

Abstract
The SOD1-G93A transgenic mouse is the most widely used animal model of amyotrophic lateral sclerosis (ALS). At ALS TDI we developed a phenotypic screening protocol, demonstrated in video herein, which reliably assesses the neuromuscular function of SOD1-G93A mice in a quick manner. This protocol encompasses a simple neurological scoring system (NeuroScore) designed to assess hindlimb function. NeuroScore is focused on hindlimb function because hindlimb deficits are the earliest reported neurological sign of disease in SOD1-G93A mice. The protocol developed by ALS TDI provides an unbiased assessment of onset of paresis (slight or partial paralysis), progression and severity of paralysis and it is sensitive enough to identify drug-induced changes in disease progression. In this report, the combination of a detailed manuscript with video minimizes scoring ambiguities and inter-experimenter variability thus allowing for the protocol to be adopted by other laboratories and enabling comparisons between studies taking place at different settings. We believe that this video protocol can serve as an excellent training tool for present and future ALS researchers.
Keywords: Medicine, Issue 104, High-throughput, in vivo screen, transgenic mice, G93A, neurological score, survival, progression, therapy
Introduction
Ever since its development in the mid 1990’s the SOD1-G93A transgenic mouse model has been the most widely used animal model of amyotrophic lateral sclerosis (ALS) 1. This transgenic mouse model was genetically engineered to overexpress a mutant form of the human Cu/Zn superoxide dismutase 1(SOD1) gene harboring the ALS-associated glycine to alanine mutation at amino acid 93 (G93A). The G93A mutation is one of many mutations in the SOD1 gene that collectively make-up approximately one fifth of familial ALS cases 2.
The SOD1-G93A model has become a workhorse of ALS drug development research because in addition to its obvious genetic link to ALS, it recapitulates many of the pathological features of ALS in humans such as motor neuron loss, progressive muscle weakness and atrophy, with eventual paralysis and death 3. Though, as is often the case with transgenic animal models, there is an inherent biological variability associated with SOD1-G93A mice. Scott et al. have determined that cohorts of at least 24 litter-matched gender-balanced mice are needed in drug-efficacy study designs in order to overcome the noise created by the biological variability 4. The large number of animals required in these studies in combination with the aggressive disease progression and the need for daily monitoring prohibits the use of elaborate, time-consuming and potentially stressful techniques for measuring neuromuscular strength and function (e.g., electromyography, grip strength, rotorod, etc.). Instead, it highlights the need for an in vivo screening tool that reliably assesses the neuromuscular function of SOD1-G93A mice in a quick manner.
At ALS TDI a protocol was developed that allows for the reliable assessment of the neuromuscular function of SOD1-G93A mice in less than 30 sec per mouse on average. This protocol encompasses daily assessment of hindlimb function using a simple neurological scoring system (NeuroScore) which is described in this report in press and in video. NeuroScore is focused on hindlimb function because hindlimb deficits are the earliest reported neurological sign of disease in SOD1-G93A mice 5,6. Further, the protocol developed by ALS TDI provides an unbiased assessment of onset of paresis (slight or partial paralysis), progression and severity of paralysis and it is sensitive enough to identify drug-induced changes in disease progression.
Protocol
All experiments are conducted in accordance with the protocols described by the National Institutes of Health Guide for the Care and Use of Animals and were approved by ALS TDI’s institutional animal care and use committee (IACUC).
1. Animals, Housing, and Study Design
Note: The SOD1-G93A mouse colony was derived from the high copy B6SJLTgN(SOD1G93A)1Gur strain originally produced by Gurney et al. 7. The colony is currently being maintained by crossing transgenic C57BL/6-SJL males with wild-type C57BL/6-SJL F1 females. The F1 animals are generated by crossing SJL males with B6 females. Mice are shipped to ALS TDI weekly at 35–45 days of age.
Allow mice at least one week to acclimate to the animal facility (12-hr light/dark cycle at a temperature of 18-23 °C and 40-60% humidity) before assigning to a study.
House male SOD1-G93A mice one mouse per cage, since group housed males tend to fight. House female SOD1-G93A mice up to two per cage. Provide environmental enrichment, in the form of plastic huts. Provide food and water ad libitum, unless the mice are part of a study requiring drug administration in drinking water/food. In these cases, ration and record food and water.
Begin a drug-efficacy study around age day 50 and include 64 litter-matched gender-balanced mice (32 per cohort). Finally, ensure that the researchers conducting the studies are blinded to the experimental conditions. Note: It is recommended that a typical drug-efficacy study starts at age day 50 however based on the type of therapeutic intervention it might be preferable to start the study earlier or later. Note: A study with 32 mice per cohort is powered to detect gender-specific drug-effects. If no gender-specific comparisons are required, 24 mice per cohort should suffice to detect overall drug-effects.
2. Body Weight
Record body weights for all mice in studies daily with a precision of one tenth of a gram. Note: Body weight is a sensitive indicator of disease progression and of any malaise that might result from chronic drug treatment.
Mouse body weights vary considerably during the course of a day, being higher in the mornings, thus record them around the same time every day.
3. Neurological Scoring System (NeuroScore)
- Assess neurological scores (NS) for each mouse daily by observing the mouse under the following three conditions performed sequentially: a) the mouse is suspended by its tail, b) the mouse is allowed to walk, and after the onset of paresis c) the mouse is placed on its side.
- For the tail suspension test, hold the mouse approximately 1.5” from the base of the tail over the wire top of their home cage for 1-2 sec, away from the food basin, while observing the hindlimbs. Repeat the suspension test 3 times and record the most consistent outcome.
- For the walking test, place the mouse on a clean surface that provides some traction and a walking distance of 25 cm (e.g., a paper towel taped down to prevent slipping). Allow the mouse to walk a total of 75 cm (3X length of the paper towel) while observing its gait.
- For the “righting reflex” test, place the mouse on its left or right side and using a stop-watch measure the time it takes to right itself up unassisted from both sides. Note: Only one trial of this test is required.
Determine the NS of each hindlimb (left or right) independently on a scale from 0 to 4. Note: For instance a score of 0, 1 indicates that the left hindlimb NS is 0, while the right hindlimb NS is 1.
- Assign neurological scores (0-4) corresponding to the following set of observations (summary provided in Table 1):
- Assign NS 0 (Pre-symptomatic) if the following is observed: When the mouse is suspended by the tail, the hindlimb presents a normal splay i.e., it is fully extended away from the lateral midline and it stays in this position for 2 sec or longer. When the mouse is allowed to walk, normal gait is observed.
- Assign NS 1 (First symptoms) if the following is observed: When the mouse is suspended by the tail, the hindlimb presents an abnormal splay, i.e., it is collapsed or partially collapsed towards lateral midline OR it trembles during tail suspension OR it is retracted/clasped. When the mouse is allowed to walk, normal OR slightly slow gait is observed.
- Assign NS 2 (Onset of paresis) if the following is observed: When the mouse is suspended by the tail, the hindlimb is partially OR completely collapsed, not extending much. (There might still be joint movement). When the mouse is allowed to walk, the hindlimb is used for forward motion however the toes curl downwards at least twice during a 90 cm walk OR any part of the foot is dragging along cage bottom/table. When the mouse is placed on its left AND right side, it is able to right itself within 10 sec from BOTH sides.
- Assign NS 3 (Paralysis) if the following is observed: When the mouse is suspended by the tail, there is rigid paralysis in the hindlimb OR minimal joint movement. When the mouse is allowed to walk there is forward motion however the hindlimb is NOT being used for forward motion. When the mouse is placed on its left AND right side, it is able to right itself within 10 sec from BOTH sides. Note: Rarely, after the onset of paralysis, urine moisture might appear on the hindlimbs. Left untreated urine moisture might result to urine “burn” and skin lesions. Treat urine moisture by clipping the hair, apply warm water soaks to remove the urine, and gently blot dry. If skin lesions are present apply antibiotic ointment.
- Assign NS 4 (Humane end-point) if the following is observed: When the mouse is suspended by the tail, there is rigid paralysis in the hindlimbs. When the mouse is allowed to walk, there is no forward motion. When the mouse is placed on its left AND right side it is NOT able to right itself within 10 sec from EITHER side. i.e., absence of righting reflex.
4. Data Entry and Analysis
During the course of a drug efficacy study, enter data collected for each mouse into a Laboratory Information Management System (LIMS) or into a commercially available spreadsheet software. Note: The ALS TDI-developed LIMS software is a central repository for all data collected from internal studies, making these data conveniently accessible over time for ALS TDI investigators. LIMS tracks any individual mouse, with its unique ID, back to its lineage, body weight or neurological score on all study days, and includes any other observations noted during its lifetime. LIMS ultimately generates a spreadsheet which is used for subsequent analyses.
Upon the completion of a drug efficacy study, i.e., when all mice reach the humane end-point, use spreadsheet software (see Table of Materials) to compute the combined NS for each mouse, for each day by averaging the left and right hindlimb NS. Next, compute the median age at each NS for each mouse and the number of days each mouse spends at a particular NS.
Analyze overall changes in NS over time by importing the data from the spreadsheet software to the statistical software (see Table of Materials). Fit these data using a model of ordinal logistic regression which estimates the expected NS, with median age at NS and drug-treatment as effects in the regression. Using the log-rank chi square test estimate the effect likelihood probability and determine whether the drug-treatment effect on NS progression is statistically significant (see Supplemental Code File for step-by-step instructions). Note: A p-value <0.05 is considered statistically significant. Note: This method of statistical analysis respects the ordinal nature of the NS data and the variable nature of the NS trajectories for individual mice. It is preferred because individual NS trajectories can be markedly different resulting in NS time-associated group averages that are not sufficiently reflective of NS responses. In addition, in some cases NS can fluctuate, particularly when in the zero-to-one range.
Finally, use graphing software (see Table of Materials) to generate “Ordinal Expected NS vs Median Age at NS” graphs for drug-treated and control animals (Figure 2; see Supplemental Code File for step-by-step instructions). Use these graphs to conveniently estimate shifts between drug-treated and control groups by interpolating from the y-axis the median age of disease onset (i.e., NS 2) on the x-axis. Note: The difference in median age between the two groups represents a drug-induced shift and is treated as a practical measure of the magnitude of effect. Note: Rightward shifts in the expected NS vs. median age curve indicate slowed symptomatic disease progression. Conversely, leftward shifts in the expected NS vs. median age curve indicate accelerated symptomatic disease progression.
Representative Results
The neurological score data for 90 male and 94 female non-treated SOD1-G93A mice studied at ALS TDI during 2014 were evaluated. The results show that male mice typically have a more aggressive disease progression than female mice, as evidenced by a greater proportion of their lifespan at NS 1 compared to females. NS 2 and NS 3 occur with virtually equal frequency across genders. NS 0 was excluded from the analysis because it is considered a “normal” phenotype. NS 4 was also excluded because by definition it can only occur once in a SOD1-G93A mouse’s lifetime (Table 2 and Figure 1).
NS data is presented from an ALS disease progression efficacy pharmacology study of test compound TDI173 in SOD1-G93A mice (n=16 per gender/treatment; 64 total mice). In Figure 2, the rate of neurological disease progression is presented as captured by ordinal regression analysis of data captured using the NeuroScore protocol detailed herein. TDI173-treated mice were compared to vehicle treated control. No statistically significant differences were observed when genders were separately analyzed but statistical significance was reached when genders were merged. The Kaplan-Meier analysis of survival, even though showing similar trends as the ordinal logistic statistical analysis, was not sensitive enough to reveal significant differences even when genders were merged (Figure 3).
| NS 1 | The hindlimb presents an abnormal splay, i.e., it is collapsed or partially collapsed towards lateral midline OR it trembles during tail suspension OR it is retracted/clasped. | Normal OR slightly slower gait. | NA |
| NS 2 | The hindlimb is partially OR completely collapsed, not extending much. There might still be joint movement. | The hindlimb is used for forward motion however the toes curl downwards at least twice during a 75 cm walk OR any part of the foot is dragging along cage bottom/table. | The mouse is able to right itself within 10 sec from BOTH sides. |
| NS 3 | Rigid paralysis in the hindlimb OR minimal joint movement. | Forward motion however the hindlimb is NOT being used for forward motion. | The mouse is able to right itself within 10 sec from BOTH sides. |
| NS 4 | Rigid paralysis in the hindlimbs. | No forward motion. | The mouse is NOT able to right itself within 10 sec from EITHER side. |
Table 1: Summary of phenotypic observations used to assign neurological scores.
| Frequency of NS1 | Frequency of NS2 | Frequency of NS3 | ||||
| Male | Female | Male | Female | Male | Female | |
| Number of Mice | 90 | 94 | 90 | 94 | 90 | 94 |
| Minimum | 12 | 5 | 1 | 1 | 0 | 0 |
| 25% | 29 | 16 | 4 | 4 | 0 | 0 |
| Median | 37 | 27.5 | 5 | 5 | 1 | 1 |
| 75% | 44 | 37.5 | 7.5 | 6.5 | 2.5 | 3 |
| Maximum | 61 | 60 | 28 | 11 | 8 | 9 |
Table 2: A frequency analysis of NS data from non-treated SOD1-G93A mice scored daily from age day 50 until death. Distributions were calculated for NS 1, 2, and 3.
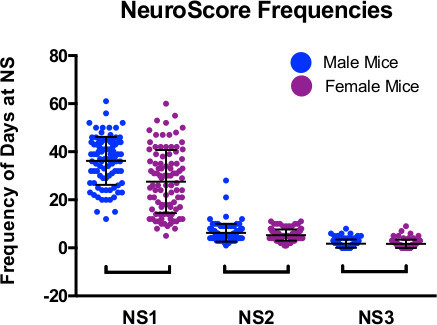 Figure 1. Graphical representation of the number of days spent at each abnormal NeuroScore for each mouse from the population of untreated SOD1-G93A mice in our colony during 2014 (male n=90, female n=94). The score frequencies for the same mice are represented for each possible NS. NS 4 was omitted because the event can only occur once in each mouse’s lifetime by definition. Please click here to view a larger version of this figure.
Figure 1. Graphical representation of the number of days spent at each abnormal NeuroScore for each mouse from the population of untreated SOD1-G93A mice in our colony during 2014 (male n=90, female n=94). The score frequencies for the same mice are represented for each possible NS. NS 4 was omitted because the event can only occur once in each mouse’s lifetime by definition. Please click here to view a larger version of this figure.
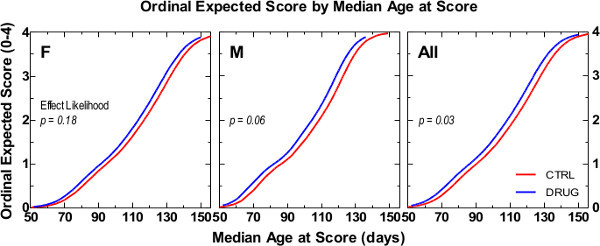 Figure 2. A typical ordinal logistic statistical analysis graph comparing neurological disease progression of TDI173-treated mice (red line) to litter matched vehicle-treated control mice (blue line; Reported p-values represent log-rank chi square test).Please click here to view a larger version of this figure.
Figure 2. A typical ordinal logistic statistical analysis graph comparing neurological disease progression of TDI173-treated mice (red line) to litter matched vehicle-treated control mice (blue line; Reported p-values represent log-rank chi square test).Please click here to view a larger version of this figure.
 Figure 3. A typical Kaplan-Meier analysis graph comparing lifespan of TDI173-treated mice (red line) to litter matched vehicle-treated control mice (blue line; Reported p-values represent log-rank test).Please click here to view a larger version of this figure.
Figure 3. A typical Kaplan-Meier analysis graph comparing lifespan of TDI173-treated mice (red line) to litter matched vehicle-treated control mice (blue line; Reported p-values represent log-rank test).Please click here to view a larger version of this figure.
Discussion
In this report, we describe a quick and simple video protocol which, if properly applied, is capable of reliably assessing the disease progression in SOD1-G93A mice and identifying drug-induced changes. While different groups have developed phenotypic scoring systems for SOD1-G93A mice 8-10, they often do not provide sufficient details on the procedure and are inadequate for replication. As a result, scoring systems are rarely used outside the specific group that develops them. In this report, the combination of a detailed manuscript with video minimizes scoring ambiguities and inter-experimenter variability thus allowing for the protocol to be adopted by other laboratories and enabling comparisons between studies taking place at different settings.
The neurological scoring system described in this report, referred to as NeuroScore, generates ordinal data where the exact numerical values ascribed to the sets of signs and symptoms have no meaning beyond establishing ranked disease severity. In this case, NS 0 is the least severe disease state and signifies a mouse with normal hindlimb function while NS 4 is the most severe disease state with mice at or approaching morbidity secondary to neuromuscular disease. Even though the scoring system makes use of symptomatic thresholds, each ordinal NS value encompasses a wide spectrum of possible clinical presentations many of which were captured on video and included in this report.
NS 1 encompasses the spectrum of early symptoms of disease in SOD1-G93A mice, which can vary widely from mouse to mouse. Some mice exhibit an abnormal splay with the hindlimbs collapsing towards the midline while others show hindlimb tremors and some even show hindlimb retraction or clasping. Furthermore, in the early stages of disease the NS of some mice might fluctuate for several days between NS 1 and 0. Invariably though, in SOD1-G93A mice the frequency of NS 1 increases with time and it includes more and more of the different phenotypes described. On average, male and female SOD1-G93A mice typically spend 37 and 28 days respectively at this stage.
NS 2 represents the onset of paresis (slight or partial paralysis) and provides undeniable evidence for the presence of pathology. While it is not uncommon for a wild-type mouse to sometimes present as NS 1, NS 2 only occurs in SOD1-G93A mice. Similarly to NS 1, NS 2 captures a spectrum of signs and symptoms. In an early stage presentation of NS 2 the mouse is ambulatory and energetic. A more advanced presentation of NS 2 reveals a mouse with a severely limited ability to move. Advanced paresis in the hindlimbs may prevent the mice from reaching the food and water at the top of the cage, so food pellets should be placed at the bottom of the cage or hydrated food should be provided. Also, longer sipper tubes should be placed on water bottles. SOD1-G93A mice typically spend 5 days at this stage.
NS 3 signifies complete hindlimb paralysis, and by all accounts is the easiest phenotype to identify and score. The paralysis symptoms encompassed by NS 3 may be either flaccid or spastic, but are typically spastic. By this point, the mice are very sick and should be monitored frequently. On average, SOD1-G93A mice spend 1 to 2 days at this stage.
NS 4 denotes the absence of “righting reflex” which does not exclusively depend on hindlimb function like NS 0-3 but instead it reflects the whole body wellness of the mouse. The ability of a mouse to demonstrate a normal “righting reflex” requires motivation, energy, but also the function of multiple muscle groups including hindlimbs, forelimbs, and abdomen. It is important to note that disease progression in these mice is often not symmetrical and a mouse that can successfully complete the righting reflex when placed on one side may be incapable of doing so when placed on the other side. It is therefore important, for the humane treatment of animals and for the integrity of the study, to perform testing on both sides and euthanize the mouse if it fails to right itself on either side.
NS 4 defines the date of euthanasia and by default directly aligns with survival data. However, the overall disease progression, as defined by ordinal regression analysis of NS, might not completely overlap with Kaplan-Meier survival curves and body weight data. Certain therapeutic interventions may influence disease onset or symptoms, but fail to impact overall lifespan.
While not discussed in depth in this report, body weight can be another measure that can be used in combination to NS to assess disease onset and progression in SOD1-G93A however it can be skewed by differences in diets and by therapeutics that cause edema or influence fat metabolism11,12.
The greatest asset of the ALS TDI protocol is its simplicity and quick execution. This can come at the expense of not capturing certain disease phenotypes. For instance, in rare cases symptoms related to ALS in SOD1-G93A mice may manifest as orolingual and forelimb motor deficits 13 and not hindlimb motor deficits, a phenotype that is not captured by NeuroScore. Another limitation of this protocol is the fact that it was designed for and applies only to high copy SOD1-G93A mice. In our experience, the clinical presentation of disease in other mouse models of ALS, like the low copy SOD1-G93A mouse 14 or the TDP-43 mouse 15,16, is different than in the high copy SOD1-G93A mice and this NS system might be ill-suited for those models. Thus, it is important to determine the transgene copy number of mice prior to assigning them to studies and verify that it is equal to that of high copy positive-control mice. Regardless, the benefits of using this neurological scoring system outweigh its limitations and this video protocol can serve as an excellent training tool for ALS researchers.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We would like to acknowledge Beth Levine for critically reviewing the manuscript, Valerie Tassinari for developing the genotyping protocol, and Matt Ferola and Carlos Maya for their exceptional animal care.
References
- Gurney ME, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264(5166):1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Rosen DR, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Gurney ME. The use of transgenic mouse models of amyotrophic lateral sclerosis in preclinical drug studies. J Neurol Sci. 152, Suppl. 1997;1(6415) doi: 10.1016/s0022-510x(97)00247-5. [DOI] [PubMed] [Google Scholar]
- Scott S, et al. Design, power, and interpretation of studies in the standard murine model of ALS. Amyotroph Lateral Scler. 2008;9(1):4–15. doi: 10.1080/17482960701856300. [DOI] [PubMed] [Google Scholar]
- Hegedus J, Putman CT, Gordon T. Time course of preferential motor unit loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2007;28(2):154–164. doi: 10.1016/j.nbd.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Gordon T, Ly V, Hegedus J, Tyreman N. Early detection of denervated muscle fibers in hindlimb muscles after sciatic nerve transection in wild type mice and in the G93A mouse model of amyotrophic lateral sclerosis. Neurol. Res. 2009;31(1):28–42. doi: 10.1179/174313208X332977. [DOI] [PubMed] [Google Scholar]
- Gurney ME, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264(5166):1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Kim K, Moore DH, Makriyannis A, Abood ME. AM1241, a cannabinoid CB2 receptor selective compound, delays disease progression in a mouse model of amyotrophic lateral sclerosis. Eur. J. Pharmacol. 2006;542(1-3):1–3. doi: 10.1016/j.ejphar.2006.05.025. [DOI] [PubMed] [Google Scholar]
- Weydt P, Hong SY, Kliot M, Moller T. Assessing disease onset and progression in the SOD1 mouse model of ALS. Neuroreport. 2003;14(7):1051–1054. doi: 10.1097/01.wnr.0000073685.00308.89. [DOI] [PubMed] [Google Scholar]
- Azari MF, et al. Behavioural and anatomical effects of systemically administered leukemia inhibitory factor in the SOD1(G93A G1H) mouse model of familial amyotrophic lateral sclerosis. Brain Res. 2003;982(1):92–97. doi: 10.1016/s0006-8993(03)02989-5. [DOI] [PubMed] [Google Scholar]
- Guan Y, et al. Thiazolidinediones expand body fluid volume through PPARgamma stimulation of ENaC-mediated renal salt absorption. Nat. Med. 2005;11(8):861–866. doi: 10.1038/nm1278. [DOI] [PubMed] [Google Scholar]
- Savcheniuk OA, et al. The effect of probiotic therapy on development of experimental obesity in rats caused by monosodium glutamate. Fiziol. Zh. 2014;60(2):63–69. [PubMed] [Google Scholar]
- Smittkamp SE, Brown JW, Stanford JA. Time-course and characterization of orolingual motor deficits in B6SJL-Tg(SOD1-G93A)1Gur/J mice. Neuroscience. 2008;151(2):613–621. doi: 10.1016/j.neuroscience.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- C M, Gurney ME. A low expressor line of transgenic mice carrying a mutant human Cu,Zn superoxide dismutase (SOD1) gene develops pathological changes that most closely resemble those in human amyotrophic lateral sclerosis. Acta Neuropathol. 1997;93(6):537–550. doi: 10.1007/s004010050650. [DOI] [PubMed] [Google Scholar]
- Wegorzewska I, Baloh RH. TDP-43-based animal models of neurodegeneration: new insights into ALS pathology and pathophysiology. Neurodegener. Dis. 2011;8(4):262–274. doi: 10.1159/000321547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzipetros T, et al. C57BL/6J congenic Prp-TDP43A315T mice develop progressive neurodegeneration in the myenteric plexus of the colon without exhibiting key features of ALS. Brain Res. 2014;1584:59–72. doi: 10.1016/j.brainres.2013.10.013. [DOI] [PubMed] [Google Scholar]