

Abstract
The calcineurin inhibitor tacrolimus is the cornerstone of most immunosuppressive treatment protocols after solid organ transplantation in the United States. Tacrolimus is a narrow therapeutic index drug and as such requires therapeutic drug monitoring and dose adjustment based on its whole blood trough concentrations. To facilitate home therapeutic drug and adherence monitoring, the collection of dried blood spots is an attractive concept. After a finger stick, the patient collects a blood drop on filter paper at home. After the blood is dried, it is mailed to the analytical laboratory where tacrolimus is quantified using high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) in combination with a simple manual protein precipitation step and online column extraction.
For tacrolimus analysis, a 6-mm disc is punched from the saturated center of the blood spot. The blood spot is homogenized using a bullet blender and then proteins are precipitated with methanol/0.2 M ZnSO4 containing the internal standard D2,13C-tacrolimus. After vortexing and centrifugation, 100 µl of supernatant is injected into an online extraction column and washed with 5 ml/min of 0.1 formic acid/acetonitrile (7:3, v:v) for 1 min. Hereafter, the switching valve is activated and the analytes are back-flushed onto the analytical column (and separated using a 0.1% formic acid/acetonitrile gradient). Tacrolimus is quantified in the positive multi reaction mode (MRM) using a tandem mass spectrometer.
The assay is linear from 1 to 50 ng/ml. Inter-assay variability (3.6%-6.1%) and accuracy (91.7%-101.6%) as assessed over 20 days meet acceptance criteria. Average extraction recovery is 95.5%. There are no relevant carry-over, matrix interferences and matrix effects. Tacrolimus is stable in dried blood spots at RT and at +4 °C for 1 week. Extracted samples in the autosampler are stable at +4 °C for at least 72 hr.
Keywords: Medicine, Issue 105, Tacrolimus, dried blood spots, high-performance liquid chromatography, tandem mass spectrometry, LC-MS/MS, column switching, online extraction, therapeutic drug monitoring, home monitoring, adherence monitoring
Introduction
Tacrolimus is a potent immonosuppressant1-7 that has a macrolide structure8 (Figure 1). Due to cis-trans isomerism of the C-N bonds it forms two rotamers in solution9 that can be separated by reversed phase high-performance liquid chromatography (HPLC) Tacrolimus is lipophilic and soluble in alcohols (methanol: 653 g/L, ethanol: 355 g/L), halogenated hydrocarbons (chloroform: 573 g/L) and ether. It is sparingly soluble in aliphatic hydrocarbons (hexane: 0.1 g/L and water (pH 3: 0.0047 g/L)9. The molecule does not contain any chromophore and its UV-absorption maximum is 192 nm. Tacrolimus acts via inhibition of calcineurin. Its mechanism of action has been reviewed in references10,11. It is currently used in more than 80% of solid-organ transplant patients in the United States12.
The therapeutic index of tacrolimus is considered to be narrow13. In addition, the correlation between tacrolimus doses and blood concentrations is poor and pharmacokinetics is variable14,15. Therapeutic drug monitoring to guide tacrolimus dosing in transplant patients is therefore general clinical practice16-20. The goal is to keep the tacrolimus blood concentrations within a pre-defined therapeutic range. Tacrolimus blood concentrations below the therapeutic range may result in increased activity of chronic or acute allo-immune reactions, while concentrations above the therapeutic window increase the risk for over-immunosuppression, cancer and toxicities, such as nephrotoxicity, neurotoxicity, hypertension, and diabetes. High pharmacokinetic intra-individual variability of tacrolimus may be detrimental to both transplant organ and patient survival21,22. While inter-individual variability of tacrolimus pharmacokinetics is mainly caused by CYP3A5 polymorphisms, reasons for intra-individual variability include, but are not limited to, drug-drug, disease-drug and food-drug interactions14,15. Also lack of adherence to the immunosuppressive therapeutic drug regimen is a contributing factor and a major reason for graft loss23,24.
These considerations suggest that frequent home therapeutic drug and adherence monitoring of tacrolimus whole blood concentrations may be beneficial to ensure that patients have tacrolimus exposure within the desired therapeutic window at all times. However, the logistics and cost of more frequent therapeutic drug monitoring as it is current clinical practice15 is prohibitive. One of the reasons is that the patient has to see a phlebotomist to have the required venous blood sample drawn. Dried blood spots have recently emerged as an attractive concept25-28. After a simple finger stick the patient collects a blood drop on a special filter paper card and after the blood spot has dried, it can be mailed to a central laboratory for analysis of tacrolimus and any other immunosuppressant that the patient may currently be taking. This has become possible due to the development of highly sensitive and specific LC-MS/MS assays for the quantification of tacrolimus and other immunosuppressants in very small blood volumes such as dried blood spots (typically 20 µl of blood)25,29-43. Another advantage is that minimally invasive, low volume sample collection strategies such as dried blood spots greatly facilitate therapeutic drug monitoring and pharmacokinetic studies in small children28.
Tacrolimus is usually measured in venous EDTA whole blood15. Reasons are that tacrolimus extensively distributes into blood cells and that clinical studies have reported better correlation between tacrolimus trough concentrations in blood than in plasma with clinical events15,18. In comparison, the analysis of tacrolimus in dried blood spots is based on capillary blood that is mixed with the filter paper matrix. This presents challenges in terms of solubilization of tacrolimus and potential interferences with the LC-MS/MS analysis. Here we present an established and validated assay based on homogenization of the dried blood spot using a bullet blender in combination with a high-flow online column sample clean up procedure and LC-MS/MS analysis. As of today, this assay has successfully been used for the quantification of more than five thousand tacrolimus dried blood spot samples for adherence monitoring in clinical trials.
Protocol
De-identified blood samples from healthy individuals were from the University of Colorado Hospital (Aurora, Colorado). The use of de-identified blood bank samples for validation studies as well as for the preparation of calibrators and quality control samples was considered “exempt” by the Colorado Multi-institutional Review Board (COMIRB, Aurora, Colorado).
1. Preparation of References and Solutions
- Purchase tacrolimus and the internal standard D2,13C-tacrolimus from the vendors listed in the Materials List.
- Prepare stock solutions in pure methanol at a concentration of 1 mg/ml for tacrolimus and a concentration of 10 µg/ml for D2,13C-tacrolimus. Make stock solutions of reference materials based on three independent weightings. Aliquot stock solutions and store at -70 °C or below.
- Prepare solution to precipitate proteins and extract tacrolimus using methanol / 0.2 M ZnSO4 in water (7:3, v:v). This solution also contains the internal standard D2,13C-tacrolimus at a concentration of 2.5 ng/ml and is used for the extraction of all samples except for the extraction of blank samples (please see 1.3.3).
- Prepare this protein precipitation solution freshly on each extraction day and set the expiration of the solution at 12 hr.
- Preparation of calibration curve and quality control (QC) samples
- Prepare stock solutions of tacrolimus by performing appropriate dilutions of the stock solution using pure methanol.
- To prepare calibrators and quality control samples, spike 20 µl of appropriately diluted stock solution into EDTA whole blood, incubate at 37 °C under gentle shaking in a water bath to allow for homogeneous distribution of tacrolimus into the cellular blood components for 20 min and aliquot into 1.5 ml polypropylene tubes with conical bottom and snap-on lids. Ensure that the relative volume of organic solvent does not exceed 5%.
- Spot 50 µl of the spiked whole blood into the middle of each circle on the filter cards using a pipette.
- Dry the blood spots on filter cards at RT for 3 hr.
- Prepare tacrolimus calibration standards in human EDTA whole blood at tacrolimus concentrations of 1, 2.5, 5, 10, 25, and 50 ng/ml. Prepare a blank sample for extraction like the calibration standards with the protein precipitation solution containing the internal standard D2,13C-tacrolimus (“zero sample”).
- Prepare QC samples in human EDTA whole blood at concentrations of 0, 2, 4, 20, 40 ng/ml. Prepare a blank sample. In contrast to the QC samples that are extracted with precipitation containing the internal standard D2,13C-tacrolimus, extract this blank sample with protein precipitation solution that does not contain the internal standard D2,13C-tacrolimus (“blank sample”).
- Collection of clinical samples
- Collect dried blood spots as described in43,44.
2. Extraction of Tacrolimus Dried Blood Spot Samples
Visually inspect the dried blood spot to ensure acceptable sample quality and volume45.
Punch center of the blood spot on the filter card with a 6-mm hole punch. Note: The quality of punches may be monitored by weighing. A punched saturated filter disc weighs in average 5.02 mg ± 0.09 mg (range: 4.83- 5.14 mg, n=12).
Place discs into 1.5 ml polypropylene tubes with conical bottom and snap-on lids.
Add 20-30 bullets to each tube.
Add 500 µl of the protein precipitation solution (methanol: 0.2 M ZnSO4, 7:3, v:v with 2.5 ng/ml internal standard) into each tube. For the extraction of blank samples, use protein precipitation solution without the internal standard.
Homogenize the discs in the bullet blender for 1 min (maximum speed, setting “10”).
Shake samples at RT on multi-tube vortex (maximum speed, setting “10”) for 10 min.
Centrifuge samples at 16,000 x g and 4 °C for 10 min.
Transfer the supernatants into glass HPLC vials equipped with a 300 µl insert. Use pre-slit Teflon seals. Note: Extracted samples may be stored at -20 °C or below until LC-MS/MS analysis.
3. LC-MS/MS Analysis
Load 100 µl of the supernatant of the extracted sample onto the C8 cartridge extraction column and wash with a 7:3 ratio of 0.1% formic acid in water: acetonitrile at a flow of 5 ml/min for 1 min. The connections of the switching valve are shown in Figure 2 and the gradient run by the extraction pump in Table 1.
Hereafter, activate the switching valve resulting in back-flush of the analytes from the pre-column onto the analytical column.
Set the column thermostat to 65 °C.
Elute the analytes from the analytical column using the flow rates and gradient shown in Table 1.
Connect the analytical column to a tandem mass spectrometer via the turbo electrospray ionization source. Adjust the key parameters of the mass spectrometer according to Table 2.
Detect positive ions ([M+Na]+) in the multiple reaction mode (MRM). Use the following ion transitions for quantification: tacrolimus: m/z (mass/charge) = 826.6 → 616.2 and D2,13C-tacrolimus: m/z= 829.6 → 619.2. Note: The total run time is 4.6 min.
4. Quantification
- For each run, generate a calibration curve based on the calibrators prepared in 1.3.5 and include in each analytical run.
- Generate a calibration curve by plotting nominal concentrations versus response factor of analyte (Peak Area [Analyte] / Peak Area [internal standard]) using the mass spectrometer software.
- Fit the calibrators using a quadratic fit in combination with 1/X weighting.
To quantity tacrolimus in the dried blood spots integrate the tacrolimus and internal standard peaks in the extracted MRM chromatograms. Calculate the response factor for tacrolimus (Peak Area [Analyte] / Peak Area [internal standard]) and compare with the calibration curve using the mass spectrometry software.
5. Validation Procedures
- Lower limit of detection (LLOD) and lower limit of quantification (LLOQ).
- Consider the lowest tacrolimus concentration with a peak-to-noise ratio of 4:1 as the lower limit of detection (LLOD). Define the lower limit of quantification (LLOQ) as the lowest concentration of the calibration curve with an accuracy equal to or better than ± 20% deviation from the nominal concentration and precision equal to or better than 20% (coefficient of variance).
- Intra- and inter-day accuracies and precisions.
- Test the accuracy and precision at four concentration levels of 2 ng/ml (QC1), 4 ng/ml (QC2), 20 ng/ml (QC3) and 40 ng/ml (QC4).
- Prepare the QC samples on each validation day in human EDTA whole blood, dry on filter cards, extract, and analyze as described above.
- Determine intra-day accuracy and precision with 6 samples per QC concentration level.
- Assess inter-day accuracy and precision over 20 days. Measure each QC level with 4 samples each day.
- Analyze two calibration curves together with the QC samples on each day.
- Calculate intra-day accuracy as the % of the nominal concentration (six samples per concentration level, please see 5.2.2). Calculate precision as the coefficient of variance (CV%).
- Consider intra-day accuracy acceptable if it falls into the acceptance limits 85% to 115% of the nominal concentration. Consider intra-day precision acceptable if it is equal to or better than a CV (coefficient of variance) of 15%.
- Calculate inter-day accuracy and precision as the mean for each QC concentration level analyzed over the 20 validation days.
- Consider mean inter-day accuracy acceptable if it falls into the acceptance limits 85% to 115% of the nominal concentration. Consider inter-day precision acceptable if it is equal to or better than a CV (coefficient of variance) of 15%.
- Exclusion of matrix interferences.
- For the exclusion of interferences that may be caused by matrix signals, analyze blank dried blood spots (8 different individuals, preferably 4 males and 4 females).
- Visually inspect ion chromatograms. If peaks within the retention time window of tacrolimus are detected, integrate and compare their areas under the curve with those of tacrolimus peaks in blank samples spiked with tacrolimus at the LLOQ. The area of peaks in the blank samples are not supposed to exceed 15% of those of tacrolimus at the LLOQ.
- Ion suppression/ion enhancement.
- Use a post-column infusion protocol as described45 to assess potential interference of ion suppression/ion enhancement caused by co-eluting matrix components.
- Infuse tacrolimus at a concentration of 10 µg/ml dissolved in 0.1% formic acid: methanol (30:70, v/v) post-column at a rate of 10 µl/min.
- Connect a syringe pump via T-piece between the analytical column and the electrospray source of the mass spectrometer.
- Monitor the MS/MS signal intensity of the MRM transitions for tacrolimus and its internal standard (m/z = 826.6 → 616.2 and m/z = 829.6 → 619.2) after injection of extracted blank samples (n = 8 samples from different individuals). Note: In the absence of ion suppression/ion enhancement the continuous signal caused by infusion of the analytes should not be affected by injection of the blank matrix, while ion suppression causes a dip of the signal and ion enhancement a peak.
- Carry-over.
- Assess potential carry-over by analyzing extracted blank samples after the highest calibrators (50 ng/ml, n = 6).
- Visually inspect ion chromatograms. If peaks within the retention time window of tacrolimus are detected, integrate and compare their areas under the curve with those of tacrolimus peaks in blank samples spiked with tacrolimus at the LLOQ. The area of peaks in the blank samples are not supposed to exceed 15% of those of tacrolimus at the LLOQ.
- Extraction recoveries.
- Determine recoveries by comparing the signals of the analytes after extraction of QC samples at all four concentration levels (n = 6 per concentration) with those of blank dried blood spots spiked with the corresponding amounts of tacrolimus after the extraction.
- Prepare four sets of QCs (concentration levels: 2, 4, 20, 40 ng/ml).
- Prepare another 4 sets of corresponding “recovery test samples” by spotting 50 µl of blank EDTA whole blood onto the filter paper cards and drying for 2 hr.
- Hereafter, for both the QC and blank “recovery test samples”, cut out the entire blood spot on the filter card with scissors and place the resulting discs into a polypropylene tube with conical bottom and snap-on lid.
- Extract all samples.
- Transfer the supernatants (400 µl) into glass HPLC vials.
- Add tacrolimus stock solution to the blank “extracted recovery test samples” to reach concentrations of 2, 4, 20 and 40 ng/ml (4 µl of 200, 400, 2,000, 4,000 ng/ml tacrolimus stock solutions to 400 µl of supernatant).
- After LC-MS/MS analysis, compare the signals in both QC samples and “recovery test samples” of the corresponding concentration (recovery% = signal samples spiked before extraction / signal samples spiked after extraction x 100).
- Dilution integrity.
- Establish dilution integrity using samples spiked with the analytes at 500, 250 and 100 ng/ml.
- After extraction, dilute samples using protein precipitation solution (1:10, n = 3 per concentration level).
- Calculate deviations from the nominal concentrations. Consider results that fall within 85%-115% of nominal acceptable.
- Stabilities.
- Investigate stabilities using the QC samples at all four concentration levels (n = 4 per concentration) analyzed at different time-points and under the different storage conditions.
- Compare results after storage with the nominal values. Consider results that fall within 85%-115% of nominal acceptable.
- Establish sample stability for 1 week at ambient temperature, 1 week at 4 °C, 1 month at -20 °C and 1 month at -80 °C.
- Test freeze-thaw stability over three cycles (-20 °C). Test extracted sample and autosampler stability by placing samples into the thermostatted autosampler adjusted to 4 °C. Inject samples after 72 hr.
Representative Results
Representative ion chromatograms of a blank sample, a sample spiked at the lower limit of quantification and a patient sample are shown in Figure 3.
Calibration Curves
The lower limit of detection was 0.5 ng/ml and the lower limit of quantification was 1.0 ng/ml. Fifty ng/ml was chosen as the highest calibrator as higher concentrations are unlikely to be reached in the clinic under normal circumstances.
Calibration curves were freshly prepared on each validation day in human EDTA whole blood, dried on filter cards and extracted with methanol / 0.2 M ZnSO4 (70:30 v/v) + internal standard (final concentration of internal standard: 2.5 ng/ml). For the day 1 validation (n = 6 for calibrators and n = 6 for QC level) and for days 2 - 20 (n = 2 for calibrators and n = 4 for each QC level) were analyzed with concentrations 1, 2.5, 5, 10, 25, 50 ng/ml for calibrators. A typical calibration curve is shown in Figure 4. Mean accuracies of 85% to 115% of nominal, within the working range for 2/3 of the calibrators (with a minimum of 6 non-zero calibrators) were considered acceptable. The mean coefficient of correlation was (r) = 0.999 (n = 40 calibration curves).
Accuracies and Precisions
The results are shown in detail in Table 3.
Extraction Recovery
Average extraction recoveries were 98.2% (2 ng/ml), 92.2 (4 ng/ml), 95.5 (20 ng/ml), 96.2 (40 ng/ml).
Matrix Interferences, Ion Suppression/Ion Enhancement Testing Using Continuous Post-Column Infusion and Carry-over
Analysis of the blank samples from eight different individuals (n = 4 female and n = 4 male) showed signals less than 15% of the LLOQ (1 ng/ml) at the retention time corresponding to the tacrolimus peak indicating that the detected tacrolimus peak can be considered specific. A representative example is shown in Figure 3. Potential interferences by ion suppression/ion enhancement were tested using blank dried blood samples from eight different healthy individuals. A representative experiment is shown in Figure 5. No indications of significant ion suppression/ion enhancement were observed. No relevant carry-over resulting in peaks exceeding 15% of the signal at LLOQ were detected.
Dilution Integrity
Dilution integrity was investigated by analyzing samples prepared at concentrations above the highest calibrator (100, 250 and 500 ng/ml) and diluted 1:10 in protein precipitation solution after extraction to reach the target concentrations of: 10, 25, and 50 ng/ml. Mean accuracies had to fall within the acceptance criteria of 85% to 115% of the nominal concentrations. All dilutions tested met acceptance criteria (Table 4).
Stabilities
Stability of tacrolimus in dried blood spots was investigated by analyzing QC samples at all four levels (n = 4 / concentration level), which were stored under varying conditions.
Mean accuracies had to fall within the acceptance criteria of 85% to 115% of the nominal concentrations. Results are shown in detail in Table 5.No losses after 1 week of storage at RT, after 1 week of storage at 4 °C, after 1 month of storage at -20 °C, after 1 month of storage at -80 °C, after 3 freeze and thaw cycles, and after 72 hr of extracted samples in auto sampler at 4 °C were apparent.
| Extraction Pump | Analytical (Elution) Pump | ||||||
| Time [min] | Water + 0.1% formic acid | Acetonitrile | Flow rate [µl/min] | Time [min] | Water + 0.1% formic acid | Acetonitrile | Flow rate [µl/min] |
| 0 | 70 | 30 | 5,000 | 0 | 13 | 87 | 1,000 |
| 1 | 70 | 30 | 5,000 | 2 | 2 | 98 | 1,200 |
| 1.1 | 2 | 98 | 100 | 3.5 | 2 | 98 | 1,200 |
| 3 | 2 | 98 | 100 | 3.6 | 13 | 87 | 1,000 |
| 3.1 | 20 | 80 | 2,000 | 4.6 | 13 | 87 | 1,000 |
| 4 | 70 | 30 | 5,000 | ||||
| 4.6 | 70 | 30 | 5,000 |
Table 1. Gradient Program for the Extraction and Analytical HPLC Pumps.
| Parameter | Setting |
| Collision gas (CAD) | 10 |
| Curtain gas (CUR) (psi) | 30 |
| Ion Source gas 1 (GS1) (psi) | 50 |
| Ion source gas 2 (GS2) (psi) | 30 |
| Nebulizer current (NC) (V) | 1 |
| Temperature (TEM) (°C) | 600 |
| IonSpray Voltage (IS) (V) | 5500 |
| Interface heater (ihe) | On |
| Declustering potential (DP) (V) | 136 |
| Entrance potential (EP) (V) | 10 |
| Collision energy (CE) (V) | 47 |
| Collision cell exit potential (CXP) (V) | 16 |
Table 2. Turbo Electrospray Interface and Mass Spectrometer Parameters. The nomenclature corresponds to that used in the mass spectrometry software (for manufacturer details, please see Materials List).
| Validation Day | QC Level [% of nominal concentration] | |||
| 2 | 4 | 20 | 40 | |
| Day 1 | 93.0 | 86.3 | 88.9 | 93.4 |
| 101 | 90.4 | 95.3 | 100 | |
| 85.6 | 95.9 | 99.0 | 97.5 | |
| 88.6 | 93.6 | 105 | 103 | |
| 85.0 | 97.4 | 97.2 | 109 | |
| 89.1 | 96.7 | 100 | 101 | |
| Intra-day Accuracy [%] | 90.4 | 93.4 | 97.6 | 100.7 |
| Intra-Day Imprecision [CV%] | 6.6 | 4.6 | 5.5 | 5.2 |
| Day 2 | 92.8 | 86.0 | 103 | 95.4 |
| 91.1 | 88.8 | 94.7 | 87.0 | |
| 88.6 | 90.9 | 92.8 | 94.2 | |
| 97.2 | 93.7 | 94.2 | 115 | |
| Intra-day Accuracy [%] | 92.4 | 89.9 | 96.2 | 97.9 |
| Intra-Day Imprecision [CV%] | 3.9 | 3.6 | 4.8 | 12.2 |
| Day 3 | 97.6 | 101 | 98.4 | 112 |
| 104 | 85.6 | 102 | 110 | |
| 99.2 | 88.4 | 99.4 | 105 | |
| 96.3 | 87.2 | 108 | 117 | |
| Intra-day Accuracy [%] | 99.3 | 90.6 | 102.0 | 111.0 |
| Intra-Day Imprecision [CV%] | 3.4 | 7.8 | 4.2 | 4.5 |
| Day 4 | 95.2 | 88.6 | 112 | 94.5 |
| 105 | 87.3 | 93.2 | 116 | |
| 99.8 | 96.2 | 103 | 103 | |
| 100 | 104 | 97.2 | 99.4 | |
| Intra-day Accuracy [%] | 100.0 | 94.0 | 101.4 | 103.2 |
| Intra-Day Imprecision [CV%] | 4.0 | 8.2 | 8.1 | 8.9 |
| Day 5 | 106 | 90.4 | 101 | 106 |
| 108 | 89.0 | 106 | 100 | |
| 102 | 101 | 96.6 | 128 | |
| 105 | 88.8 | 105 | 107 | |
| Intra-day Accuracy [%] | 105.3 | 92.3 | 102.2 | 110.3 |
| Intra-Day Imprecision [CV%] | 2.4 | 6.3 | 4.2 | 11.1 |
| Day 6 | 90.9 | 93.7 | 119 | 106 |
| 98.8 | 88.1 | 96.4 | 110 | |
| 94.6 | 96.3 | 99.1 | 108 | |
| 108 | 100 | 102 | 102 | |
| Intra-day Accuracy [%] | 98.1 | 94.5 | 104.1 | 106.5 |
| Intra-Day Imprecision [CV%] | 7.5 | 5.3 | 9.8 | 3.2 |
| Day 7 | 85.1 | 87.5 | 99.5 | 95.4 |
| 86.4 | 85.4 | 94.7 | 101 | |
| 94.5 | 87.3 | 98.9 | 94.6 | |
| 85.5 | 97.0 | 101 | 99.6 | |
| Intra-day Accuracy [%] | 87.9 | 89.3 | 98.5 | 97.7 |
| Intra-Day Imprecision [CV%] | 5.1 | 5.8 | 2.7 | 3.2 |
| Day 8 | 86.1 | 92.5 | 91.9 | 102 |
| 87.5 | 91.5 | 95.2 | 88.5 | |
| 115 | 85.6 | 92.1 | 102 | |
| 85.8 | 85.9 | 95.4 | 108 | |
| Intra-day Accuracy [%] | 93.6 | 88.9 | 93.7 | 100.1 |
| Intra-Day Imprecision [CV%] | 15.3 | 4.1 | 2.0 | 8.2 |
| Day 9 | 88.9 | 91.4 | 96.9 | 100 |
| 90.0 | 89.8 | 95.0 | 100 | |
| 69.7 | 85.9 | 95.8 | 109 | |
| 91.9 | 87.0 | 105 | 101 | |
| Intra-day Accuracy [%] | 85.1 | 88.5 | 98.2 | 102.5 |
| Intra-Day Imprecision [CV%] | 12.2 | 2.9 | 4.7 | 4.3 |
| Day 10 | 90.9 | 91.3 | 96.2 | 100 |
| 97.7 | 89.5 | 94.4 | 100 | |
| 99.9 | 109 | 98.7 | 96.8 | |
| 99.1 | 90.0 | 95.7 | 96.1 | |
| Intra-day Accuracy [%] | 96.9 | 95.0 | 96.3 | 98.2 |
| Intra-Day Imprecision [CV%] | 4.2 | 9.9 | 1.9 | 2.1 |
| Day 11 | 92.7 | 91.9 | 88.2 | 104 |
| 96.6 | 91.2 | 97.0 | 110 | |
| 109.0 | 92.8 | 97.6 | 102 | |
| 98.3 | 107 | 93.7 | 111 | |
| Intra-day Accuracy [%] | 99.2 | 95.7 | 94.1 | 106.8 |
| Intra-Day Imprecision [CV%] | 7.0 | 7.9 | 4.6 | 4.1 |
| Day 12 | 87.7 | 85.5 | 105 | 95.3 |
| 112 | 88.1 | 101 | 96.1 | |
| 102 | 89.1 | 89.7 | 97.5 | |
| 106 | 92.5 | 102 | 104 | |
| Intra-day Accuracy [%] | 101.9 | 88.8 | 99.4 | 98.2 |
| Intra-Day Imprecision [CV%] | 10.1 | 3.3 | 6.7 | 4.0 |
| Day 13 | Failed | 85.7 | 93.3 | 102 |
| 101 | 105 | 88.0 | 93.9 | |
| 112 | 98.0 | 91.4 | 102 | |
| 104 | 113 | 104 | 101 | |
| Intra-day Accuracy [%] | 105.7 | 100.4 | 94.2 | 99.7 |
| Intra-Day Imprecision [CV%] | 5.4 | 11.5 | 7.3 | 3.9 |
| Day 14 | 91.5 | 89.1 | 97.4 | 93.1 |
| 90.4 | 87.1 | 93.9 | 99.8 | |
| 89.7 | 97.0 | 94.8 | 106 | |
| 97.4 | 86.8 | 89.9 | Failed | |
| Intra-day Accuracy [%] | 92.3 | 90.0 | 94.0 | 99.6 |
| Intra-Day Imprecision [CV%] | 3.8 | 5.3 | 3.3 | 6.5 |
| Day 15 | 92.8 | 92.8 | 92.5 | 95.4 |
| 97.5 | 96.3 | 96.2 | 95.5 | |
| 95.5 | 108 | 97.3 | 99.3 | |
| 110 | 109 | 115 | 113 | |
| Intra-day Accuracy [%] | 99.0 | 101.5 | 100.3 | 100.8 |
| Intra-Day Imprecision [CV%] | 7.7 | 8.1 | 10.0 | 8.3 |
| Day 16 | 93.7 | 97.8 | 90.7 | 112 |
| 90.3 | 87.1 | Failed | 101 | |
| 97.9 | 88.3 | 95.5 | 107 | |
| 91.4 | 85.7 | 89.3 | 96.7 | |
| Intra-day Accuracy [%] | 93.3 | 89.7 | 91.8 | 104.2 |
| Intra-Day Imprecision [CV%] | 3.6 | 6.1 | 3.5 | 6.4 |
| Day 17 | 88.0 | 86.0 | 93.7 | 103 |
| 89.8 | 90.8 | 94.8 | 93.2 | |
| 85.9 | 91.1 | 99.7 | 94.8 | |
| 86.7 | 88.1 | 95.6 | 91.7 | |
| Intra-day Accuracy [%] | 87.6 | 89.0 | 96.0 | 95.7 |
| Intra-Day Imprecision [CV%] | 1.9 | 2.7 | 2.7 | 5.3 |
| Day 18 | 89.6 | 85.8 | 91.0 | 98.3 |
| 89.6 | 86.2 | 88.3 | 93.6 | |
| Failed | 86.7 | 96.8 | 104 | |
| 88.1 | 85.8 | 95.2 | 111 | |
| Intra-day Accuracy [%] | 89.1 | 86.1 | 92.8 | 101.7 |
| Intra-Day Imprecision [CV%] | 1.0 | 0.5 | 4.2 | 7.4 |
| Day 19 | 98.0 | 89.7 | 94.2 | 102 |
| 88.3 | 86.0 | 97.6 | 102 | |
| 91.6 | 88.1 | 95.8 | 97.5 | |
| 90.7 | 90.1 | 92.8 | 88.3 | |
| Intra-day Accuracy [%] | 92.2 | 88.5 | 95.1 | 97.5 |
| Intra-Day Imprecision [CV%] | 4.5 | 2.1 | 2.2 | 6.6 |
| Day 20 | 93.0 | 87.0 | 99.4 | 101 |
| 97.3 | 87.6 | 95.5 | 91.9 | |
| 89.0 | 88.4 | 91.2 | 93.5 | |
| 104 | 90.7 | 97.7 | 115 | |
| Intra-day Accuracy [%] | 95.8 | 88.4 | 96.0 | 100.4 |
| Intra-Day Imprecision [CV%] | 6.7 | 1.8 | 3.7 | 10.5 |
| Inter-Day Accuracy and Imprecision | ||||
| Inter-day Accuracy | 95.2 | 91.7 | 97.2 | 101.6 |
| Inter-Day Imprecision | 6.1 | 4.5 | 3.6 | 4.2 |
Table 3. Results of Quality Control Samples over 20 Days. Data is presented as % of nominal. Samples listed as “failed” are samples that were lost to laboratory/instrument errors. In most cases, no peaks were detected at all or the internal standard peak was missing.
| Dilution | 1:10 |
| Nominal target concentration after dilution | 50 ng/ml |
| 98.6 | |
| 94.5 | |
| 91.4 | |
| Accuracy [%] | 94.8 |
| Imprecision [CV%] | 3.6 |
| Dilution | 1:10 |
| Nominal target concentration after dilution | 10 ng/ml |
| 103 | |
| 99.5 | |
| 101 | |
| Accuracy [%] | 101.2 |
| Imprecision [CV%] | 1.8 |
| Dilution | 1:10 |
| Nominal target concentration after dilution | 25 ng/ml |
| 91.7 | |
| 98.2 | |
| 103 | |
| Accuracy [%] | 97.6 |
| Imprecision [CV%] | 5.7 |
Table 4. Results of Dilution Integrity Testing. Data is presented as % of nominal.
| A | ||||
| Stability at RT, Day 1 | ||||
| QC level [ng/ml] | 2 | 4 | 20 | 40 |
| 91.9 | 85.8 | 86.0 | 102 | |
| 86.7 | 85.7 | 88.5 | 102 | |
| 86.0 | 85.9 | 90.1 | 103 | |
| 89.2 | 98.0 | 90.8 | 112 | |
| % of nominal concentration | 88.5 | 88.9 | 88.9 | 104.8 |
| Imprecision [%CV] | 2.7 | 6.1 | 2.1 | 4.9 |
| Stability at RT, Day 3 | ||||
| QC level [ng/ml] | 2 | 4 | 20 | 40 |
| 88.6 | 105 | 101 | 113 | |
| 94.1 | 100 | 98.5 | 103 | |
| 100 | 101 | 106 | 109 | |
| 99.5 | 102 | 102 | 108 | |
| % of nominal concentration | 95.6 | 102.0 | 101.9 | 108.3 |
| Imprecision [%CV] | 5.3 | 2.2 | 3.1 | 4.1 |
| Stability at RT, Day 7 | ||||
| QC level [ng/ml] | 2 | 4 | 20 | 40 |
| 105 | 103 | 91.7 | 109 | |
| 101 | 107 | 100 | 110 | |
| 102 | 108 | 107 | 105 | |
| 93.8 | 105 | 109 | 111 | |
| % of nominal concentration | 100.5 | 105.8 | 101.9 | 108.8 |
| Imprecision [%CV] | 4.7 | 2.2 | 7.8 | 2.6 |
| B | ||||
| Stability at +4 °C, Day 1 | ||||
| QC level [ng/ml] | 2 | 4 | 20 | 40 |
| 101 | 89.9 | 95.8 | 100 | |
| 88.9 | 91.0 | 94.1 | 99.0 | |
| 96.2 | 100 | 102 | 96.7 | |
| 89.5 | 87.8 | 95.4 | 88.6 | |
| % of nominal concentration | 93.9 | 92.2 | 96.8 | 96.1 |
| Imprecision [%CV] | 5.8 | 5.4 | 3.5 | 5.2 |
| Stability at +4 °C, Day 3 | ||||
| QC level [ng/ml] | 2 | 4 | 20 | 40 |
| 87.3 | 85.2 | 105 | 95.3 | |
| Failed | 87.8 | 101 | 96 | |
| 101 | 88.8 | 89.6 | 97.5 | |
| 106 | 92.2 | 102 | 104 | |
| % of nominal concentration | 98.1 | 88.5 | 99.4 | 98.2 |
| Imprecision [%CV] | 9.7 | 2.9 | 6.8 | 4.0 |
| Stability at +4 °C, Day 7 | ||||
| QC level [ng/ml] | 2 | 4 | 20 | 40 |
| 94.0 | 98.5 | 96.1 | 110 | |
| 92.9 | 96.4 | 109 | 109 | |
| 91.7 | 96.3 | 97.9 | 115 | |
| 94.7 | 96.9 | 99.8 | 113 | |
| % of nominal concentration | 93.3 | 97.0 | 100.7 | 111.8 |
| Imprecision [%CV] | 1.3 | 1.0 | 5.7 | 2.8 |
| C | ||||
| Stability at -20 °C, Day 3 | ||||
| QC level [ng/ml] | 2 | 4 | 20 | 40 |
| 87.3 | 98.7 | 111 | 111 | |
| 93.5 | 89.6 | 108 | 105 | |
| 89.5 | 91.5 | 107 | 112 | |
| 88.2 | 99.1 | 108 | 92.4 | |
| % of nominal concentration | 89.6 | 94.7 | 108.5 | 105.1 |
| Imprecision [%CV] | 2.7 | 4.9 | 1.7 | 9.0 |
| Stability at -20 °C, Day 7 | ||||
| QC level [ng/ml] | 2 | 4 | 20 | 40 |
| 96.5 | 98.7 | 102 | 109 | |
| 94.8 | 94.6 | 114 | 106 | |
| 95.5 | 102 | 98.1 | 108 | |
| 107 | 99.5 | 115 | 105 | |
| % of nominal concentration | 98.5 | 98.7 | 107.3 | 107 |
| Imprecision [%CV] | 5.7 | 3.1 | 8.5 | 1.8 |
| Stability at -20 °C, Day 30 | ||||
| QC level [ng/ml] | 2 | 4 | 20 | 40 |
| 82.3 | 83.1 | 90.4 | 93.2 | |
| 87.9 | 85.8 | 85.3 | 97.9 | |
| 85.7 | 88.6 | 98.3 | 98.0 | |
| 92.0 | 95.6 | 110 | 103 | |
| % of nominal concentration | 87.0 | 88.3 | 96.0 | 98.0 |
| Imprecision [%CV] | 4.1 | 5.4 | 10.8 | 4.0 |
| D | ||||
| Stability at -80 °C, Day 3 | ||||
| QC level [ng/ml] | 2 | 4 | 20 | 40 |
| 87.5 | 96.5 | 96.7 | Failed | |
| Failed | 97.6 | 98.7 | 110 | |
| 88.8 | 96.4 | 106 | 109 | |
| 87.3 | 101 | 96.5 | 109 | |
| % of nominal concentration | 87.9 | 97.9 | 99.5 | 109.3 |
| Imprecision [%CV] | 0.8 | 2.2 | 4.5 | 0.6 |
| Stability at -80 °C, Day 7 | ||||
| QC level [ng/ml] | 2 | 4 | 20 | 40 |
| Failed | 98.0 | 105 | 99.8 | |
| 97.1 | 106 | 104 | 105 | |
| 99.7 | 102 | 99.3 | 102 | |
| 101 | 99.9 | 109 | 106 | |
| % of nominal concentration | 99.3 | 101.5 | 104.3 | 103.2 |
| Imprecision [%CV] | 2.0 | 3.4 | 4.0 | 2.8 |
| Stability at -80 °C, Day 30 | ||||
| QC level [ng/ml] | 2 | 4 | 20 | 40 |
| 88.2 | 85.3 | 89.6 | 96.7 | |
| 96.2 | 92.6 | 85.8 | 94.1 | |
| 83.9 | 93.7 | 91.0 | 105 | |
| 95.6 | 94.0 | 98.9 | 102 | |
| % of nominal concentration | 91.0 | 91.4 | 91.3 | 99.5 |
| Imprecision [%CV] | 6.0 | 4.1 | 5.5 | 4.9 |
| E | ||||
| Freeze thaw stability, -20 °C, 1 cycle | ||||
| QC level [ng/ml] | 2 | 4 | 20 | 40 |
| 92.5 | 86.2 | 90.2 | 94.3 | |
| 89.8 | 90.5 | 85.4 | 104 | |
| 94.7 | 88.6 | 93.4 | 104 | |
| 101 | 89.2 | 93.7 | 96.3 | |
| % of nominal concentration | 94.5 | 88.6 | 90.7 | 99.7 |
| Imprecision [%CV] | 4.8 | 1.8 | 3.9 | 5.1 |
| Freeze thaw stability, -20 °C, 2 cycles | ||||
| QC level [ng/ml] | 2 | 4 | 20 | 40 |
| 99.1 | 97.6 | Failed | 85.3 | |
| 93.1 | 88.1 | 93.4 | 92.0 | |
| 94.9 | 91.5 | 85.8 | 93.9 | |
| Failed | 90.5 | 86.8 | 85.4 | |
| % of nominal concentration | 95.7 | 91.9 | 88.7 | 89.2 |
| Imprecision [%CV] | 3.1 | 4.0 | 4.1 | 4.5 |
| Freeze thaw stability, -20 °C, 3 cycles | ||||
| QC level [ng/ml] | 2 | 4 | 20 | 40 |
| 95.7 | Failed | 86.0 | 93.3 | |
| 95.5 | 87.4 | 85.0 | 91.3 | |
| 90.5 | 89.1 | 86.0 | 86.0 | |
| 96.9 | 85.7 | 90.2 | Failed | |
| % of nominal concentration | 94.7 | 87.4 | 86.8 | 90.2 |
| Imprecision [%CV] | 2.8 | 1.7 | 2.3 | 3.8 |
| F | ||||
| Extracted sample stability at +4 °C, 24 hr | ||||
| QC level [ng/ml] | 2 | 4 | 20 | 40 |
| 122 | 112 | 91.0 | 104 | |
| 93.5 | 106 | 91.8 | 96.1 | |
| 111 | 94.8 | 98.2 | 93.5 | |
| 98.4 | 97.6 | 91.3 | 89.8 | |
| % of nominal concentration | 106.2 | 102.6 | 93.1 | 95.9 |
| Imprecision [%CV] | 12.8 | 7.9 | 3.4 | 6.0 |
| Extracted sample stability at +4 °C, 48 hr | ||||
| QC level [ng/ml] | 2 | 4 | 20 | 40 |
| 106 | 108 | 93.7 | 110 | |
| 105 | 98.1 | 94.6 | 98.9 | |
| 103 | 98.4 | 95.0 | 92.6 | |
| 108 | 93.1 | 89.7 | 85.5 | |
| % of nominal concentration | 105.5 | 99.4 | 93.3 | 96.8 |
| Imprecision [%CV] | 2.1 | 6.2 | 2.4 | 10.4 |
| Extracted sample stability at +4 °C, 72 hr | ||||
| QC level [ng/ml] | 2 | 4 | 20 | 40 |
| 96.5 | 112 | 96.4 | 104 | |
| 101 | 95.3 | 103 | 95.2 | |
| 94.2 | 105 | 93.5 | 99.5 | |
| 100 | 96.9 | 92.6 | 89.3 | |
| % of nominal concentration | 97.9 | 102.3 | 96.4 | 97.0 |
| Imprecision [%CV] | 3.1 | 7.7 | 4.7 | 6.3 |
Table 5. Results of Stability Testing. A: Stability of tacrolimus on dried blood spots at RT over 7 days, B: Stability of tacrolimus on dried blood spots in the refrigerator (+4 °C) over 7 days, C: Stability of tacrolimus on dried blood spots at -20 °C over 1 month, D: Stability of tacrolimus on dried blood spots at -80 °C over 1 month, E: Stability of tacrolimus on dried blood spots over three freeze-thaw cycles (-20 °C), F: Extracted sample/autosampler stability at +4 °C over 72 hr. Data is presented as % of nominal concentration. Samples listed as “failed” are samples that were lost to laboratory/instrument errors. In most cases no peaks were detected at all or the internal standard peak was missing.
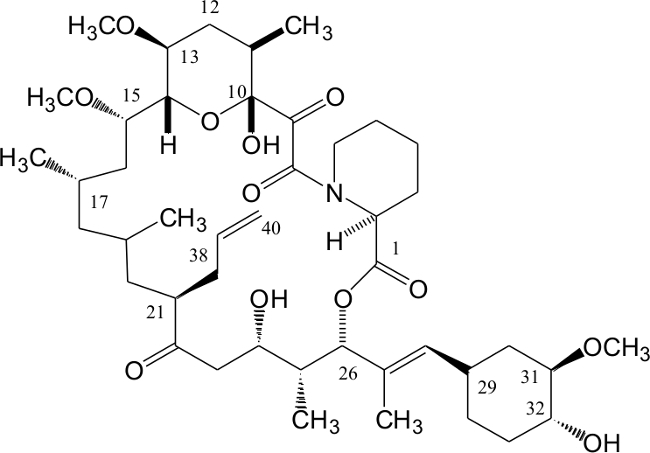 Figure 1. Structure of Tacrolimus. Atom numbering follows the International Union of Pure and Applied Chemistry (IUPAC) nomenclature. Please click here to view a larger version of this figure.
Figure 1. Structure of Tacrolimus. Atom numbering follows the International Union of Pure and Applied Chemistry (IUPAC) nomenclature. Please click here to view a larger version of this figure.
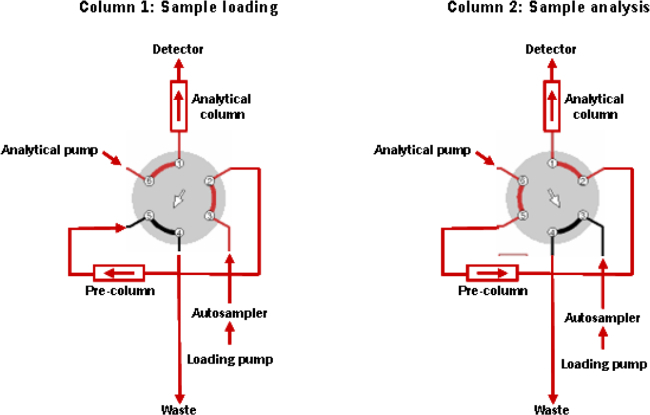 Figure 2. Connection of the Switching Valve.
Please click here to view a larger version of this figure.
Figure 2. Connection of the Switching Valve.
Please click here to view a larger version of this figure.
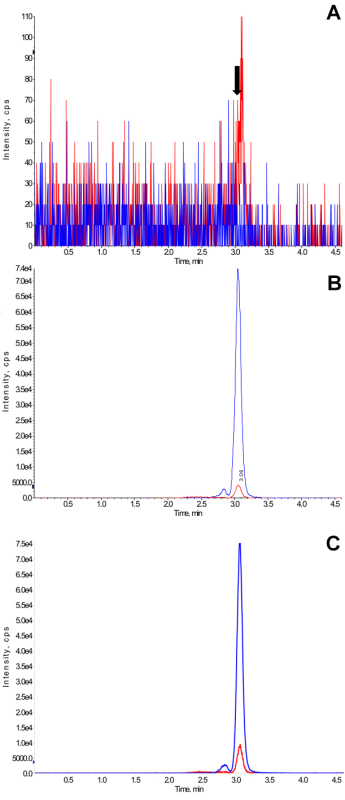 Figure 3.Representative Ion Chromatograms. (A) Representative ion chromatogram of a blank blood samples spotted onto filter paper (for manufacturer details, please see Materials List) and dried. The arrow marks the retention time of the tacrolimus peak, (B) Representative ion chromatogram of a blank blood samples spiked at the lower limit of quantification (1 ng/ml) spotted onto the filter paper and dried, and (C) Representative ion chromatogram of a sample collected by a transplant patient on filter paper. This is a trough sample and the measured tacrolimus concentration was 2.1 ng/ml. This sample was collected by the patient at home and is from a clinical trial that was approved by the University of Cincinnati Institutional Review Board (Cincinnati, OH). All patients gave their appropriate written consent. Ion chromatograms are original print-outs as generated by the mass spectrometry software (for manufacturer details, please see Materials List). Blue and red lines in ion chromatograms represent the internal standard D2,13C-tacrolimus and tacrolimus, respectively. The peak eluting in front of the main tacrolimus and internal standard peaks are the rotamers. Please click here to view a larger version of this figure.
Figure 3.Representative Ion Chromatograms. (A) Representative ion chromatogram of a blank blood samples spotted onto filter paper (for manufacturer details, please see Materials List) and dried. The arrow marks the retention time of the tacrolimus peak, (B) Representative ion chromatogram of a blank blood samples spiked at the lower limit of quantification (1 ng/ml) spotted onto the filter paper and dried, and (C) Representative ion chromatogram of a sample collected by a transplant patient on filter paper. This is a trough sample and the measured tacrolimus concentration was 2.1 ng/ml. This sample was collected by the patient at home and is from a clinical trial that was approved by the University of Cincinnati Institutional Review Board (Cincinnati, OH). All patients gave their appropriate written consent. Ion chromatograms are original print-outs as generated by the mass spectrometry software (for manufacturer details, please see Materials List). Blue and red lines in ion chromatograms represent the internal standard D2,13C-tacrolimus and tacrolimus, respectively. The peak eluting in front of the main tacrolimus and internal standard peaks are the rotamers. Please click here to view a larger version of this figure.
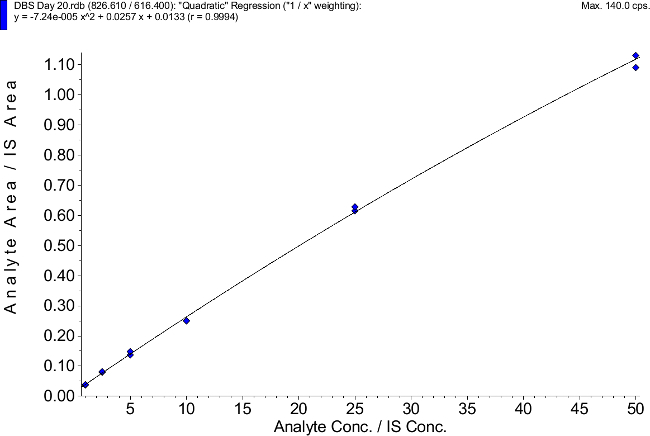 Figure 4. Representative Calibration Curve. An original print-out as generated by the mass spectrometry software is shown. Please click here to view a larger version of this figure.
Figure 4. Representative Calibration Curve. An original print-out as generated by the mass spectrometry software is shown. Please click here to view a larger version of this figure.
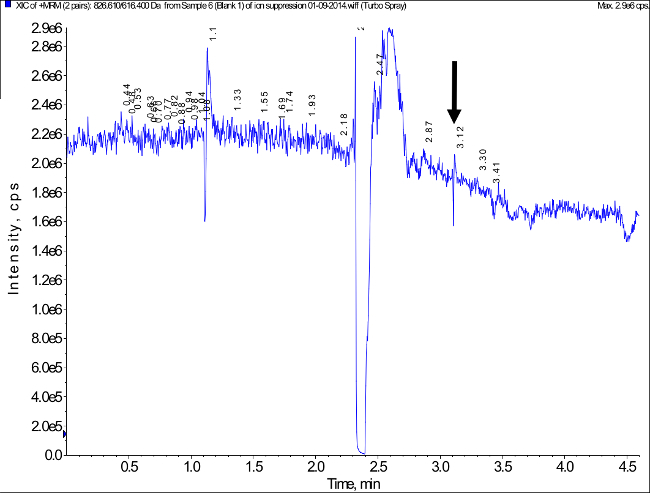 Figure 5. Representative Ion Chromatogram Measured During Post-Column Infusion of Tacrolimus and Injection of an Extracted Blank Blood Sample to Assess a Potential Matrix Effect (Ion Suppression/Ion Enhancement). The arrow marks the retention time of the tacrolimus peak. Matrix effect testing was based on the procedure described in46. No relevant matrix effect was detected. Please click here to view a larger version of this figure.
Figure 5. Representative Ion Chromatogram Measured During Post-Column Infusion of Tacrolimus and Injection of an Extracted Blank Blood Sample to Assess a Potential Matrix Effect (Ion Suppression/Ion Enhancement). The arrow marks the retention time of the tacrolimus peak. Matrix effect testing was based on the procedure described in46. No relevant matrix effect was detected. Please click here to view a larger version of this figure.
Discussion
Although, as aforementioned, the concept of therapeutic drug and adherence monitoring of tacrolimus based on dried blood spots is attractive, there are analytical challenges that go beyond those typically associated with the LC-MS/MS analysis of tacrolimus in venous EDTA whole blood samples. These include, but are not limited to, the fact that the matrix is capillary whole blood soaked into the cotton linters material of the filter card material used here and the low blood volume (20 µl). Nevertheless, high-throughput analysis in a central laboratory requires a fast and reliable extraction method that results in samples that lack matrix interferences and matrix effects in combination with a robust, specific and highly sensitive LC-MS/MS assay. Reliability of the assay is critical as there is usually not enough material on the already punched filter card left for re-analysis in case the extraction / LC-MS/MS analysis fails.
The filter cards used in the present study (for manufacturer details, please see Materials List) were chosen as these are an FDA approved class II device, are in compliance with NCCLS guidance LA4-A544 and are CE-marked in Europe. If completely filled, one circle on the Whatman 903 filter card holds ≈50 µl blood46. However, the size of the blood drops collected by individual patients vary and training in the proper sampling technique is essential46.
The first important step of extracting a punched-out dried blood spot sample is homogenization. Based on our experience, the use of a bullet blender is more efficient and better reproducible than other methods used to enhance extraction efficiency such as sonication. The use of the bullet blender was essential to consistently achieve extraction recoveries above 90%. For reliability of the extraction procedure, it was also important to ensure that all centrifuges were temperature controlled (4 °C) and that the vortexing step was not shorter than 10 min, which resulted in more variable and lower extraction recoveries. In addition, it is important that the methanol/ZnSO4 ratio is not altered as tacrolimus recovery is very sensitive to the correct composition of the protein precipitation solution.
The next challenge is to obtain a clean extract ideally devoid of materials that may cause matrix interferences and effects. Thus, a simple one-step protein precipitation step as often used for the extraction of tacrolimus from EDTA blood samples was not considered a viable option. After protein precipitation using ZnSO4 (including addition of an isotope-labeled internal standard), vortexing and centrifugation, the supernatants were injected into a 2D HPLC system and onto an online extraction column. Online column extraction using high flows of 5 ml/min on conventional pre-column cartridges using a simple 6-port switching valve for the analysis of tacrolimus have been described before47. The mobile phase was chosen so that tacrolimus and its internal standard concentrated in the front of the extraction column and did not migrate over the column during the cleanup step. The online extraction used in the present protocol had several advantages including injection of relatively large sample volumes without negatively affecting HPLC analysis. The back-flush after enriching the analytes on top of the extraction column (“peak focusing”) resulted in sharper peaks allowing for more reliable integration by the software algorithm especially for samples with low tacrolimus concentrations.48 The online cleanup step did not only remove potentially interfering matrix compounds, but also de-salted the sample. One important yet rarely discussed problem for high-volume, high throughput LC-MS/MS assays is the gradual loss of sensitivity of the LC-MS/MS system due to increasing contamination of the electrospray source during analysis of large batches. No significant matrix effects (ion suppression/ion enhancement) were observed. The negative effect of potential matrix effects were reduced/avoided by combination of the following: effective protein precipitation using methanol/ZnSO4, centrifugation after protein precipitation at 16,000 x g, high-flow online extraction, clear chromatographic separation of tacrolimus from potential interferences early eluting from the analytical column and the use of isotope-labeled tacrolimus as internal standard instead of structurally related internal standards such as ascomycin.
The lower limit of quantification was 1 ng/ml, and thus lower than that of most immunoassays that are currently frequently used for therapeutic drug monitoring of tacrolimus in EDTA blood samples. This lower limit of quantification is sufficient even for so-called low calcineurin inhibitor long-term immunosuppressive maintenance protocols.
In comparison with previously described LC-MS/MS assays to quantify tacrolimus in dried bloods spots29-34,36,39,41,42, the present assay matches or exceeds their performance in terms of lower limit of quantification, extraction recovery, accuracy and precision while avoiding potentially risky concepts such as one-step protein precipitation procedures and ultra-short chromatography times, which usually give acceptable results during the validation based on blood samples from healthy individuals. However, transplant patients are a highly complex group of patients who have diseases that affect the composition of blood and who take multiple medications. This makes it virtually impossible to exclude all potential interferences that may be present in individual patients during the validation and the only viable strategy is to set up the assay in a way that it minimizes the risk of such potential interferences. Dried blood spots have challenges such as the effect of the hematocrit on blood viscosity and thus the diffusion properties of the blood applied on filter paper49. This was not tested here again as such effects have already been described not to affect tacrolimus analysis in dried blood spots at hematocrits and tacrolimus concentrations within clinically reasonable limits29,32. Also stability of tacrolimus in dried blood spots at elevated temperatures has already been studied by others and tacrolimus in dried blood spots was found to be stable for 5 days at 37 °C and even 60 °C32, which is important for shipment of dried blood spots under not temperature-controlled conditions especially during summer.
This assay based on a combination of bullet blender homogenization, high-flow online column cleanup and LC-MS/MS analysis may provide a platform strategy for the development of bioanalytical assays for the quantification of other immunosuppressants, alone or simultaneous, as well as of other drugs in dried blood spot samples.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by the United States Federal Drug Administration (FDA) contract HHSF223201310224C and the United States National Institutes of Health/FDA grant 1U01FD004573-01.
References
- Goto T, et al. Discovery of FK506, a novel immunosuppressant isolated from Streptomyces Tsukubaensis. Transplant Proc. 1987;19(5 Suppl 6):4–8. [PubMed] [Google Scholar]
- Kino T, Hatanaka H, Miyata S. FK506, a novel immunosuppressant isolated from a streptomyces. I: Fermentation, isolation and physico-chemical and biological characteristics. J. Antibiotics. 1987;40(9):1249–1255. doi: 10.7164/antibiotics.40.1249. [DOI] [PubMed] [Google Scholar]
- Starzl TE, et al. FK506 for liver, kidney and pancreas transplantation. Lancet. 1989;2(8670):1000–1004. doi: 10.1016/s0140-6736(89)91014-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randomised trial comparing tacrolimus (FK506) and cyclosporin in prevention of liver allograft rejection. European FK506 Multicentre Liver Study Group. Lancet. 1994;344(8920):423–428. [PubMed] [Google Scholar]
- A comparison of tacrolimus (FK 506) and cyclosporine for immunosuppression in liver transplantation. The U.S. Multicenter FK506 Liver Study Group. N. Engl. J. Med. 1994;331(17):1110–1115. doi: 10.1056/NEJM199410273311702. [DOI] [PubMed] [Google Scholar]
- Mayer AD, et al. Multicenter randomized trial comparing tacrolimus (FK506) and cyclosporine in the prevention of renal allograft rejection: a report of the European Tacrolimus Multicenter Renal Study Group. Transplantation. 1997;64(3):436–443. doi: 10.1097/00007890-199708150-00012. [DOI] [PubMed] [Google Scholar]
- Pirsch JD, Miller J, Deierhoi MH, Vincenti F, Filo RS. A comparison of tacrolimus (FK506) and cyclosporine for immunosuppression after cadaveric renal transplantation. FK506 Kidney Transplant Study Group. Transplantation. 1997;15(7):977–983. doi: 10.1097/00007890-199704150-00013. [DOI] [PubMed] [Google Scholar]
- Tanaka H, et al. Physicochemical properties of FK506 a novel immunosuppressant isolated from Streptomyces Tsukubaensis. Transplant Proc. 1987;14((5 Suppl 6)):11–16. [PubMed] [Google Scholar]
- Spencer CM, Goa KL, Gills JC. Tacrolimus. An update of its pharmacology and clinical efficacy in the management of organ transplantation. Drugs. 1997;54(6):925–975. doi: 10.2165/00003495-199754060-00009. [DOI] [PubMed] [Google Scholar]
- Clipstone NA, Crabtree GR. Identification of calcineurin as a key signalling enzyme in T-lymphocyte activation. Nature. 1992;357(6380):695–697. doi: 10.1038/357695a0. [DOI] [PubMed] [Google Scholar]
- Barbarino JM, Staatz CE, Venkataramanan R, Klein TE, Altman RB. PharmGKB summary: cyclosporine and tacrolimus pathways. Pharmacogenet. Genomics. 2013;23(10):563–585. doi: 10.1097/FPC.0b013e328364db84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annual Data Report. Department of Health and Human Services, Health Resources and Services Administration, Healthcare Systems Bureau, Division of Transplantation; 2014. Available from: http://optn.transplant.hrsa.gov/data/annualreport.asp. [Google Scholar]
- Draft Guidance on Tacrolimus. Food and Drug Administration, Office of Generic Drugs; 2012. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM181006.pdf. [Google Scholar]
- Christians U, Benet LZ, Lampen A. Mechanisms of clinically significant drug interactions associated with tacrolimus. Clin. Pharmacokinet. 2002;41(11):813–851. doi: 10.2165/00003088-200241110-00003. [DOI] [PubMed] [Google Scholar]
- Christians U, Pokaiyavananichkul T, Chan L. Tacrolimus In: Pharmacokinetics and Pharmacodynamics. In: Burton ME, Shaw LM, Schentag JJ, Evans Webb, editors. Principles of Therapeutic Drug Monitoring. 4th Edition. =Baltimore: Lipincott, Wiliams, and Wilkins; 2005. pp. 529–562. [Google Scholar]
- Holt DW, et al. International Federation of Clinical Chemistry/ International Association of Therapeutic Drug Monitoring and Clinical Toxicology working group on immunosuppressive drug monitoring. Ther. Drug Monit. 2002;24(1):59–67. doi: 10.1097/00007691-200202000-00011. [DOI] [PubMed] [Google Scholar]
- Holt DW, Jones K, Lee T, Stadler P, Johnston A. Quality assessment issues of new immunosuppressive drugs and experimental experience. Ther. Drug Monit. 1996;18(4):362–367. doi: 10.1097/00007691-199608000-00008. [DOI] [PubMed] [Google Scholar]
- Jusko WJ, et al. Consensus document: therapeutic drug monitoring of tacrolimus (FK-506) Ther. Drug Monit. 1995;17(6):606–614. doi: 10.1097/00007691-199512000-00011. [DOI] [PubMed] [Google Scholar]
- Oellerich M, et al. Therapeutic drug monitoring of cyclosporine and tacrolimus. Update on Lake Louise Conference on cyclosporine and tacrolimus. Clin. Biochem. 1998;31(5):309–316. doi: 10.1016/s0009-9120(98)00049-6. [DOI] [PubMed] [Google Scholar]
- Wong SH. Therapeutic drug monitoring for immunosuppressants. Clin. Chim. Acta. 2001;313(1-2):241–253. doi: 10.1016/s0009-8981(01)00678-7. [DOI] [PubMed] [Google Scholar]
- Kahan BD, et al. Low intraindividual variability of cyclosporin A exposure reduces chronic rejection incidence and health care costs. J. Am. Soc. Nephrol. 2000;11(6):1122–1131. doi: 10.1681/ASN.V1161122. [DOI] [PubMed] [Google Scholar]
- Kahan BD, et al. Variable oral absorption of cyclosporine. A biopharmaceutical risk factor for chronic renal allograft rejection. Transplantation. 1996;62(5):599–606. doi: 10.1097/00007890-199609150-00010. [DOI] [PubMed] [Google Scholar]
- Kelly DA. Current issues in pediatric transplantation. Pediatr. Transplant. 2006;10(6):712–720. doi: 10.1111/j.1399-3046.2006.00567.x. [DOI] [PubMed] [Google Scholar]
- Spivey CA, Chisholm-Burns MA, Damadzadeh B, Billheimer D. Determining the effect of immunosuppressant adherence on graft failure risk among renal transplant recipients. Clin. Transplant. 2014;28(1):96–104. doi: 10.1111/ctr.12283. [DOI] [PubMed] [Google Scholar]
- Taylor PJ, Tai CH, Franklin ME, Pillans PI. The current role of liquid chromatography-tandem mass spectrometry in therapeutic drug monitoring of immunosuppressant and antiretroviral drugs. Clin. Biochem. 2011;44(1):14–20. doi: 10.1016/j.clinbiochem.2010.06.012. [DOI] [PubMed] [Google Scholar]
- Edelbroek PM, van der Heijden J, Stolk LM. Dried blood spot methods in therapeutic drug monitoring: methods, assays, and pitfalls. Ther. Drug Monit. 2009;31(3):327–336. doi: 10.1097/FTD.0b013e31819e91ce. [DOI] [PubMed] [Google Scholar]
- Meesters RJ, Hooff GP. State-of-the-art dried blood spot analysis: an overview of recent advances and future trends. Bioanalysis. 2013;5(17):2187–2208. doi: 10.4155/bio.13.175. [DOI] [PubMed] [Google Scholar]
- Pandya HC, Spooner N, Mulla H. Dried blood spots, pharmacokinetic studies and better medicines for children. Bioanalysis. 2011;3(7):779–786. doi: 10.4155/bio.11.19. [DOI] [PubMed] [Google Scholar]
- Koster RA, Alffenaar JW, Greijdanus B, Uges DR. Fast LC-MS/MS analysis of tacrolimus, sirolimus, everolimus and cyclosporin A in dried blood spots and the influence of the hematocrit and immunosuppressant concentration on recovery. Talanta. 2013;115(Oct 15):47–54. doi: 10.1016/j.talanta.2013.04.027. [DOI] [PubMed] [Google Scholar]
- Hinchliffe E, Adaway J, Fildes J, Rowan A, Keevil BG. Therapeutic drug monitoring of ciclosporin A and tacrolimus in heart lung transplant patients using dried blood spots. Ann Clin. Biochem. 2014;51(Pt 1):106–109. doi: 10.1177/0004563213488759. [DOI] [PubMed] [Google Scholar]
- Koop DR, Bleyle LA, Munar M, Cherala G, Al-Uzri A. Analysis of tacrolimus and creatinine from a single dried blood spot using liquid chromatography tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2013;926((May 1)):54–61. doi: 10.1016/j.jchromb.2013.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadilkova K, Busby B, Dickerson JA, Rutledge JC, Jack RM. Clinical validation and implementation of a multiplexed immunosuppressant assay in dried blood spots by LC-MS/MS. Clin. Chim. Acta. 2013;421((Jun 5)):152–156. doi: 10.1016/j.cca.2013.02.009. [DOI] [PubMed] [Google Scholar]
- Li Q, Cao D, Huang Y, Xu H, Yu C, Li Z. Development and validation of a sensitive LC-MS/MS method for determination of tacrolimus on dried blood spots. Biomed. Chromatogr. 2013;27(3):327–334. doi: 10.1002/bmc.2795. [DOI] [PubMed] [Google Scholar]
- Hinchliffe E, Adaway JE, Keevil BG. Simultaneous measurement of cyclosporin A and tacrolimus from dried blood spots by ultra-high performance liquid chromatography tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2012;883-884((Feb 1)):102–107. doi: 10.1016/j.jchromb.2011.05.016. [DOI] [PubMed] [Google Scholar]
- Webb NJ, Roberts D, Preziosi R, Keevil BG. Fingerprick blood samples can be used to accurately measure tacrolimus levels by tandem mass spectrometry) Pediatr. Transplant. 2005;9(6):729–733. doi: 10.1111/j.1399-3046.2005.00367.x. [DOI] [PubMed] [Google Scholar]
- Keevil BG, Fildes J, Baynes A, Yonan N. Liquid chromatography-mass spectrometry measurement of tacrolimus in finger-prick samples compared with venous whole blood samples. Ann. Clin. Biochem. 2009;46(Pt 2):144–145. doi: 10.1258/acb.2008.008147. [DOI] [PubMed] [Google Scholar]
- Yonan N, Martyszczuk R, Machaal A, Baynes A, Keevil BG. Monitoring of cyclosporine levels in transplant recipients using self-administered fingerprick sampling. Clin. Transpl. 2006;20(2):221–225. doi: 10.1111/j.1399-0012.2005.00472.x. [DOI] [PubMed] [Google Scholar]
- Keevil BG, et al. Simultaneous and rapid analysis of cyclosporin A and creatinine in finger prick blood samples using liquid chromatography tandem mass spectrometry and its application in C2 monitoring. Ther Drug Monit. 2002;24(6):757–767. doi: 10.1097/00007691-200212000-00013. [DOI] [PubMed] [Google Scholar]
- Hoogtanders K, et al. Dried blood spot measurement of tacrolimus is promising for patient monitoring. Transplantation. 2007;83(2):237–238. doi: 10.1097/01.tp.0000250730.30715.63. [DOI] [PubMed] [Google Scholar]
- Heijden J, et al. Therapeutic drug monitoring of everolimus using the dried blood spot method in combination with liquid chromatography-mass spectrometry. J. Pharm. Biomed. Anal. 2009;50(4):664–670. doi: 10.1016/j.jpba.2008.11.021. [DOI] [PubMed] [Google Scholar]
- Cheung CY, et al. Dried blood spot measurement: application in tacrolimus monitoring using limited sampling strategy and abbreviated AUC estimation. Transpl. Int. 2008;21(2):140–145. doi: 10.1111/j.1432-2277.2007.00584.x. [DOI] [PubMed] [Google Scholar]
- Hoogtanders K, et al. Therapeutic drug monitoring of tacrolimus with the dried blood spot method. J. Pharm. Biomed. Anal. 2007;44(3):658–664. doi: 10.1016/j.jpba.2006.11.023. [DOI] [PubMed] [Google Scholar]
- Wilhelm AJ, den Burger CJ, Vos RM, Chahbouni A, Sinjewel A. Analysis of cyclosporin A in dried blood spots using liquid chromatography tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009;877(14-15):1595–1598. doi: 10.1016/j.jchromb.2009.03.024. [DOI] [PubMed] [Google Scholar]
- Ostler MW, Porter JH, Buxton MO. Dried blood spot collection of health biomarkers to maximize participation in population studies. J. Vis. Exp. 2014. p. e50973. [DOI] [PMC free article] [PubMed]
- Hannon HW, et al. Blood collection on filter paper for neonatal screening programs, approved standard LA4-A5. Clinical Laboratory and Standards Institute; 2007. Available from: http://www.clsi.org. [Google Scholar]
- Schäfer P, Störtzel M, Vogt S, Weinmann W. Ion suppression effects in liquid chromatography-electrospray-ionisation transport-region collision induced dissociation mass spectrometry with different serum extraction methods for systematic toxicological analysis with mass spectra libraries. J. Chromatogr. B. 2002;773(1):47–52. doi: 10.1016/s1570-0232(02)00142-3. [DOI] [PubMed] [Google Scholar]
- Peck HR, Timko DM, Landmark JD, Stickle DF. A survey of apparent blood volumes and sample geometries among filter paper bloodspot samples submitted for lead screening. Clin. Chim. Acta. 2009;400(1-2):103–106. doi: 10.1016/j.cca.2008.10.020. [DOI] [PubMed] [Google Scholar]
- Christians U, et al. Automated, fast and sensitive quantification of drugs in blood by liquid chromatography-mass spectrometry with on-line extraction: immunosuppressants. J. Chromatogr. B. 2000;748(1):41–53. doi: 10.1016/s0378-4347(00)00380-7. [DOI] [PubMed] [Google Scholar]
- Clavijo C, et al. Development and validation of a semi-automated assay for the highly sensitive quantification of Biolimus A9 in human whole blood using high performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. B. Analyt. Technol. Biomed. Life Sci. 2009;877(29):3506–3514. doi: 10.1016/j.jchromb.2009.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei JV, Alexander JR, Adam BW, Hannon WH. Use of filter paper for the collection and analysis of human whole blood specimens. J. Nutr. 2001;131(5):S1631–S1636. doi: 10.1093/jn/131.5.1631S. [DOI] [PubMed] [Google Scholar]