

Abstract
Digital assays are powerful methods that enable detection of rare cells and counting of individual nucleic acid molecules. However, digital assays are still not routinely applied, due to the cost and specific equipment associated with commercially available methods. Here we present a simplified method for readout of digital droplet assays using a conventional real-time PCR instrument to measure bulk fluorescence of droplet-based digital assays.
We characterize the performance of the bulk readout assay using synthetic droplet mixtures and a droplet digital multiple displacement amplification (MDA) assay. Quantitative MDA particularly benefits from a digital reaction format, but our new method applies to any digital assay. For established digital assay protocols such as digital PCR, this method serves to speed up and simplify assay readout.
Our bulk readout methodology brings the advantages of partitioned assays without the need for specialized readout instrumentation. The principal limitations of the bulk readout methodology are reduced dynamic range compared with droplet-counting platforms and the need for a standard sample, although the requirements for this standard are less demanding than for a conventional real-time experiment. Quantitative whole genome amplification (WGA) is used to test for contaminants in WGA reactions and is the most sensitive way to detect the presence of DNA fragments with unknown sequences, giving the method great promise in diverse application areas including pharmaceutical quality control and astrobiology.
Keywords: Molecular Biology, Issue 103, Whole Genome Amplification, digital assay, MDA, PCR, single-molecule, single cell, nucleic acids, micro-droplets, microfluidics, emulsion
Introduction
Digital assays for nucleic acid quantification (digital PCR)1-4 and base order (sequencing) are strongly impacting the life sciences and medicine. Digital assays provide quantification of molecular counts on an absolute scale (not relative to a control), providing high sensitivity, enabling facile comparisons across experiments, and crucially, enabling the construction of large databases containing comparable data5 (Table 1).
Over the last 15 years, whole genome amplification (WGA) emerged alongside PCR as a general tool for nucleic acid amplification. Like PCR, WGA is useful for analytical and preparative applications by amplifying minute sample elements up to a level that can be easily detected or used for subsequent analyses like base sequencing. Unlike PCR, WGA is not specific to a particular DNA locus, rather allowing amplification of all sequences in the sample, including unknown sequences. This fundamental difference between PCR and WGA makes the methods complementary to one another and gives rise to different challenges in their application.
The high yield of WGA reactions6 enables routine amplification of genomic DNA from single molecules7, single cells8 and other low-biomass samples9 for quantification or further analysis. The major challenges associated with WGA chemistry are its extreme sensitivity to contaminants and the uneven amplification across individual template molecules6. However, WGA is gaining popularity as single-cell sequencing has emerged as the “killer application” of WGA technology10, and template quantification by WGA is important in many fields of application7.
Instrumentation for digital nucleic acid quantification has been previously described in a variety of valved and valveless microfluidic formats11-14, including droplet-based assays15,16 (Table 2). However, commercial microfluidic systems for digital analysis require specialized equipment for reaction setup and product detection17. Custom valved microfluidics are flexible, but require precision microfabrication and pneumatic control systems18. While it is relatively simple to make monodisperse micro-droplets for emulsion-based digital assays19,20, digital readout is technically burdensome, requiring either large-scale wide-field imaging (similar to popular next-generation sequencing technologies)21,22 or high-speed flow-based droplet detection23-25. Ideally, a digital assay would be simple from set-up to readout, reducing the need for complex instrumentation and allowing large numbers of samples to be read out quickly. Here we describe a simplified method for readout of digital droplet assays that uses a conventional real-time PCR instrument to measure bulk fluorescence of droplet-based digital assays.
While the new approach can be applied to digital PCR assays, it is particularly advantageous for digital WGA assays for which analog real-time amplification assays (that give excellent results for PCR) are problematic. WGA is commonly applied to samples with template molecules that are heterogeneous in sequence, length, and base content. These different template molecules are amplified at different rates6, necessitating the use of a reference (“standard”) sample with matching characteristics. Often, no such standard is available, or the characteristics of the incoming samples are unknown. The heterogeneity of the input material and length-dependent property of WGA chemistries26 also complicate the interpretation of results by creating ambiguity in what is being quantified--the input mass, input number of molecules, a combination of the two, or neither. Finally, the sequence-non-specificity renders quantitative multiple displacement amplification (MDA) more sensitive to contamination than quantitative PCR since contaminant molecules of any sequence have a potential to interfere. Microfluidic digital assays address contamination by segregating template molecules and reducing reaction volumes such that fewer contaminants are sampled.
Here we use a popular isothermal WGA method, MDA27. Of note, several other WGA chemistries including PicoPlex and MALBAC28 depend crucially on initial isothermal strand displacement steps. Isothermal steps exacerbate the challenge of applying analog real-time assays for quantitative WGA. WGA can neither be entirely prevented during setup, leading to unwanted variable pre-amplification, nor discretized (“cycled”) in a way where replication of heterogeneous molecules can be driven to completion and stopped prior to the next cycle like PCR11 (Figure 1). A digital assay format for WGA accomodates typical reaction setup procedures due to the segregation of each molecule for enumeration at the assay endpoint (so pre-amplification does not affect the results) original readout means accuracy would be largely independent of variation in amplification efficiency.
The assay depends on droplets produced in oil with uniform volumes, as the signal level per droplet at the assay endpoint will depend on the droplet volume, and we do not want the consistency of results to depend on averaging across a distribution of droplet sizes. Making monodisperse droplets is now a standard procedure (about 3,500 monodisperse droplet papers have been published since 2013), but requires microfluidic instrumentation25. In fact, more than six companies have developed independent commercial products that rely on production of such droplets, and droplet-making microfluidic chips are commercially available23,29. For this study, we used custom microfluidic devices produced in-house (see Protocol). Syringe pumps to drive flow through the devices are also commercially available, but alternatively, can be substituted with a single disposable syringe for vacuum-driven flow to reduce costs30.
Protocol
Note: Fabricating the microfluidic device is not necessary for this assay, as droplets for this protocol can be formed with existing commercial droplet makers23,29.
1. Make the Droplet-forming Microfluidic Device
Prepare master mold for the channels with the SU-8 Master Fabrication protocol outlined previously31, but with a droplet generator mask pattern32.
Fabricate devices in PDMS using the techniques of soft lithography32,33.
2. Prepare Reaction Mix for Bulk Droplet Readout
Note: The protocol can be used to quantify nucleic acids using many types of amplification reactions. As an example, the reagents needed for MDA of several Lambda DNA concentrations are given. Reagent details are listed in the Table of Specific Reagents. The distribution of template within the droplets depends on sufficient mixing of the sample.
- Obtain or Purify Phi29 DNA polymerase.
- Purify Phi29 as previously described7, or purchase from supplier. The Phi29 used for the shown experiments was purified.
Prepare 10 ml of Denaturation Buffer consisting of 65 mM KOH, 1.65 mM EDTA, and 14 mM DTT.
Prepare 10 ml of Neutralization Buffer consisting of 65 mM HCl, 0.21 M Tris-Cl pH 7.0, and 9 mM Tris-Cl pH 8.0.
Prepare two standards, one no template control (NTC) such as nuclease-free water, and one high template concentration (around 4 pg/µl Lambda DNA). Denature 3.3 µl of each standard with 3.3 µl of Denaturation Buffer in a qPCR tube. Incubate at RT for 3 min. Quench each with 3.3 µL of Neutralization Buffer.
Denature 3.3 µl of the template of interest with 3.3 µl of Denaturation Buffer in a qPCR tube. Incubate at RT for 3 min. Quench with 3.3 µl of Neutralization Buffer.
Prepare 11 µl of master mixture per sample on ice. The 2x MDA reaction master mixture consists of 2x double-stranded DNA (dsDNA) binding dye, 2x qPCR reference dye, 50 µM randomized oligo, 2 mg/ml BSA, 2x Phi29 DNA Polymerase Reaction Buffer, 4.8 mM dNTPs, nuclease-free water, and 40 µg/ml Phi29 DNA polymerase. Add Phi29 DNA polymerase last to ensure the polymerase does not encounter pH levels much higher or lower than 7.5.Mix well.
- Optional: For fewer false positives, expose master mix with UV light prior to forming droplets34.
- Prepare 10 µl of pre-mixture per sample in a 0.5 ml or 1.5 ml clear tube on ice. The pre-mix consists of 55 µM randomized oligo, 2.2 mg/ml BSA, 1.1x Phi29 DNA Polymerase Reaction Buffer, 5.3 mM dNTPs, and nuclease-free water.
- Place the tube in water on ice as described34 and expose to UV light (254 nm) to an accumulated dose of 5.7 J/cm2.
- Add dsDNA-binding dye to 1x, reference dye to 1x, and Phi29 to 40 µg/ml in the pre-mixture. Mix well.
Combine 10 µl of denatured template or standard and 10 µl of master mixture. Mix well.
3. Droplet Formation
Note: Droplets can be very sensitive to static electricity and shear stress. Remove clothes that may cause static electricity, and ground yourself before forming droplets. Handle tubes of emulsion from the top of the tube, as far from the emulsion as possible. Pipette emulsions very slowly, preferably with a wide bore pipet tip. When dispensing from a pipette tip, watch the emulsion on the side of the tip to determine pipetting speed.
Form droplets. For the experiments shown, the microfluidic device has an oil inlet and two aqueous inlets with a flow-focusing junction for generating droplets. Use two syringe pumps to control the flow rates of 55 µl of oil with surfactant and a 20 µl of reaction mixture through the microfluidic chip to generate 20,000 1 nl droplets.
Note: It is possible to produce droplets with many types of oil with surfactant, but the composition will greatly affect droplet stability at high temperatures and over time. One possible combination is fluorinated oil HFE 7500 with an anionic surfactant19,35.
Produce droplets at any size with any method13,36, but the size variability in the droplet population will affect the bulk fluorescence, and therefore the accuracy of the assay. For the experiments shown, droplets were monodisperse with a volume of 1 nl.
Collect all of the droplets and oil into PCR tubes. For each sample, aliquot 30 µl of oil into fresh PCR tubes with optical caps, and transfer 20 µl of droplets on top of the oil. The consistent volumes ensure the assay is as accurate as possible.
Close tube caps tightly, as loose caps will allow the oil to evaporate.
4. Isothermal Amplification
Using a PCR thermocycler, qPCR thermocycler, or hot plate, incubate the samples at 30 °C for 7 hr, and inactivate for 1 min at 75 °C. The tubes of droplets may be left in a fridge covered with foil for a few hours at this point.
5. Data Acquisition and Analysis
Immediately following reaction inactivation, measure dye fluorescence levels for all samples using the qPCR thermocycler. Optimal filter settings will depend on dye excitation and emission spectra. For this example, use the 492 nm-516 nm filter set for the dsDNA-binding dye, and the 585 nm-610 nm filter set for the reference dye. Other fluorescent measurement techniques may be used to quantify the fluorescence.
For each sample, divide the fluorescence intensity of the dsDNA-binding dye by the fluorescence intensity of the reference dye. Subtract the background fluorescence, or the normalized fluorescence of the no template control, from all samples.
Create a linear standard curve using the corrected fluorescence measurements from the standards. The NTC standard represents the fluorescence for a sample with 0% fluorescent droplets (“all negative”), and the high template concentration fluorescence measurement is the expected fluorescence for a sample with 100% fluorescent droplets (“all positive”). Using the corresponding standard curve, predict the intermediate ratios of positive and negative droplets based on their bulk fluorescence.
6. Producing Droplets for Proof-of-Principle Measurements
Make fluorescent (positive) droplets: Prepare a PCR reaction mixture consisting of 0.1 ng Lambda DNA, 1x dsDNA-binding dye, 1x reference dye, 1.5 units Taq DNA polymerase, 10 mM Tris-HCl, 50mM KCl, 1.5 mM MgCl2, 0.2 mM dNTP, and 0.5 µM primers. Thermocycle the mixture 30 times (95 °C for 20 sec, 55 °C for 20 sec, 72 °C for 1 min), then form droplets as described above.
Make non-fluorescent (negative) droplets: Prepare a PCR reaction mixture consisting of 1x dsDNA-binding dye, 1x reference dye, 1.5 units Taq DNA polymerase, 10 mM Tris-HCl, 50mM KCl, 1.5 mM MgCl2, 0.2 mM dNTP, and 0.5 µM primers. Thermocycle the mixture 30 times (95 °C for 20 sec, 55 °C for 20 sec, 72 °C for 1 min), then form droplets.
- Forming pre-mixed droplet ratios
- Distribute 20 µl of fluorinated oil into qPCR tubes, cover with an optical cap to prevent evaporation.
- Gently pipette 10 µl of well-mixed droplets in desired positive:negative ratios on top of the oil. In the results shown, positive:negative droplet ratios were 1 (all positive), 0.8, 0.5, 0.2, 0.1, 0.01, 0.001, 0.0001, and 0 (all negative).
- Measure dye fluorescence levels for all samples using the qPCR thermocycler. For this protocol, use the 492 nm-516 nm filter set for the dsDNA-binding dye, and the 585 nm-610 nm filter set for the reference dye.
- Normalize dsDNA binding dye fluorescence using reference dye fluorescence. Further normalize by the fluorescence of the “all positive” sample.
Create a linear standard curve using the normalized fluorescence measurements from the “all positive” and “all negative” samples. Using the corresponding standard curve, predict the intermediate ratios of positive and negative droplets based on their bulk fluorescence.
Repeat the experiment for each ratio (n = 3) with independent droplet formation and mixing for each replicate.
7. Proof-of-Principle MDA Experiment
- Measure the concentration of Lambda DNA stock solution with a spectrophotometer.
- Calculate the template concentration necessary to average 10 template molecules per droplet. For this protocol, assume droplet volumes are 1 nl and 1 ng of Lambda DNA contains approximately 1.9 x 107 copies. Therefore, 10 templates per droplet will be 10 copies per nl, or 526 fg Lambda DNA per µl. The template will be diluted ~6x when droplets are formed, therefore the initial highest template concentration will be 3.2 pg/µl.
Serially dilute the Lambda DNA to 3.2 pg/µl with Denaturation Buffer and equal volume Neutralization Buffer. For a dilution factor of 0.1, combine 10 µl sample with 45 µl of Denaturation Buffer and incubate at RT for 3 min. Add 45 lL of Neutralization Buffer to quench.
Further dilute the sample as described in step 7.2 to make the rest of the samples for the experiment. The initial template concentrations in the reactions shown were 3.2 pg, 320 fg, 160 fg, 32 fg, 16 fg, 3.2 fg, and 1.6 fg per microliter. Note: The final template concentrations after mastermix addition were 526 fg, 52.6 fg, 26.3 fg, 5.26 fg, 2.63 fg, 526 ag, and 263 ag per microliter. 10, 1, 0.5, 0.1, 0.05, 0.01, and 0.005 expected Lambda copies per droplet, respectively.
Generate and analyze droplets from each sample as described in steps 2.5-5.3.
Acquire the fluorescence images shown (Figure 2,3) using a digital camera mounted to an epi-fluorescence microscope with a 10x objective at RT. Acquire twelve fields of view for each sample. Correct fluorescence images by a background image obtained with a fluorescent slide.
Use a contained chamber for imaging37 as the emulsion oil will evaporate quickly.
- Analyze individual droplet intensities using an ImageJ macro (Supplemental code file).
- Choose a threshold fluorescence intensity that best separates non-fluorescent droplets in the NTC images from the fluorescent droplets in the high template concentration images. For each unknown sample, calculate the fraction of droplets with a fluorescence intensity above the threshold.
8. PCR and MDA bulk reactions
- Prepare PCR reaction.
- Serially dilute template DNA with a dilution factor of 0.1. For each dilution, combine 10 µl of template with 45 µl of Denaturation Buffer and incubate at RT for 3 min. Combine the dilution with 45 µl of Neutralization Buffer.
- Prepare 20 µl of PCR mastermix per sample. The 1.1x PCR reaction master mixture consists of 60 units/µl Taq DNA polymerase, 11 mM Tris-HCl, 55mM KCl, 1.65 mM MgCl2, 0.22 mM dNTP, 1.1x dsDNA-binding dye, 1.1x reference dye, and 550 nM each primer.
- Combine 2 µl of diluted template and 20 µl of mastermix. Note: The final template concentrations in the PCR reactions shown were 50 pg, 5 pg, 500 fg, 50 fg, 5 fg, 500 ag, and 0 g Lambda DNA per microliter, and were performed in triplicate.
- Thermocycle PCR reaction in qPCR machine with the following program. Run 40 cycles of 95 °C for 30 sec, 57 °C for 20 sec, 72 °C for 30 sec before 2 min at 72 °C for 2 min and holding at 4 °C.
- Normalize dsDNA binding dye fluorescence using reference dye fluorescence prior to further analysis.
- Prepare MDA reaction.
- Dilute samples as described in step 8.1.1.
- Prepare 11 µl of master mixture per sample on ice. The 2x MDA reaction master mixture consists of 2x dsDNA-binding dye, 2x reference dye, 50 µM randomized oligo, 2 mg/ml BSA, 2x phi29 DNA Polymerase Reaction Buffer, 4.8 mM dNTPs, nuclease-free water, and 40 µg/ml Phi29 DNA Polymerase.
- Add the Phi29 DNA polymerase to 40 µg/ml last to ensure the polymerase does not encounter pH levels much higher or lower than 7.5. Mix well.
- Denature 3.3 µL of diluted template with 3.3 µl of Denaturation Buffer in a qPCR tube. Incubate at RT for 3 min. Add 3.3 µl of Neutralization Buffer.
- Combine 10 µL of denatured template and 10 µl of master mixture. Mix well.
- Run MDA reaction in qPCR machine with the following program.
- Hold at 30 °C for 4 hr, measure fluorescence every 7.5 min.
- Inactivate at 75 °C for 1 min.
- Normalize dsDNA binding dye fluorescence using reference dye fluorescence. Further normalize by the maximum fluorescence for each sample.
Representative Results
While conventional bulk/real-time readouts can be used for both quantitative PCR and quantitative WGA assays (Figure 1), digital quantitative assays provide advantages (Table 1). In the method described, we read out digital assays in micro-droplet format with a simple bulk endpoint measurement (Figure 2). While this method is broadly applicable, we focus on quantitative WGA (MDA) because this method presents special challenges for conventional real-time assays.
To check whether our real-time PCR instrument could detect varying fractions of fluorescent microdroplets dispersed in oil, we measured the bulk fluorescence of synthetic mixtures of fluorescent and non-fluorescent droplets (Figure 2). The bulk fluorescence and positive droplet counts (independently assessed by fluorescence microscopy) scale linearly with the input fraction of fluorescent droplets as expected (Figure 3A). The experiment was performed three times, with independent droplet formation and mixing for each set.
To evaluate the theoretical quantification performance for “unknown” samples, we established linear standard curves using entirely positive and entirely negative control samples. With such droplet-lot-specific standard curves, we calculated the fraction of synthetic positive and negative droplet mixtures based on each sample’s bulk fluorescence. The results show good performance in droplet ratio quantification across two logs of dynamic range (R2 = 0.984; Figure 3B). The input analyte concentration is easily calculated from the fraction of positive or negative droplets38.
To test our method in a real quantitative WGA assay, we measured fluorescence levels and fraction of positive droplets of a digital droplet MDA assay with Lambda DNA across a wide range of concentrations (Figure 4). In Figure 4B, representative fluorescent images of the droplets are shown. Both bulk fluorescence and positive droplet fraction from the digital MDA samples scale as expected with the average template per droplet, indicating that the bulk readout can faithfully capture the result of a digital assay (R2 = 0.927). We repeated the experiment for each concentration, independent forming droplets and carrying out WGA for each replicate.
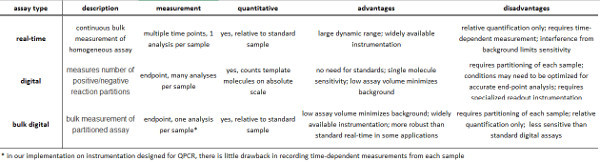 Table 1. Summary of real-time and digital assays.
Please click here to view a larger version of this figure.
Table 1. Summary of real-time and digital assays.
Please click here to view a larger version of this figure.
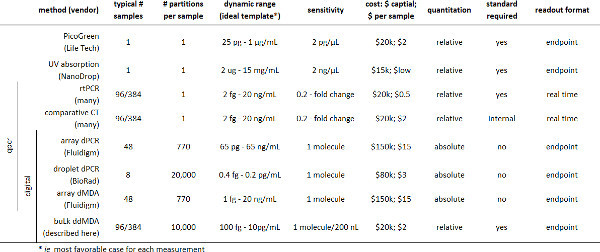 Table 2. Summary of existing methods for quantitating nucleic acids.
Please click here to view a larger version of this figure.
Table 2. Summary of existing methods for quantitating nucleic acids.
Please click here to view a larger version of this figure.
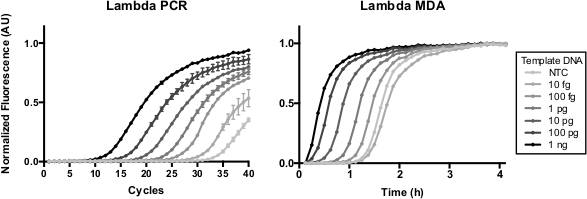 Figure 1. Real-time quantitative PCR and MDA of Lambda DNA. MDA is not discretized through temperature cycling like PCR, and is prone to preamplification if not prepared carefully on ice. Here, quantitative PCR and MDA were performed with increasing concentrations of Lambda DNA. Please click here to view a larger version of this figure.
Figure 1. Real-time quantitative PCR and MDA of Lambda DNA. MDA is not discretized through temperature cycling like PCR, and is prone to preamplification if not prepared carefully on ice. Here, quantitative PCR and MDA were performed with increasing concentrations of Lambda DNA. Please click here to view a larger version of this figure.
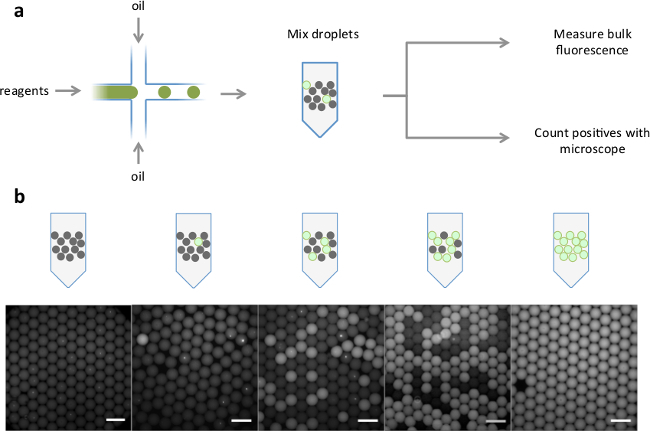 Figure 2. Schematic of analog readout of droplets and representative images. (A) Positive and negative droplets were generated from fluorescent and non-fluorescent reagents in a microfluidic device. The droplets were pre-mixed in different positive:negative ratios. Bulk fluorescence levels were measured with a standard real-time thermocycler, and fraction of positive droplets was determined by fluorescence microscopy. (B) Representative fluorescent overlaid images of increasing ratios of positive droplets (scale bar = 200 µm). Please click here to view a larger version of this figure.
Figure 2. Schematic of analog readout of droplets and representative images. (A) Positive and negative droplets were generated from fluorescent and non-fluorescent reagents in a microfluidic device. The droplets were pre-mixed in different positive:negative ratios. Bulk fluorescence levels were measured with a standard real-time thermocycler, and fraction of positive droplets was determined by fluorescence microscopy. (B) Representative fluorescent overlaid images of increasing ratios of positive droplets (scale bar = 200 µm). Please click here to view a larger version of this figure.
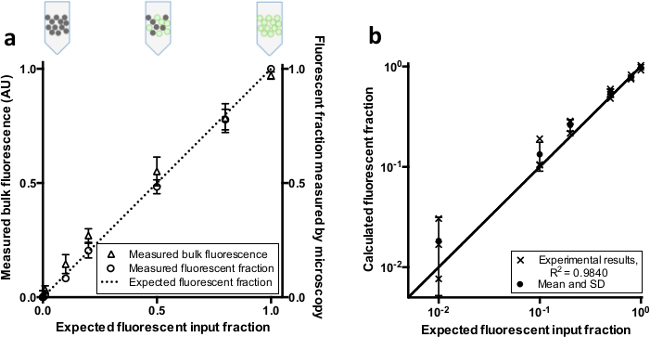 Figure 3. Bulk fluorescence and fluorescent fraction of combinations of separately prepared positive and negative droplets. (A) Fluorescent (positive) and non-fluorescent (negative) droplets were pre-mixed in different ratios. Comparison of the input fraction of positive droplets with the measured bulk fluorescence and measured positive droplet fraction (n = 3, bars represent +/- SD). The dotted line represents the expected fluorescent fraction given a linear relationship. (B) Comparison of predicted ratios based on standards to the expected fluorescent input fraction. The line indicates the expected value given a linear relationship. Please click here to view a larger version of this figure.
Figure 3. Bulk fluorescence and fluorescent fraction of combinations of separately prepared positive and negative droplets. (A) Fluorescent (positive) and non-fluorescent (negative) droplets were pre-mixed in different ratios. Comparison of the input fraction of positive droplets with the measured bulk fluorescence and measured positive droplet fraction (n = 3, bars represent +/- SD). The dotted line represents the expected fluorescent fraction given a linear relationship. (B) Comparison of predicted ratios based on standards to the expected fluorescent input fraction. The line indicates the expected value given a linear relationship. Please click here to view a larger version of this figure.
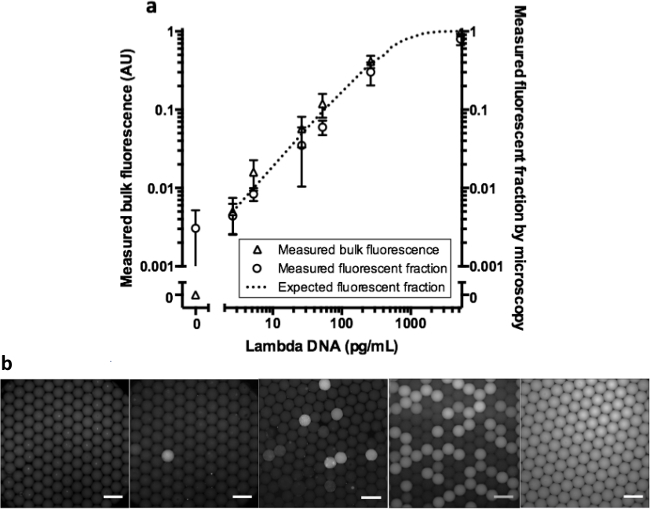 Figure 4. Bulk fluorescence and fluorescent fraction of a droplet digital MDA assay. Digital MDA was performed with increasing concentrations of template Lambda DNA (n = 3, error bars represent SD) and measured as described in Figure 2A. The line indicates the expected fluorescent fraction using the Poisson distribution to model the data from our experiment. b) Representative fluorescent overlaid images of droplets after reaction inactivation (scale bar = 200 µm) for increasing expected Lambda DNA molecules per droplet (NTC, 0.005, 0.05, 0.5, and 10). Please click here to view a larger version of this figure.
Figure 4. Bulk fluorescence and fluorescent fraction of a droplet digital MDA assay. Digital MDA was performed with increasing concentrations of template Lambda DNA (n = 3, error bars represent SD) and measured as described in Figure 2A. The line indicates the expected fluorescent fraction using the Poisson distribution to model the data from our experiment. b) Representative fluorescent overlaid images of droplets after reaction inactivation (scale bar = 200 µm) for increasing expected Lambda DNA molecules per droplet (NTC, 0.005, 0.05, 0.5, and 10). Please click here to view a larger version of this figure.
Discussion
Digital assays are powerful methods that enable detection of rare cells and counting of individual of nucleic acid molecules. However, digital assays are still not routinely applied in analytical laboratories, due in part to the cost of specialized equipment associated with commercially available methods. Here we describe an endpoint digital assay for quantifying nucleic acids with a simplified analog readout using a standard real-time quantitative PCR machine. This method is quick to set-up, and readout is much simpler than droplet counting methods.
Our assay forfeits the single-molecule (single-droplet) sensitivity of individual droplet measurements for the simplicity of en masse endpoint measurement using standard instrumentation. In our digital MDA experiment, we could not distinguish less than 1 positive droplet per 200 negative droplets from background, despite confirmation by fluorescence microscopy that the actual fraction of positive droplets was as expected (Figure 4). The sensitivity and background in the bulk fluorescence measurement limit the sensitivity of the bulk measurement assay for low positive droplet fractions. If higher sensitivity is required, a digital readout is recommended. Differences in oil levels or bulk droplet volumes can greatly affect the sample fluorescence data. Even normalization with a reference dye cannot make up for the differences oil volumes make in fluorescence reads. It is possible that optimizing assay volume, instrument acquisition parameters, microwell geometry, or optical filters may improve assay performance.
The sensitivity and dynamic range of bulk digital assays can also be improved by optimizing the partition size. Larger droplets yield a greater quantity of fluorescent products from each template molecule, which can help overcome instrumental background and improve sensitivity for low input concentrations. On the other hand, smaller droplets allow the isolation of more molecules per volume assayed. If template concentration is highly variable across samples, both small and large droplet reactions can be carried out in parallel to expand the assay dynamic range39.
Consistent droplet volumes are also necessary for accurate measurement. Many custom microfluidic solutions exist for rapidly forming monodisperse droplets from a sample, both in series19,40 and in parallel41,42. While few enable the droplet generation from several separate samples in parallel, simple modifications to existing designs would allow for simultaneous droplet generation of any number of samples. For example, the Bio-Rad ddPCR system23 applies uniform pressure to the inlets of a series of independent droplet makers, enabling simultaneous droplet formation from multiple samples.
Our bulk readout methodology accesses key advantages of partitioned digital assays without the need for specialized readout instrumentation and is well-suited for quantitative WGA. Here, we focused on quantitative WGA, which is highly susceptible to background contaminants due to its lack of sequence specificity7. In addition, the unknown and potentially broad molecular weight distribution of products from real-time quantitative WGA assays make interpretation difficult in applications where the number of original templates is of interest. Digital assay formatting addresses both these problems. Contaminants are contained within individual reaction partitions, preventing the dominance of low-molecular-weight analytes by high-molecular-weight contaminants as would occur in a conventional real-time assay. In Figures 2B and 4B, possible contaminants, seen as bright spots within the droplets, are segregated and do not amplify or contribute substantially to overall fluorescence. Furthermore, the signal generated in digital assays is proportional to the number of templates, not the size of templates or their amplification rate. Although our method relies on standards to calibrate the bulk digital measurement, the requirement to match sample and standard characteristics can be relaxed significantly.
Quantitative WGA is commonly used to quantify contaminants in WGA reactions7,34, and is the most sensitive method known to quantify the presence of DNA of unknown sequence, giving the method great promise in application areas as diverse as pharmaceutical quality control, forensics, and astrobiology. Bulk digital readout can benefit other digital assay protocols such as digital PCR, by speeding up, parallelizing, and simplifying assay readout.
Disclosures
The Broad Institute may move to file a patent application that includes aspects of this work.
Acknowledgments
The authors acknowledge Liyi Xu for providing MDA reagents and protocols. We also thank David Feldman and Navpreet Ranu for discussions relating to this work. This work was supported in part by a Burroughs Wellcome Career Award at the Scientific Interface to PCB.
References
- Sykes PJ, Neoh SH, Brisco MJ, Hughes E, Condon J, Morley AA. Quantitation of targets for PCR by use of limiting dilution. Biotechniques. 1992;13(3):444–449. [PubMed] [Google Scholar]
- Kalinina O, Lebedeva I, Brown J, Silver J. Nanoliter scale PCR with TaqMan detection. Nucleic Acids Res. 1997;25(10):1999–2004. doi: 10.1093/nar/25.10.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Kinzler KW. Digital PCR. Proc. Natl. Acad. Sci. U.S.A. 1999;96(16):9236–9241. doi: 10.1073/pnas.96.16.9236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day E, Dear PH, McCaughan F. Digital PCR strategies in the development and analysis of molecular biomarkers for personalized medicine. Methods. 2013;59(1):101–107. doi: 10.1016/j.ymeth.2012.08.001. [DOI] [PubMed] [Google Scholar]
- Tatusova T, Ciufo S, Fedorov B, O'Neill K, Tolstoy I. RefSeq microbial genomes database: new representation and annotation strategy. Nucleic Acids Res. 2014;42(Database issue):D553–D559. doi: 10.1093/nar/gkt1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blainey PC. The future is now: single-cell genomics of bacteria and archaea. FEMS Microbiol. Rev. 2013;37(3):407–427. doi: 10.1111/1574-6976.12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blainey PC, Quake SR. Digital MDA for enumeration of total nucleic acid contamination. Nucleic Acids Res. 2011;39(4):e19. doi: 10.1093/nar/gkq1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blainey PC, Quake SR. Dissecting genomic diversity, one cell at a time. Nat. Methods. 2014;11(1):19–21. doi: 10.1038/nmeth.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binga EK, Lasken RS, Neufeld JD. Something from (almost) nothing: the impact of multiple displacement amplification on microbial ecology. ISME J. 2008;2(3):233–241. doi: 10.1038/ismej.2008.10. [DOI] [PubMed] [Google Scholar]
- Method of the Year 2013. Nature Methods. 2014;11(1):1. doi: 10.1038/nmeth.2801. [DOI] [PubMed] [Google Scholar]
- Ottesen EA, Hong JW, Quake SR, Leadbetter JR. Microfluidic digital PCR enables multigene analysis of individual environmental bacteria. Science. 2006;314(5804):1464–1467. doi: 10.1126/science.1131370. [DOI] [PubMed] [Google Scholar]
- OpenArray Technology Overview. Life Technologies; 2014. http://www.lifetechnologies.com/us/en/home/life-science/pcr/real-time-pcr/real-time-openarray/open-array-technology.html. [Google Scholar]
- Shen F, Du W, Kreutz JE, Fok A, Ismagilov RF. Digital PCR on a SlipChip. Lab Chip. 2010;10(20):2666–2672. doi: 10.1039/c004521g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyries KA, et al. Megapixel digital PCR. Nat. Methods. 2011;8(8):649–651. doi: 10.1038/nmeth.1640. [DOI] [PubMed] [Google Scholar]
- Dressman D, Yan H, Traverso G, Kinzler KW, Vogelstein B. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc. Natl. Acad. Sci. U.S.A. 2003;100(15):8817–8822. doi: 10.1073/pnas.1133470100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss MM, et al. High-throughput quantitative polymerase chain reaction in picoliter droplets. Anal. Chem. 2008;80(23):8975–8981. doi: 10.1021/ac801276c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M. Digital PCR hits its stride. Nat. Methods. 2012;9(6):3. [Google Scholar]
- Marcus JS, Anderson WF, Quake SR. Parallel picoliter rt-PCR assays using microfluidics. Anal. Chem. 2006;78(3):956–958. doi: 10.1021/ac0513865. [DOI] [PubMed] [Google Scholar]
- Lee M, et al. Synchronized reinjection and coalescence of droplets in microfluidics. Lab Chip. 2014;14(3):509–513. doi: 10.1039/c3lc51214b. [DOI] [PubMed] [Google Scholar]
- Rhee M, et al. Pressure stabilizer for reproducible picoinjection in droplet microfluidic systems. Lab Chip. 2014;14(23):4533–4539. doi: 10.1039/c4lc00823e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch AC, et al. 1-Million droplet array with wide-field fluorescence imaging for digital PCR. Lab Chip. 2011;11(22):3838–3845. doi: 10.1039/c1lc20561g. [DOI] [PubMed] [Google Scholar]
- Illumina. Technology; 2014. http://www.illumina.com/technology.html. [Google Scholar]
- Droplet digital PCR applications guide. Bio-Rad; 2014. Available from: http://www.bio-rad.com/en-us/category/digital-pcr. [Google Scholar]
- Hindson BJ, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem. 2011;83(22):8604–8610. doi: 10.1021/ac202028g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo MT, Rotem A, Heyman JA, Weitz DA. Droplet microfluidics for high-throughput biological assays. Lab Chip. 2012;12(12):2146–2155. doi: 10.1039/c2lc21147e. [DOI] [PubMed] [Google Scholar]
- Lage JM, et al. Whole genome analysis of genetic alterations in small DNA samples using hyperbranched strand displacement amplification and array-CGH. Genome Res. 2003;13(2):294–307. doi: 10.1101/gr.377203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean FB, et al. Comprehensive human genome amplification using multiple displacement amplification. Proc. Natl. Acad. Sci. U.S.A. 2002;99(8):5261–5266. doi: 10.1073/pnas.082089499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong C, Lu S, Chapman AR, Xie XS. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science. 2012;338(6114):1622–1626. doi: 10.1126/science.1229164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitos Dropix Systems. Dolomite; 2014. http://www.dolomite-microfluidics.com/webshop/mitos_dropix_system. [Google Scholar]
- Abate AR, Weitz DA. Syringe-vacuum microfluidics: A portable technique to create monodisperse emulsions. Biomicrofluidics. 2011;5:14107. doi: 10.1063/1.3567093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyer K, Kuhn P, Stratz S, Dittrich PS. A microfluidic chip for the versatile chemical analysis of single cells. J Vis Exp. 2013. p. e50618. [DOI] [PMC free article] [PubMed]
- Fainman Y. Optofluidics : fundamentals, devices, and applications. McGraw-Hill; 2010. [Google Scholar]
- Xia YW. G.M. Softlithographie. Angew Chem. 1998;110(5):26. [Google Scholar]
- Woyke T, et al. Decontamination of MDA reagents for single cell whole genome amplification. PLoS One. 2011;6(10):e26161. doi: 10.1371/journal.pone.0026161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindson BJ, inventor. System for hot-start amplification via a multiple emulsion. US patent. 2014.
- Utada AS, et al. Monodisperse double emulsions generated from a microcapillary device. Science. 2005;308(5721):537–541. doi: 10.1126/science.1109164. [DOI] [PubMed] [Google Scholar]
- Eastburn DJ, Sciambi A, Abate AR. Picoinjection enables digital detection of RNA with droplet rt-PCR. PLoS One. 2013;8(4):e62961. doi: 10.1371/journal.pone.0062961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube S, Qin J, Ramakrishnan R. Mathematical analysis of copy number variation in a DNA sample using digital PCR on a nanofluidic device. PLoS One. 2008;3(8):e2876. doi: 10.1371/journal.pone.0002876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen F, et al. Multiplexed quantification of nucleic acids with large dynamic range using multivolume digital RT-PCR on a rotational SlipChip tested with HIV and hepatitis C viral load. J Am. Chem. Soc. 2011;133(44):17705–17712. doi: 10.1021/ja2060116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link DR, Anna SL, Weitz DA, Stone HA. Geometrically mediated breakup of drops in microfluidic devices. Phys. Rev. Lett. 2004;92(5):054503. doi: 10.1103/PhysRevLett.92.054503. [DOI] [PubMed] [Google Scholar]
- Nisisako T, Torii T. Microfluidic large-scale integration on a chip for mass production of monodisperse droplets and particles. Lab Chip. 2008;8(2):287–293. doi: 10.1039/b713141k. [DOI] [PubMed] [Google Scholar]
- Romanowsky MB, Abate AR, Rotem A, Holtze C, Weitz DA. High throughput production of single core double emulsions in a parallelized microfluidic device. Lab Chip. 2012;12(4):802–807. doi: 10.1039/c2lc21033a. [DOI] [PubMed] [Google Scholar]