

Abstract
Cell-based systems are useful for discovering antiviral agents. Dissecting the viral life cycle, particularly the early entry stages, allows a mechanistic approach to identify and evaluate antiviral agents that target specific steps of the viral entry. In this report, the methods of examining viral inactivation, viral attachment, and viral entry/fusion as antiviral assays for such purposes are described, using hepatitis C virus as a model. These assays should be useful for discovering novel antagonists/inhibitors to early viral entry and help expand the scope of candidate antiviral agents for further drug development.
Keywords: Immunology, Issue 104, Drug development, antiviral activity, early viral entry, viral inactivation, viral attachment/binding, viral entry/fusion.
Introduction
Viral infections are a constant threat to the public health and a significant cause of epidemic diseases, morbidity, and deaths worldwide. Specific modes of control against viral infections include vaccine development and antiviral therapies. While vaccine efforts have proven successful in immunizing against several viruses, many viral pathogens remain without a protective vaccine including dengue virus (DENV), human cytomegalovirus (HCMV), hepatitis C virus (HCV), human immunodeficiency virus (HIV), and respiratory syncytial virus (RSV)1-5. Antivirals, on the other hand, play an important role for the management of these viral infections when prophylactic vaccines are unavailable. However, to date, only few licensed and cost-effective antiviral drugs are available compared to the number of viral pathogens that threatens the public health. More importantly, due to an increase in global travel and rapid urbanization, the situation is aggravated by risks of epidemic outbreaks from emerging/re-emerging viral infections that are being introduced into non-indigenous areas6. Recent outbreaks caused by severe acute respiratory syndrome (SARS) virus, influenza viruses (H1N1, H5N1, H3N2, and H7N9), DENV, West Nile virus (WNV), measles virus (MV), Middle East Respiratory Syndrome (MERS) virus, and Ebola virus6-12 are among the examples reflecting the need for antivirals development when immunization and/or therapeutics are unavailable. In addition, there is always a potential risk of generating drug-resistant infections with currently used antivirals. Thus, the continuous development and expansion of the scope of antivirals to these emerging/re-emerging viral infections are necessary to provide better management strategies and safeguard the public health.
Most antiviral therapies consist of direct acting antivirals (DAAs) which target a specific viral protein or cofactor that mediates important steps in the viral life cycle. For example, the nucleoside analogue Acyclovir inhibits herpesvirus DNA polymerase, protease inhibitors Boceprevir and Telaprevir antagonize the HCV NS3, and Oseltamivir and Zanamivir are neuraminidase inhibitors that block the release of influenza virus particles from infected cells13-15. There are however very few licensed viral entry inhibitors including Enfuvirtide, which targets HIV gp41 to prevent fusion, and Maraviroc, which blocks the HIV co-receptor CCR5, thereby preventing viral entry16. Exploring novel antagonists/inhibitors to viral entry could help provide additional therapeutics for prophylactic or therapeutic use, such as in combination with other antivirals with a different mechanism of action to better manage viral infections17-19.
Identification of antivirals can involve structure-guided drug design and candidate drug screening-based strategy. Methods for assessing antiviral activity of test agents include biochemical assays of enzymatic activity and evaluation by cell-based systems20-23. In cell-based systems, the viral life cycle can be dissected into distinct stages of infection, such as entry events (attachment, fusion, uncoating), the replication phase (viral genome replication and protein translation), and virion egress (assembly, maturation, and release). Since the assays can be adapted to investigate each specific stage using various tools and methods, this approach allows identification/examination of potential candidate antivirals with a specific mechanism of action targeting the distinct stage analyzed. For instance, to analyze drug effect specifically on the free virus particles prior to binding to the host cell, a ‘viral inactivation assay’ can be performed. In this assay, the virus is allowed to incubate with the test drug and then diluted to titrate out the drug before infecting a cell monolayer. Additional steps such as viral attachment and entry/fusion stages can also be analyzed individually, by shifting the temperature during the infection. For many enveloped viruses, viral entry/fusion at the host cell membrane is greatly facilitated at 37 °C, but is typically precluded at 4 °C which does not affect virus binding24-29. Finally, the use of reporter viruses or cell systems could facilitate these studies and permit a high-throughput analysis.
We previously employed the cell-based approach and dissected the early entry of various enveloped viruses for the examination of antiviral compounds that potentially antagonize viral entry30,31. Herein, the various methods used, including viral inactivation, viral attachment, and viral entry/fusion assays, are described.
Protocol
Note: Ensure that all procedures involving cell culture and virus infection are conducted in certified biosafety hoods that are appropriate for the biosafety level of the samples being handled. For the purpose of describing the protocols, Gaussia luciferase reporter-tagged HCV is used as a model virus32. In the context of the representative results, the compounds chebulagic acid (CHLA) and punicalagin (PUG) are used as candidate antivirals that target viral glycoprotein interactions with cell surface glycosaminoglycans during the early viral entry steps31. Heparin, which is known to interfere with the entry of many viruses30,31,33,34, is used as a positive control treatment in such context. For basic background on virology techniques, propagation of viruses, determination of virus titer, and expression of infectious dose in plaque forming units (PFU), focus forming units (FFU), or multiplicity of infection (MOI), the reader is referred to reference35. For prior examples and optimized conditions used for viruses shown in the representative results, the reader is referred to references30-32,36-39 as well as details listed in Table 1, Figure 1A, and Figure 2A.
1. Cell Culture, Compound Preparation, and Compound Cytotoxicity
Grow the respective cell line for the virus infection system to be analyzed (Table 1). For HCV, grow the Huh-7.5 cells in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 200 U/ml penicillin G, 200 µg/ml streptomycin, and 0.5 µg/ml amphotericin B.
Prepare the test compounds and controls using their respective solvents: for example, dissolve CHLA and PUG in dimethyl sulfoxide (DMSO); prepare heparin in sterile double-distilled water. For all subsequent dilutions, use culture media. Note: The final concentration of DMSO in the test compound treatments is less than 1% in the experiments; 1% DMSO is included as a negative control treatment in the assays for comparison.
- Determine the cytotoxicity of the test compounds (e.g., CHLA and PUG) on the cells for the viral infection by using a cell viability determining reagent such as XTT (2,3-bis[2-methoxy-4-nitro-5-sulfophenyl]-5-phenylamino)-carbonyl]-2H-tetrazolium hydroxide):
- For HCV, seed Huh-7.5 cells in a 96-well plate (1 × 104 cells per well) and incubate at 37 °C in a 5% CO2 incubator O/N to obtain a monolayer.
- Apply DMSO control (1%) or increasing concentrations of the test compounds CHLA and PUG (ex. 0, 10, 50, 100, and 500 μM) to the culture wells in triplicate.
- Incubate at 37 °C for 72 hr, then discard the medium in the plate and wash the cells with 200 μl of phosphate buffered saline (PBS) twice.
- Add 100 µl of assaying solution from the XTT-based in vitro toxicology assay kit to each well and incubate the plates at 37 °C for another 3 hr to allow XTT formazan production.
- Determine the absorbance with a microplate reader at a test wavelength of 492 nm and a reference wavelength of 690 nm.
- Calculate the percentage of surviving cells using the following formula: cell viability (%) = At / As × 100%, where ‘At’ and ‘As’ refer to the absorbance of the test compounds and the solvent control (ex. 1% DMSO) treatments, respectively. Determine the concentration of 50% cellular cytotoxicity (CC50) of the test compounds from an analytical software such as GraphPad Prism according to manufacturer’s protocol.
2. Readout of Viral Infection
Note: The readout of viral infection depends on the virus system used and can involve methods such as plaque assays or measuring reporter signals from reporter-tagged viruses. The method for detecting reporter-HCV infection based on the luciferase reporter activity is described below.
Collect the supernatants from the infected wells and clarify at 17,000 x g in a microcentrifuge for 5 min at 4 °C.
Mix 20 μl of test supernatant to 50 μl of luciferase substrate from the Gaussia luciferase assay kit and directly measure with a luminometer according to the manufacturer’s instructions.
Express HCV infectivity as log10 of relative light units (RLU) for determining viral inhibition (%) and calculate the 50% effective concentration (EC50) of the test compounds against HCV infection using algorithms from GraphPad Prism software according to manufacturer’s protocol.
3. Viral Inactivation Assay
Note: Examples of incubation period and viral dose for various viruses are listed in Figure 1A. Higher concentrations of the virus can also be tested by increasing the MOI/PFU.
Seed Huh-7.5 cells in a 96-well plate (1 × 104 cells per well) and incubate at 37 °C in a 5% CO2 incubator O/N to obtain a monolayer.
Incubate the test compounds or controls (final concentrations are: CHLA = 50 μM; PUG = 50 μM; heparin = 1,000 μg/ml; DMSO = 1%) with the HCV particles at 37 °C (Figure 1A, ‘Long-Term’) in a 1:1 ratio. For example, to a 100 μl virus inoculum containing 1 x 104 FFU, add 100 μl of a 100 μM CHLA working dilution; this yields CHLA treatment at a final concentration of 50 μM.
Dilute the virus-drug mixture to “sub-therapeutic” (ineffective) concentration of the test compounds. For example, the ineffective concentration of CHLA and PUG against HCV is at 1 μM31; therefore this requires a 50-fold dilution of the virus-drug mixture which can be accomplished with 9.8 ml of basal medium (cell culturing medium with 2% FBS). Note: The dilution to sub-therapeutic concentration prevents significant interaction between the test compounds and the host cell surface and allows examination of treatment effect on the cell-free virions. Note that this dilution is dependent on the antiviral dose response of the test compounds against the particular viral infection, and is determined prior to performing this particular assay31.
For comparison, mix the virus with the test compounds and immediately dilute (no incubation period) to sub-therapeutic concentration prior to infection (Figure 1A, ‘Short-Term’).
Add 100 μl of the diluted HCV-drug mixture onto the Huh-7.5 cell monolayer (the amount of virus is now at 1 x 102 FFU; final MOI = 0.01) and incubate for 3 hr at 37 °C to allow viral adsorption.
Following the infection, remove the diluted inocula and gently wash the wells with 200 μl of PBS twice. Note: Perform the washes gently to avoid lifting the cells.
Apply 100 μl of basal medium to each well and incubate at 37 °C for 72 hr.
Analyze the resulting infection by assaying the supernatant for luciferase activity as described in ‘2. Readout of Viral Infection’.
4. Viral Attachment Assay
Note: Examples of incubation period and viral dose for various viruses are listed in Figure 2A, ‘Attachment’. Higher concentrations of the virus can also be tested by increasing the MOI/PFU.
Seed Huh-7.5 cells in a 96-well plate (1 × 104 cells per well) and incubate at 37 °C in a 5% CO2 incubator O/N to obtain a monolayer.
Pre-chill the cell monolayers in plates at 4 °C for 1 hr.
Co-treat the cells with HCV inoculum (MOI = 0.01) and test compounds or controls (final concentrations are: CHLA = 50 μM; PUG = 50 μM; heparin = 1,000 μg/ml; DMSO = 1%) at 4 °C for 3 hr. For example, to a 90 μl virus inoculum containing 1 x 102 FFU, add 10 μl of a 500 μM CHLA working dilution; this yields CHLA treatment at a final concentration of 50 μM and an infection by HCV at MOI = 0.01 on the cell monolayer. Note: It is important to carry out the experiment at 4 °C since it allows for virus binding but precludes entry which most efficiently occurs at 37 °C. Perform the addition of virus and test compounds on ice and the ensuing incubation in a 4 °C refrigerator to ensure that the temperature is maintained at 4 °C.
Remove the supernatant and gently wash the cell monolayer with 200 μl of ice-cold PBS twice. Note: Perform the washes gently to avoid lifting the cells.
Apply 100 μl of basal medium to each well and incubate at 37 °C for 72 hr.
Analyze the resulting infection by assaying the supernatant for luciferase activity as described in ‘2. Readout of Viral Infection’.
5. Viral Entry/Fusion Assay
Note: Example of incubation periods and viral dose for various viruses are listed in Figure 2A ‘Entry/Fusion’. Higher concentrations of the virus can also be tested by increasing the MOI/PFU.
Seed Huh-7.5 cells in a 96-well plate (1 × 104 cells per well) and incubate at 37 °C in a 5% CO2 incubator O/N to obtain a monolayer.
Pre-chill the cell monolayers in plates at 4 °C for 1 hr.
Infect the cells with HCV (MOI = 0.01) at 4 °C for 3 hr. For example, use a 100 μl virus inoculum containing 1 x 102 FFU. Note: Perform the addition of the viral inoculum on ice and the ensuing incubation in a 4 °C refrigerator to maintain the temperature at 4 °C, which permits viral binding but not entry.
Remove the supernatant and gently wash the cell monolayers with 200 μl of ice-cold PBS twice. Note: Perform the washes gently to avoid lifting the cells.
Treat the wells with the test compounds or controls (final concentrations are: CHLA = 50 μM; PUG = 50 μM; heparin = 1,000 μg/ml; DMSO = 1%) and incubate at 37 °C for 3 hr. For example, add 10 μl of a 500 μM CHLA working dilution to 90 μl of media, mix, and treat the wells; this yields CHLA treatment at a final concentration of 50 μM. Note: The shift from 4 °C to 37 °C now facilitates the viral entry/fusion event and therefore allows assessment of test compounds’ effect on this particular step.
Aspirate the drug-containing supernatant and remove non-internalized extracellular viruses by either washing with 200 μl of citrate buffer (50 mM sodium citrate, 4 mM potassium chloride, pH 3.0) or PBS. Apply 100 μl of the basal medium before incubating at 37 °C for 72 hr.
Analyze the resulting infection by assaying the supernatant for luciferase activity as described in ‘2. Readout of Viral Infection’.
Representative Results
In Figure 1, the ‘viral inactivation assay’ was performed to examine whether two specific natural compounds CHLA and PUG could inactivate the different enveloped viruses in cell-free state and prevent subsequent infection. The cytotoxicity and antiviral dose response of these compounds have been determined prior to performing the mechanistic study31. The viruses were pre-treated with the test compounds and then the virus-drug mixtures were diluted to sub-therapeutic concentrations before inoculation onto the respective cell monolayer for each virus system. As shown in Figure 1, both CHLA and PUG appeared to interact with the cell-free virions, resulting in irreversible effects that protected the cell monolayer from the subsequent infection. The two test compounds achieved a near 100% inhibition against HCMV, HCV, and DENV-2, whereas a 60 – 80% block was observed against MV and RSV. These results suggest that CHLA and PUG have direct impact on these free virus particles by inactivating them and neutralizing their infectivity.
In Figure 2, the attachment and entry/fusion assays were carried out to explore the effect of CHLA and PUG against these early viral entry-related events from HCMV, HCV, DENV-2, MV, and RSV. Both CHLA and PUG effectively prevented binding of the investigated viruses onto the respective host cell as shown by inhibition on the resulting viral infection (Figure 2, ‘Attachment’: light gray bars). The inhibitory effect on virus attachment by both compounds was similar against HCMV (Figure 2B), HCV (Figure 2C), DENV-2 (Figure 2D), and RSV (Figure 2F), ranging from 90 – 100%. On the other hand, PUG appeared to be more effective than CHLA against MV binding (Figure 2E), with the inhibition rate from the two compounds varying between 50 – 80%. The control treatment heparin, which is known to block entry of many viruses, also inhibited attachment of HCMV, DENV-2, RSV, ad MV, but was less efficient against HCV. The ensuing ‘viral entry/fusion assay’ examined whether CHLA and PUG retained their activity during the virus entry/fusion phase (Figure 2, ‘Entry/Fusion’: dark gray bars). Again, both CHLA and PUG were observed to effectively impair the viral entry/fusion step of the viruses examined (Figure 2B – F), yielding a 50 – 90% protective effect on the respective cell monolayer. Heparin also potently inhibited entry/fusion in DENV-2 and RSV infections, but was less efficacious against HCMV, HCV, and MV (< 40% inhibition on average).
| Virus | Cell Type |
| HCMV | HEL |
| HCV | Huh-7.5 |
| DENV-2 | Vero |
| MV | CHO-SLAM |
| RSV | HEp-2 |
Table 1: Host cell type for viral infection. The cell type used for each viral infection system described in the representative results is indicated. Additional details regarding the cells can be found in reference31.
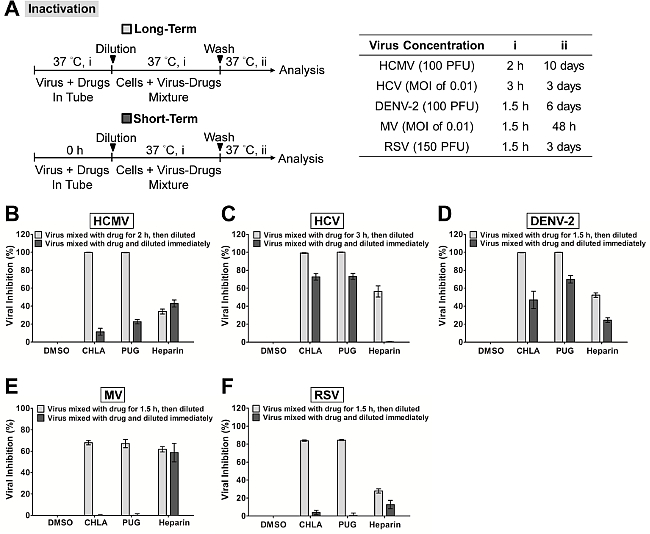 Figure 1. Inactivation of viral infections by the test compounds CHLA and PUG. Different viruses were treated with the test compounds for a long period (incubated for 1.5 – 3 hr before titration; light gray bars) or short period (immediately diluted; dark gray bars) at 37 °C before a dilution to sub-therapeutic concentration and subsequent analysis of infection on the respective host cells. (A) Schematics of the experiment (shown on the left) with the final virus concentration (PFU/well or MOI), long-term virus-drug incubation period (i), and subsequent incubation time (ii) indicated for each virus in the table on the right. Analyses for (B) HCMV, (C) HCV, (D) DENV-2, (E) MV, and (F) RSV are indicated in each additional panel. Results are plotted against the DMSO negative control treatment for virus infection and the data shown are the means ± standard errors of the mean (SEM) from three independent experiments. This figure has been modified from reference31. Please click here to view a larger version of this figure.
Figure 1. Inactivation of viral infections by the test compounds CHLA and PUG. Different viruses were treated with the test compounds for a long period (incubated for 1.5 – 3 hr before titration; light gray bars) or short period (immediately diluted; dark gray bars) at 37 °C before a dilution to sub-therapeutic concentration and subsequent analysis of infection on the respective host cells. (A) Schematics of the experiment (shown on the left) with the final virus concentration (PFU/well or MOI), long-term virus-drug incubation period (i), and subsequent incubation time (ii) indicated for each virus in the table on the right. Analyses for (B) HCMV, (C) HCV, (D) DENV-2, (E) MV, and (F) RSV are indicated in each additional panel. Results are plotted against the DMSO negative control treatment for virus infection and the data shown are the means ± standard errors of the mean (SEM) from three independent experiments. This figure has been modified from reference31. Please click here to view a larger version of this figure.
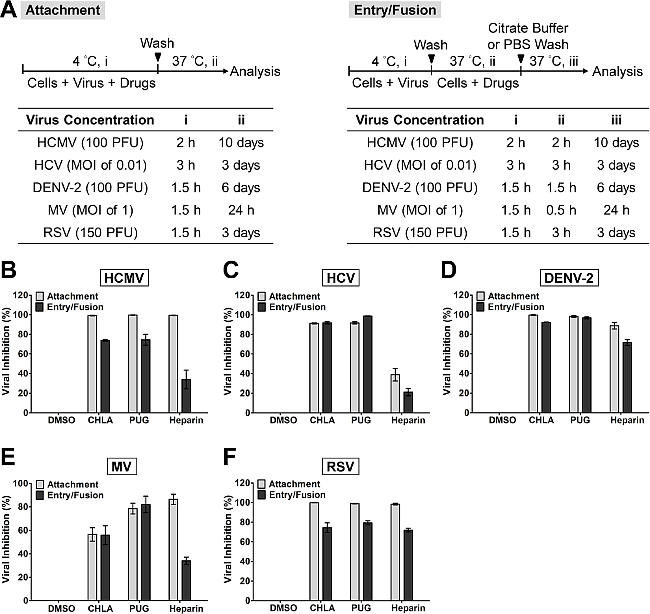 Figure 2. Evaluation of antiviral activities of the test compounds CHLA and PUG against virus attachment and entry/fusion. (A) The experimental procedure, virus concentration (PFU/well or MOI), and the time of addition and treatment with the test compounds (i, ii, iii) are presented for each virus in the schematics and the associated tables. In virus attachment analysis (light gray bars), monolayers of different cell types were pre-chilled at 4 °C for 1 hr, then co-treated with the respective viruses and test compounds at 4 °C (1.5 – 3 hr; i) before washing off the inoculates and test compounds for subsequent incubation (37 °C; ii) and examination of virus infection. In virus entry/fusion analysis (dark gray bars), seeded cell monolayers were pre-chilled at 4 °C for 1 hr and then challenged with the respective viruses at 4 °C for 1.5 – 3 hr (i). Cells were then washed and treated with the test compounds for an additional incubation period (ii) during which the temperature was shifted to 37 °C to facilitate the viral entry/fusion event. At the end of the incubation, extracellular viruses were removed by either citrate buffer (pH 3.0) or PBS washes and the cells were further incubated (iii) for analysis of virus infection. Results for (B) HCMV, (C) HCV, (D) DENV-2, (E) MV, and (F) RSV are indicated in each additional panel. Data are plotted against the DMSO negative control treatment of virus infection and are presented as means ± SEM from three independent experiments. This figure has been modified from reference31. Please click here to view a larger version of this figure.
Figure 2. Evaluation of antiviral activities of the test compounds CHLA and PUG against virus attachment and entry/fusion. (A) The experimental procedure, virus concentration (PFU/well or MOI), and the time of addition and treatment with the test compounds (i, ii, iii) are presented for each virus in the schematics and the associated tables. In virus attachment analysis (light gray bars), monolayers of different cell types were pre-chilled at 4 °C for 1 hr, then co-treated with the respective viruses and test compounds at 4 °C (1.5 – 3 hr; i) before washing off the inoculates and test compounds for subsequent incubation (37 °C; ii) and examination of virus infection. In virus entry/fusion analysis (dark gray bars), seeded cell monolayers were pre-chilled at 4 °C for 1 hr and then challenged with the respective viruses at 4 °C for 1.5 – 3 hr (i). Cells were then washed and treated with the test compounds for an additional incubation period (ii) during which the temperature was shifted to 37 °C to facilitate the viral entry/fusion event. At the end of the incubation, extracellular viruses were removed by either citrate buffer (pH 3.0) or PBS washes and the cells were further incubated (iii) for analysis of virus infection. Results for (B) HCMV, (C) HCV, (D) DENV-2, (E) MV, and (F) RSV are indicated in each additional panel. Data are plotted against the DMSO negative control treatment of virus infection and are presented as means ± SEM from three independent experiments. This figure has been modified from reference31. Please click here to view a larger version of this figure.
Discussion
In this report the methods to identify and evaluate antiviral compounds based on a mechanistic approach of dissecting the early viral entry events were described. Specifically, the assays allowed us to examine the effect of test compounds on free virus particles, viral attachment, and viral entry/fusion. Critical steps were implemented to distinctly evaluate the drug effect on the specific stage of early viral entry. For instance, in the ‘viral inactivation assay’, the dilution of the virus-drug mixture to sub-therapeutic concentration prevents significant interaction between the test compound and the host cell surface by ‘titrating out’ the drug. This ensures that the inhibitory effect observed on the subsequent infection of the host cell is due to a direct interaction between the test compound and the cell-free virions, rather than an effect from the test compound on host cell membrane or membrane-associated molecules, including viral receptors30. Similarly, the shift in temperature between 4 °C (which allows for virus binding but not entry) and 37 °C (which facilitates virus entry/fusion) in the ‘viral attachment assay’ and ‘viral entry/fusion assay’ are crucial to determine the test compound’s effect on each of these specific events. This is feasible due to the temperature sensitivity of enveloped viruses during these steps in the infection24-29. It is therefore important that the assays are performed at the indicated temperature to ensure the accuracy of the results; for example, by carrying out the experiment on ice to maintain at 4 °C and by placing the sample directly in a 37 °C incubator for the temperature shift. In addition, the use of negative (ex. DMSO solvent for drug preparation) and positive (ex. heparin treatment) controls also help further establish the assays’ accuracy. The utility and applicability of such methods have been demonstrated in many antiviral studies26,30,31,40,41. Note that while heparin is included as a control for all three assays in the context of the representative results, it typically blocks the initial virus binding rather than the ensuing fusion/entry step (as reflected by the data in Figure 2). Additional controls could also be used, such as neutralizing antibodies directed against the virus (for viral inactivation assay), antibodies that mask the cell surface receptors for the virus (for viral attachment assay), and membrane fusion inhibitors (for viral fusion/entry assay).
The assays described in this report, which are specific to the early stages of the viral infection, are useful in terms of application as secondary tests to characterize the mechanism of action of candidate drugs from primary screens which typically target the viral infection more broadly. Alternatively, they could also be incorporated in primary screens if one is specifically looking for inhibitors of early viral entry, including virus inactivating agents, viral attachment antagonists, and inhibitors to viral entry/fusion. In this case, their use allows a more focused and precise screen analysis for the identification of mechanism-specific antiviral candidates, which, in turn, would expedite downstream drug development.
The use of cell-based assays in identifying antiviral agents provides several important advantages compared to biochemical assays, including revealing potential off-target effects (such as cytotoxicity) and adding physiological relevance to the bioactivity of the test agents42. These issues are important considerations for deciding whether a candidate agent is of value for continuation in subsequent phases of drug development. Similarly, the early viral entry-specific assays described in this report allow examination of the drug effect on the distinct viral entry stage at the cellular level, and more specifically in the context of an authentic viral infection in vitro. The results obtained from such assays would therefore help better predict the antiviral efficacy of the test compounds and also identify potentially unwanted off-target effects against the host cell. One potential limitation though, is that an in vitro cell-based assay may not completely reflect the actual in vivo entry step in the context of a natural viral infection. Nonetheless, the assays presented in this protocol do serve as an analytical platform for mechanism-based identification and evaluation of novel antiviral agents.
The development of reporter viruses or reporter cell systems to quantitate the amount of viral infection has greatly facilitated cell-based screening and evaluation of antiviral compounds. Examples include the use of recombinant viruses carrying a reporter gene or by means of recombinant human cell lines containing a reporter gene driven by the specific virus promoter31,43. In this report, the infection from luciferase-tagged HCV can be easily monitored by quantitating the reporter signal, thus facilitating data analysis. By incorporating these useful reporter-based tools, the early viral entry assays described here can essentially be adapted into high-throughput format for mechanism-based screening of small molecule libraries.
In conclusion, a protocol was described for assays dissecting the early viral entry as a means of identifying and evaluating mechanism-specific antiviral compounds. Such assays would be useful for discovering novel antagonists/inhibitors to viral entry and help expand the scope of antiviral agents for development as prophylactic and/or therapeutic treatments.
Disclosures
The authors declare that no competing interests exist.
Acknowledgments
This study is supported by funding from Taipei Medical University Hospital (102TMU-TMUH-19) and the Ministry of Science and Technology of Taiwan (MOST103-2320-B-038-031-MY3).
References
- Munier CM, Andersen CR, Kelleher AD. HIV vaccines: progress to date. Drugs. 2011;71:387–414. doi: 10.2165/11585400-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Rothman AL. Immunity to dengue virus: a tale of original antigenic sin and tropical cytokine storms. Nat Rev Immunol. 2011;11:532–543. doi: 10.1038/nri3014. [DOI] [PubMed] [Google Scholar]
- Sung H, Schleiss MR. Update on the current status of cytomegalovirus vaccines. Expert Rev Vaccines. 2010;9:1303–1314. doi: 10.1586/erv.10.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torresi J, Johnson D, Wedemeyer H. Progress in the development of preventive and therapeutic vaccines for hepatitis C virus. J Hepatol. 2011;54:1273–1285. doi: 10.1016/j.jhep.2010.09.040. [DOI] [PubMed] [Google Scholar]
- Wright M, Piedimonte G. Respiratory syncytial virus prevention and therapy: past, present, and future. Pediatr Pulmonol. 2011;46:324–347. doi: 10.1002/ppul.21377. [DOI] [PubMed] [Google Scholar]
- Christou L. The global burden of bacterial and viral zoonotic infections. Clin Microbiol Infect. 2011;17:326–330. doi: 10.1111/j.1469-0691.2010.03441.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascio A, Bosilkovski M, Rodriguez-Morales AJ, Pappas G. The socio-ecology of zoonotic infections. Clin Microbiol Infect. 2011;17:336–342. doi: 10.1111/j.1469-0691.2010.03451.x. [DOI] [PubMed] [Google Scholar]
- Grais RF. Measles vaccination in humanitarian emergencies: a review of recent practice. Confl Health. 2011;5:21. doi: 10.1186/1752-1505-5-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautret P. Emerging viral respiratory tract infections-environmental risk factors and transmission. Lancet Infect Dis. 2014;14:1113–1122. doi: 10.1016/S1473-3099(14)70831-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampathkumar P. Middle East respiratory syndrome: what clinicians need to know. Mayo Clin Proc. 2014;89:1153–1158. doi: 10.1016/j.mayocp.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd EM. Ebola Virus: a Clear and Present Danger. J Clin Microbiol. 2015;53:4–8. doi: 10.1128/JCM.03115-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop BM. Potential and Emerging Treatment Options for Ebola Virus Disease. Ann Pharmacother. 2014. [DOI] [PubMed]
- Arduino PG, Porter SR. Oral and perioral herpes simplex virus type 1 (HSV-1) infection: review of its management. Oral Dis. 2006;12:254–270. doi: 10.1111/j.1601-0825.2006.01202.x. [DOI] [PubMed] [Google Scholar]
- Mitrasinovic PM. Advances in the structure-based design of the influenza A neuraminidase inhibitors. Curr Drug Targets. 2010;11:315–326. doi: 10.2174/138945010790711932. [DOI] [PubMed] [Google Scholar]
- Soriano V. Directly acting antivirals against hepatitis C virus. J Antimicrob Chemother. 2011;66:1673–1686. doi: 10.1093/jac/dkr215. [DOI] [PubMed] [Google Scholar]
- Haqqani AA, Tilton JC. Entry inhibitors and their use in the treatment of HIV-1 infection. Antiviral Res. 2013;98:158–170. doi: 10.1016/j.antiviral.2013.03.017. [DOI] [PubMed] [Google Scholar]
- Melby T, Westby M. Inhibitors of viral entry. Handb Exp Pharmacol. 2009. pp. 177–202. [DOI] [PubMed]
- Vanderlinden E, Naesens L. Emerging antiviral strategies to interfere with influenza virus entry. Med Res Rev. 2014;34:301–339. doi: 10.1002/med.21289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoine TE, Park PJ, Shukla D. Glycoprotein targeted therapeutics: a new era of anti-herpes simplex virus-1 therapeutics. Rev Med Virol. 2013;23:194–208. doi: 10.1002/rmv.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlotsky JM, Chevaliez S, McHutchison JG. The hepatitis C virus life cycle as a target for new antiviral therapies. Gastroenterology. 2007;132:1979–1998. doi: 10.1053/j.gastro.2007.03.116. [DOI] [PubMed] [Google Scholar]
- Beyleveld G, White KM, Ayllon J, Shaw ML. New-generation screening assays for the detection of anti-influenza compounds targeting viral and host functions. Antiviral Res. 2013;100:120–132. doi: 10.1016/j.antiviral.2013.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilianski A, Baker SC. Cell-based antiviral screening against coronaviruses: developing virus-specific and broad-spectrum inhibitors. Antiviral Res. 2014;101:105–112. doi: 10.1016/j.antiviral.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caillet-Saguy C, Lim SP, Shi PY, Lescar J, Bressanelli S. Polymerases of hepatitis C viruses and flaviviruses: structural and mechanistic insights and drug development. Antiviral Res. 2014;105:8–16. doi: 10.1016/j.antiviral.2014.02.006. [DOI] [PubMed] [Google Scholar]
- Frey S. Temperature dependence of cell-cell fusion induced by the envelope glycoprotein of human immunodeficiency virus type 1. J Virol. 1995;69:1462–1472. doi: 10.1128/jvi.69.3.1462-1472.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tscherne DM. Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J Virol. 2006;80:1734–1741. doi: 10.1128/JVI.80.4.1734-1741.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan RP. Molecular umbrellas: a novel class of candidate topical microbicides to prevent human immunodeficiency virus and herpes simplex virus infections. J Virol. 2007;81:7636–7646. doi: 10.1128/JVI.02851-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haywood AM, Boyer BP. Time and temperature dependence of influenza virus membrane fusion at neutral pH. J Gen Virol. 1986;67(Pt 12):2813–2817. doi: 10.1099/0022-1317-67-12-2813. [DOI] [PubMed] [Google Scholar]
- Haywood AM, Boyer BP. Sendai virus membrane fusion: time course and effect of temperature, pH, calcium, and receptor concentration) Biochemistry. 1982;21:6041–6046. doi: 10.1021/bi00267a003. [DOI] [PubMed] [Google Scholar]
- Wang G, Hernandez R, Weninger K, Brown DT. Infection of cells by Sindbis virus at low temperature. Virology. 2007;362:461–467. doi: 10.1016/j.virol.2006.12.036. [DOI] [PubMed] [Google Scholar]
- Lin LT. Hydrolyzable tannins (chebulagic acid and punicalagin) target viral glycoprotein-glycosaminoglycan interactions to inhibit herpes simplex virus 1 entry and cell-to-cell spread. J Virol. 2011;85:4386–4398. doi: 10.1128/JVI.01492-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin LT. Broad-spectrum antiviral activity of chebulagic acid and punicalagin against viruses that use glycosaminoglycans for entry. BMC Microbiol. 2013;13:187. doi: 10.1186/1471-2180-13-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marukian S. Cell culture-produced hepatitis C virus does not infect peripheral blood mononuclear cells. Hepatology. 2008;48:1843–1850. doi: 10.1002/hep.22550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba M, Snoeck R, Pauwels R, de Clercq E. Sulfated polysaccharides are potent and selective inhibitors of various enveloped viruses, including herpes simplex virus, cytomegalovirus, vesicular stomatitis virus, and human immunodeficiency virus. Antimicrob Agents ChemotheR. 1988;32:1742–1745. doi: 10.1128/aac.32.11.1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth H. Cellular binding of hepatitis C virus envelope glycoprotein E2 requires cell surface heparan sulfate. J Biol CheM. 2003;278:41003–41012. doi: 10.1074/jbc.M302267200. [DOI] [PubMed] [Google Scholar]
- Flint SJ, Enquist LW, Racaniello VR, Skalka AM. Principles of Virology. 3rd edn. ASM Press; 2008. [Google Scholar]
- Brown MG. Dramatic caspase-dependent apoptosis in antibody-enhanced dengue virus infection of human mast cells. J Leukoc Biol. 2009;85:71–80. doi: 10.1189/jlb.0308167. [DOI] [PubMed] [Google Scholar]
- Huang Y, Cyr SL, Burt DS, Anderson R. Murine host responses to respiratory syncytial virus (RSV) following intranasal administration of a Protollin-adjuvanted, epitope-enhanced recombinant G protein vaccine. J Clin Virol. 2009;44:287–291. doi: 10.1016/j.jcv.2009.01.009. [DOI] [PubMed] [Google Scholar]
- Isaacson MK, Compton T. Human cytomegalovirus glycoprotein B is required for virus entry and cell-to-cell spread but not for virion attachment, assembly, or egress. J Virol. 2009;83:3891–3903. doi: 10.1128/JVI.01251-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard VH, et al. Measles virus blind to its epithelial cell receptor remains virulent in rhesus monkeys but cannot cross the airway epithelium and is not shed. J Clin Invest. 2009;118:2448–2458. doi: 10.1172/JCI35454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciesek S. The green tea polyphenol, epigallocatechin-3-gallate, inhibits hepatitis C virus entry. Hepatology. 2011;54:1947–1955. doi: 10.1002/hep.24610. [DOI] [PubMed] [Google Scholar]
- Lin LT. Saikosaponin b2 is a naturally occurring terpenoid that efficiently inhibits hepatitis C virus entry. J Hepatol. 2015;62:541–548. doi: 10.1016/j.jhep.2014.10.040. [DOI] [PubMed] [Google Scholar]
- Atkins C, Evans CW, White EL, Noah JW. Screening methods for influenza antiviral drug discovery. Expert Opin Drug Discov. 2012;7:429–438. doi: 10.1517/17460441.2012.674510. [DOI] [PubMed] [Google Scholar]
- Zhang J. Identification of novel virus inhibitors by influenza A virus specific reporter cell based screening. Antiviral Res. 2012;93:48–54. doi: 10.1016/j.antiviral.2011.10.014. [DOI] [PubMed] [Google Scholar]