sample preservation is key to metaproteomic studies and previous work demonstrated that an RNA stabilization solution is a useful storage buffer for storing cells prior to protein extraction 28. Ideally, samples would be preserved in situ to negate shifts in protein expression during handling 33,34. In fact, in situ sampling and fixation technologies have been developed, which allow for the autonomous collection and preservation of samples by ship-deployed instruments. However, access to these technologies is not always feasible. In the common case that it is not, samples should be preserved as soon as possible after collection.
Here we present a protocol for extracting protein from RNA stabilization solution stored cells collected on a cartridge filter unit, which is commonly used in aquatic microbiology. The protocol includes cell lysis using an SDS-lysis solution and heating, followed by a protein concentration step using ultracentrifugal filter units that doubled as a necessary desalting step. It must be noted that the concentrating and desalting steps cannot be overlooked. We found that a minimum of three buffer exchange steps was required to desalt our concentrate. Due to the high salt concentration of the RNA stabilization solution, if proper desalting does not occur, too much salt will be precipitated during the overnight protein precipitation step and the desalting and precipitation step will have to be repeated. Additionally, if desalination is not properly performed the 1D-PAGE will not work and the samples will be lost.
Next, the concentrated lysate was divided so that both protein precipitation and DNA precipitation could be performed. This is useful as it is often desirable that metagenomic and metaproteomic data be generated from the same samples. If a protein is not represented in the protein sequence database then the peptide will not be identified. Including the genomic data from the same sample as the proteomic data reduces the risk of not being able to identify a protein due to its absence from the database.
Although this protocol was optimized for use with cartridge filter units and validated to work on coastal ocean microbial communities, it can be adapted for use with other types of environmental samples and filters. However, it should be clearly stated that the success of this protocol is dependent on an adequate amount of starting biomass. Therefore in aquatic ecosystems where biomass may be very low, we recommend increasing the volume of water filtered accordingly.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors would like to acknowledge Marcos Di Falco for his expertise and advice with the preparation of the samples for nano-LC MS/MS as well as Dr. Zoran Minic from the University of Regina for the LC MS/MS analysis. This work was supported by NSERC (DG402214-2011) and CRC (950-221184) funding. D.C. was supported by Concordia Institute for Water, Energy, and Sustainable Systems and FQRNT.
References
- Madsen EL. Microorganisms and their roles in fundamental biogeochemical cycles. Current opinion in biotechnology. 2011;22(3):456–464. doi: 10.1016/j.copbio.2011.01.008. [DOI] [PubMed] [Google Scholar]
- El-Swais H, Dunn KA, Bielawski JP, Li WKW, Walsh DA. Seasonal assemblages and short-lived blooms in coastal northwest Atlantic Ocean bacterioplankton. Environmental microbiology. 2014. [DOI] [PubMed]
- Galand PE, Potvin M, Casamayor EO, Lovejoy C. Hydrography shapes bacterial biogeography of the deep Arctic Ocean. The ISME journal. 2010;4(4):564–576. doi: 10.1038/ismej.2009.134. [DOI] [PubMed] [Google Scholar]
- Lane DJ, Pace B, Olsen GJ, Stahlt DA, Sogint ML, Pace NR. Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proceedings of the National Academy of Sciences of the United States of America. 1986;83:4972. doi: 10.1073/pnas.82.20.6955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams TJ, Long E, et al. A metaproteomic assessment of winter and summer bacterioplankton from Antarctic Peninsula coastal surface waters. The ISME journal. 2012;6(10) doi: 10.1038/ismej.2012.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheik CS, Jain S, Dick GJ. Metabolic flexibility of enigmatic SAR324 revealed through metagenomics and metatranscriptomics. Environmental microbiology. 2014;16(1):304–317. doi: 10.1111/1462-2920.12165. [DOI] [PubMed] [Google Scholar]
- Venter JC, Remington K, et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science (New York, N.Y.) 2004;304:66–74. doi: 10.1126/science.1093857. [DOI] [PubMed] [Google Scholar]
- Tyson GW, Chapman J, et al. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature. 2004;428:37–43. doi: 10.1038/nature02340. [DOI] [PubMed] [Google Scholar]
- Schneider T, Riedel K. Environmental proteomics: analysis of structure and function of microbial communities. Proteomics. 2010;10(4):785–798. doi: 10.1002/pmic.200900450. [DOI] [PubMed] [Google Scholar]
- Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nature reviews. Genetics. 2012;13(4):227–232. doi: 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hettich RL, Pan C, Chourey K, Giannone RJ. Metaproteomics: harnessing the power of high performance mass spectrometry to identify the suite of proteins that control metabolic activities in microbial communities. Analytical chemistry. 2013;85(9):4203–4214. doi: 10.1021/ac303053e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Bergen M, Jehmlich N, et al. Insights from quantitative metaproteomics and protein-stable isotope probing into microbial ecology. The ISME journal. 2013;7(10):1877–1885. doi: 10.1038/ismej.2013.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannoni SJ, Bibbs L, et al. Proteorhodopsin in the ubiquitous marine bacterium SAR11. Nature. 2005;438(7064):82–85. doi: 10.1038/nature04032. [DOI] [PubMed] [Google Scholar]
- Georges AA, El-Swais H, Craig SE, Li WK, Walsh DA. Metaproteomic analysis of a winter to spring succession in coastal northwest Atlantic Ocean microbial plankton. The ISME journal. 2014. pp. 1–13. [DOI] [PMC free article] [PubMed]
- Morris RM, Nunn BL, Frazar C, Goodlett DR, Ting YS, Rocap G. Comparative metaproteomics reveals ocean-scale shifts in microbial nutrient utilization and energy transduction. The ISME journal. 2010;4(5):673–685. doi: 10.1038/ismej.2010.4. [DOI] [PubMed] [Google Scholar]
- Williams TJ, Cavicchioli R. Marine metaproteomics: deciphering the microbial metabolic food web. Trends in microbiology. 2014;22(5):248–260. doi: 10.1016/j.tim.2014.03.004. [DOI] [PubMed] [Google Scholar]
- Saito Ma, McIlvin MR, et al. Multiple nutrient stresses at intersecting Pacific Ocean biomes detected by protein biomarkers. Science. 2014;345(6201):1173–1177. doi: 10.1126/science.1256450. [DOI] [PubMed] [Google Scholar]
- Bertrand E, Moran D, McIlvin M, Hoffman J, Allen A, Saito M. Methionine synthase interreplacement in diatom cultures and communities: Implications for the persistence of B12 use by eukaryotic phytoplankton. Limnology and Oceanography. 2013;58(4):1431–1450. [Google Scholar]
- Hawley AK, Kheirandish S, et al. Methods in enzymology. Vol. 531. Elsevier Inc; 2013. Molecular tools for investigating microbial community structure and function in oxygen-deficient marine waters; pp. 305–329. [DOI] [PubMed] [Google Scholar]
- Da Walsh , Zaikova E, Hallam SJ. Small volume (1-3L) filtration of coastal seawater samples. Journal of visualized experiments: JoVE. 2009. pp. 1–2. [DOI] [PMC free article] [PubMed]
- Thompson M, Chourey K. Experimental approach for deep proteome measurements from small-scale microbial biomass samples. Analytical. 2008;80(24):9517–9525. doi: 10.1021/ac801707s. [DOI] [PubMed] [Google Scholar]
- Lu X, Digestion Zhu H. Tube-Gel. 2005. pp. 1948–1958. [DOI] [PMC free article] [PubMed]
- Sharma R, Dill BD, Chourey K, Shah M, VerBerkmoes NC, Hettich RL. Coupling a detergent lysis/cleanup methodology with intact protein fractionation for enhanced proteome characterization. Journal of proteome research. 2012;11(12):6008–6018. doi: 10.1021/pr300709k. [DOI] [PubMed] [Google Scholar]
- Santoro AE, Dupont CL, et al. Genomic and proteomic characterization of "Candidatus Nitrosopelagicus brevis": An ammonia-oxidizing archaeon from the open ocean. Proceedings of the National Academy of Sciences. 2015;112(4):1173–1178. doi: 10.1073/pnas.1416223112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusch DB, Halpern AL, et al. The Sorcerer II Global Ocean Sampling expedition: northwest Atlantic through eastern tropical Pacific. PLoS biology. 2007;5(3):e77. doi: 10.1371/journal.pbio.0050077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swan BK, Martinez-Garcia M, et al. Potential for chemolithoautotrophy among ubiquitous bacteria lineages in the dark ocean. Science (New York, N.Y.) 2011;333(6047):1296–1300. doi: 10.1126/science.1203690. [DOI] [PubMed] [Google Scholar]
- Rinke C, Schwientek P, et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature. 2013;499(7459):431–437. doi: 10.1038/nature12352. [DOI] [PubMed] [Google Scholar]
- Saito MA, Bulygin VV, Moran DM, Taylor C, Scholin C. Examination of microbial proteome preservation techniques applicable to autonomous environmental sample collection. Frontiers in microbiology. 2011;2:215. doi: 10.3389/fmicb.2011.00215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A, Tomas JH, Olsen J, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nature protocols. 2007;1(6):2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- Thomas T, Gilbert J, Meyer F. Metagenomics - a guide from sampling to data analysis. Microbial Informatics and Experimentation. 2012;2(1):3. doi: 10.1186/2042-5783-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson DH, Auch AF, Qi J, Schuster SC. MEGAN analysis of metagenomic data. Genome research. 2007;17(3):377–3786. doi: 10.1101/gr.5969107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson DH, Mitra S, Ruscheweyh H-J, Weber N, Schuster SC. Integrative analysis of environmental sequences using MEGAN4. Genome research. 2011;21(9):1552–1560. doi: 10.1101/gr.120618.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ea Ottesen , Marin R, et al. Metatranscriptomic analysis of autonomously collected and preserved marine bacterioplankton. The ISME journal. 2011;5(12):1881–1895. doi: 10.1038/ismej.2011.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feike J, Jürgens K, Hollibaugh JT, Krüger S, Jost G, Labrenz M. Measuring unbiased metatranscriptomics in suboxic waters of the central Baltic Sea using a new in situ fixation system. The ISME journal. 2012;6(2):461–470. doi: 10.1038/ismej.2011.94. [DOI] [PMC free article] [PubMed] [Google Scholar]