

Abstract
Despite decades of eradication efforts, malaria remains a global burden. Recent renewed interest in regional elimination and global eradication has been accompanied by increased genomic information about Plasmodium parasite species responsible for malaria, including characteristics of geographical populations as well as variations associated with reduced susceptibility to anti-malarial drugs.
One common genetic variation, single-nucleotide polymorphisms (SNPs), offers attractive targets for parasite genotyping. These markers are useful not only for tracking drug resistance markers but also for tracking parasite populations using markers not under drug or other selective pressures.
SNP genotyping methods offer the ability to track drug resistance as well as to fingerprint individual parasites for population surveillance, particularly in response to malaria control efforts in regions nearing elimination status.
While informative SNPs have been identified that are agnostic to specific genotyping technologies, high-resolution melting (HRM) analysis is particularly suited to field-based studies. Compared to standard fluorescent-probe based methods that require individual SNPs in a single labeled probe and offer at best 10% sensitivity to detect SNPs in samples that contain multiple genomes (polygenomic), HRM offers 2-5% sensitivity. Modifications to HRM, such as blocked probes and asymmetric primer concentrations as well as optimization of amplification annealing temperatures to bias PCR towards amplification of the minor allele, further increase the sensitivity of HRM. While the sensitivity improvement depends on the specific assay, we have increased detection sensitivities to less than 1% of the minor allele.
In regions approaching malaria eradication, early detection of emerging or imported drug resistance is essential for prompt response. Similarly, the ability to detect polygenomic infections and differentiate imported parasite types from cryptic local reservoirs can inform control programs.
This manuscript describes modifications to high resolution melting technology that further increase its sensitivity to identify polygenomic infections in patient samples.
Keywords: Immunology, Issue 105, Plasmodium, malaria, eradication, resistance, high-resolution melting analysis, genotyping
Introduction
Despite renewed interest in malaria control and eradication, malaria remains a worldwide burden, with nearly half of the world’s population at risk of infection and more than 550,000 deaths annually, particularly children in sub-Saharan Africa1.
These new control and eradication programs have been supported by the genomic renaissance, with large numbers of malaria parasites sequenced and analyzed for mutations associated with reduced drug sensitivity, increased virulence, and for population characteristics2,3. Single-nucleotide polymorphisms (SNPs) are among the most commonly identified genetic variants 4-7.
Portable SNP genotyping methods offer on-site and real-time population surveillance and tracking8. In addition to fingerprinting individual parasites, the ‘molecular barcode’ is also used to detect dramatic temporal shifts in allele frequency as well as variance effective population size and complexity of infection9.
While this set of informative SNPs is easily adapted to many genotyping platforms, high resolution melting (HRM) analysis is particularly well-suited to field-based studies, where sensitive and simple operation and detection of novel mutations at low costs compared to sequencing and other approaches are attractive in resource-poor settings.
HRM starts with standard polymerase chain reaction (PCR) that incorporates a fluorescent dye. Post-PCR melting analysis determines the peak amplicon melting temperature; a single SNP difference in a short amplicon can result in substantial peak melting temperature (Tm) differences.
Several refinements to this method offer better genotyping resolution to differentiate SNPs including class IV (A-T) SNPs, and detect minor mutant alleles in samples with multiple alleles present (polygenomic infections). First, the assays incorporate short probes centered over the SNP region in addition to forward and reverse primers that are present at different concentrations. These blocked probes are not amplified during PCR, but bind to the strand produced in excess due to asymmetric primer concentrations that increase production of the probe-template product. These separate double-stranded amplicons consisting of probe bound to the excess template strand are ~20-30 bp; their significantly decreased length compared to the entire amplicon (80-150 bp) lead to much larger Tm differences associated with single or multiple base probe-template mismatches10.
Second, mutant allele amplification bias (MAAB) lowers the reaction annealing temperature to bias the reaction towards mutant alleles present at low ratios in polygenomic infections. The annealing temperature is set between the Tms of perfectly matched (wild-type) and mismatched (mutant) probes. At this temperature, the binding of the wild-type allele is stable enough that amplification is hindered compared to its mismatch equivalent, thus biasing the amplification towards the mutant allele when both are present in a single sample10.
With these HRM refinements, this technology has allowed tracking of origins and identity of parasites associated with epidemic infections in South America11 and detection of new mutations, differentiation of multiple SNPs located immediately next to each other in sequence, and mutations present in less than 1% of the alleles in polygenomic samples10.
Increased sensitivity is particularly important in regions approaching pre-elimination status and those at risk for emerging drug resistance. The ability to quickly and easily identify imported parasites and SNPs associated with reduced sensitivity on-site informs surveillance efforts and control programs about the effectiveness of their implementations and identifies hotspots for increased malaria eradication and elimination efforts. This protocol describes the methods for blocked-probe-based and MAAB for increased HRM genotyping sensitivity.
Protocol
Note: This protocol includes volumes and concentrations for 96-well plate, 8-tube strips, and capillary-based systems (10 µl reaction volume); however, HRM can also be performed in 384-well plate-based systems with a 5 µl reaction volume by scaling all volumes correspondingly. The range of detection for most systems is 10 pg to 10 ng template DNA.
1. Prepare Templates
Obtain Plasmodium falciparum samples directly from blood obtained from patients participating in field site studies and stored as whole blood, pelleted red blood cells, or on filter paper. Note: Samples may also be culture-adapted to grow in vitro; some standard laboratory strains for analysis can be ordered from the Malaria Research and Reference Reagent Resource Center (MR4, Samples may be used directly from pelleted red blood cells or culture or extracted from whole blood, red blood cells, or filter paper.
- DNA extracted from filter paper or culture-adapted strains Note: HRM is sensitive to salt concentration; concentration differences due to variable extraction methods can significantly affect melting temperatures. While specific final buffers are not a significant factor for success, use a standard common elution or resuspension buffer (i.e., water, 1x Te [‘low Te’ or ‘DNA suspension buffer’: 10 mM Tris-Cl, 0.10 mM EDTA] or 1x TE [10 mM Tris-Cl, 1 mM EDTA]) for all samples to avoid anomalous or inconsistent melt curves.
- Prepare template dilutions of 10 pg –10 ng per µl in standard Te, TE, or water for each 10 µl reaction.
- Direct amplification from pelleted red blood cells
- Determine parasitemia of red blood cells using thin-smear microscopy Note: Methods for determining parasitemia of infected red blood cells are available from the Centers for Disease Control and Prevention (CDC): http://www.cdc.gov/dpdx/diagnosticProcedures/blood/microexam.html
- Dilute parasitemias above 0.5% 1:400 in PCR-grade water, TE, or Te. Use 3 µl for each 5– 10 µl reaction.
- Controls Note: HRM can be used for gene scanning to detect sequence variants in a population12; however, accurate genotyping requires standards sequence-verified to contain the SNP being interrogated. Plasmid constructs or malaria isolates with known genotypes can be used. For most applications, 3D7 (MR4 MRA-151) is used as a wild-type control. Due to between-run variability, always include positive and negative controls.
- Positive controls: Prepare 10 pg – 10 ng per 1 µl dilutions of plasmid or standards with sequence-verified SNP genotypes.
- Negative control: Use PCR grade water as a no-template control (NTC) for the amplification reactions.
2. PCR Amplification
- Prepare reaction mixture
- (Optional) Mineral oil overlay. Note: Some HRM systems are standalone melting platforms and do not have a heated lid to prevent condensation and product evaporation during amplification.
- Add 20 µl light mineral oil to each well plate before loading the reaction mixture.
- Prepare 10x working stocks of all assays
- Refer to Table 1 for recommended 10x working stock concentrations as well as final concentrations for blocked-probe-based HRM genotyping.
- Prepare reaction mixtures
- To each well in plate or tube, add 1 µl 10x working stock forward and reverse primers and probes, 4.0 – 5.0 µl 2.5x or 2.0x HRM master mix, 1 µl template, and PCR grade water for a total reaction volume of 10 µl.
- Cover plates or tubes
- Use optical plate seals for plate-based amplification, optical caps for strip-tube-based systems, and plastic caps for capillary-based systems. Be sure coverage is complete and plates and caps are completely sealed and secured to prevent evaporation during amplification.
- Spin samples
- Spin plates 3 min at 1,800 x g to remove air bubbles and to separate oil and aqueous phases if mineral oil is used.
- Amplification protocol
- Place reaction plates or tubes in thermal cycler. Run standard blocked-probe or MAAB amplification protocols (Tables 2 and 3, respectively). Note the difference in annealing temperature between these methods.
3. HRM Analysis
- Melt
- After PCR amplification, proceed to melting analysis. For standalone melting systems that do not have built-in amplification (e.g., BioFire Defense LightScanner systems), remove amplified plate from thermal cycler and place in HRM instrument. From the system software interface, melt the plate from 40 – 80 °C to produce both probe and amplicon melting peaks. Note: To save melting and analysis time, the melting window can be shortened to cover temperature ranges for just probe or amplicon melting regions. After melting, plates and tubes can be re-melted if necessary and stored at -20 °C or 4 °C. Store glass tubes only at 4 °C to prevent tubes from cracking.
- Melt analysis
- Remove negative samples
- Within the analysis software, remove from further analysis samples that did not amplify or that have jagged melting peaks. Note: After selecting the mode of analysis for an individual run file, the instrument automatically groups the positive and negative samples. Before genotyping each subset, the user can manually alter the calls.
- From either the melting curve or melting peak view, right-click on the curves to be removed to select them. After selecting the misclassified samples, go to “New Call,” select the appropriate grouping, and click “Apply Change.”
- Normalize
- From the negative derivative of normalized fluorescence with respect to temperature (–dF/dT) (‘Difference Curves’) view, move normalization bars to surround the probe melt region. Note: The probe melt region is the lower temperature of the two melting regions. The normalization bars should be at the base of the melt peaks.
- Calculate groups
- After normalization, select ‘Calculate groups’ to automatically assign samples to groups. If necessary, manually re-assign samples to specific groups. Note: Some analysis software has high sensitivity thresholds that cannot be changed; in these cases, the software might assign samples to different groups that visually belong to another.
- Assign genotypes
- Assign genotypes for each group based on standards (positive controls).
- After the user confirms that the computed groups are correct (if not, that correct changes have been made under the Grouping tab by selecting misclassified samples and selecting the right group from the drop-box beside “New Call”), select “Edit Group Names” under the “Grouping” tab.
- Enter the genotypes of the standards into the corresponding colored box (for example, if standard 3d7 has a known A genotype and is in the red group, the red group has genotype A). Press “OK” and save the results.
- Export results
- Export genotype calls as a spreadsheet for further sample population analysis.
Representative Results
Data can be visualized in several different ways, depending on the analysis software and instrument. Typically, a plot of the negative derivative of normalized fluorescence with respect to temperature (–dF/dT) against temperature results is most straightforward for visualizing melting peaks and determining genotypes.
A full melting window (40°C–80°C) will result in both amplicon and probe melting regions. Figure 1 shows an example of normalized melting peaks for both regions. Setting the analysis window to just the probe region results in clear differentiation of peaks corresponding to specific SNPs (Figure 2).
Probe-based analysis clearly shows the presence of both alleles as individual peaks with melting temperatures that match homozygous SNP peaks (Figure 3).
Figure 4 shows that progressively reducing the annealing temperature during amplification (MAAB) results in a bias towards the mutant allele (left-side peak) (Figure 4A), resulting in increased sensitivity for the mutant allele in a population of mixed alleles (Figure 4B).
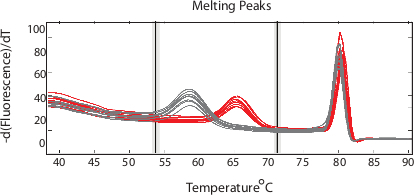
Figure 1. Probe and amplicon melting regions. Plotting the negative derivative of the normalized fluorescence with respect to temperature over a wide melting window results in both probe (lower temperature) and amplicon melting peaks that can be analyzed. (Adapted from Daniels et al. DOI: 10.1128/AAC.05737-11) Please click here to view a larger version of this figure.
 Figure 2. Probe melting peaks. Perfect matches (typically wild-type alleles) result in higher melting peaks (red), while SNP mismatches have lower melting temperatures (grey). Please click here to view a larger version of this figure.
Figure 2. Probe melting peaks. Perfect matches (typically wild-type alleles) result in higher melting peaks (red), while SNP mismatches have lower melting temperatures (grey). Please click here to view a larger version of this figure.
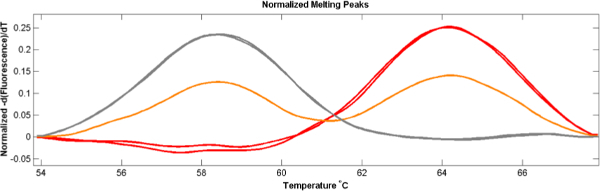 Figure 3. Polygenomic melting peaks. When both alleles are present in a sample, probe-based HRM analysis represents both alleles as two-peak curves (orange) with peaks that match single-allele samples (red and grey). (Adapted from Daniels et al. DOI: 10.1128/AAC.05737-11) Please click here to view a larger version of this figure.
Figure 3. Polygenomic melting peaks. When both alleles are present in a sample, probe-based HRM analysis represents both alleles as two-peak curves (orange) with peaks that match single-allele samples (red and grey). (Adapted from Daniels et al. DOI: 10.1128/AAC.05737-11) Please click here to view a larger version of this figure.
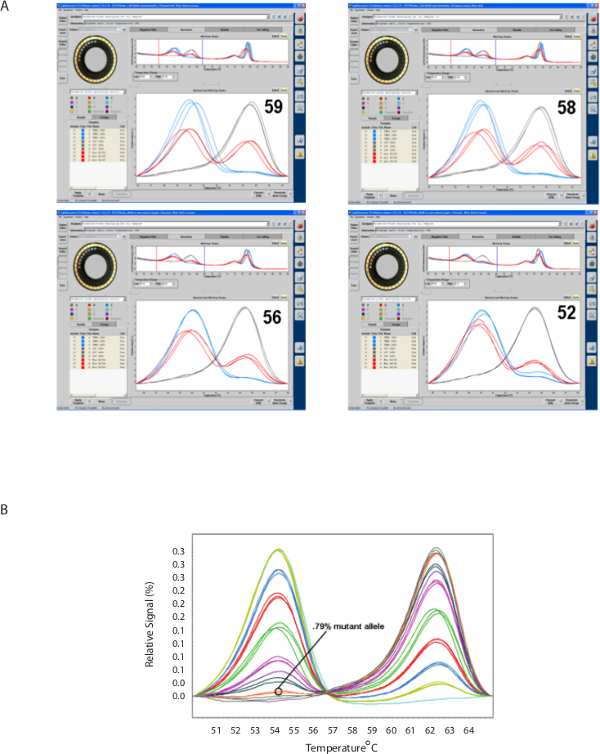 Figure 4. Mutant allele amplification bias (MAAB). A. Progressively lowering the amplification annealing temperature biases the melting peak reaction towards peak corresponding to the mutant (lower temperature) allele in a polygenomic or polyallelic sample. B. MAAB results in HRM sensitivity to detect minor alleles present at less than 1% in polygenomic or polyallelic samples. (Part B adapted from Daniels et al. DOI: 10.1128/AAC.05737-11) Please click here to view a larger version of this figure.
Figure 4. Mutant allele amplification bias (MAAB). A. Progressively lowering the amplification annealing temperature biases the melting peak reaction towards peak corresponding to the mutant (lower temperature) allele in a polygenomic or polyallelic sample. B. MAAB results in HRM sensitivity to detect minor alleles present at less than 1% in polygenomic or polyallelic samples. (Part B adapted from Daniels et al. DOI: 10.1128/AAC.05737-11) Please click here to view a larger version of this figure.
Table 1. Set-up of probe-based high resolution melting amplification reactions.
| Component | Volume per reaction (96-well plate and capillary tube) | Volume per reaction (384-well plate) | Final concentration |
| HRM Master Mix | 4–5 µl | 2-2.5 µl | 1 x |
| 10 x Excess primer | 1 µl | 0.5 µl | 0.50 µM |
| 10 x Other primer | 1 µl | 0.5µl | 0.1 µM |
| 10x Probe | 1 µl | 0.5 µl | 0.40 µM |
| PCR-grade water | 1–2 µl | 0.5-1 µl | |
| Template DNA | 1 µl | 0.5 µl | 0.01-10 ng/µl |
| 10 µl | 5 µl |
Table 2. Standard high resolution melting amplification conditions.
| Temperature | Time | |
| Hold | 95 °C | 120 sec |
| 45 cycles | 95 °C | 30 sec |
| 68 °C* | 30 sec | |
| 1 cycle | 95 °C | 30 sec |
| 28 °C | 30 sec |
* this temperature is amplicon-dependent
Table 3. Amplification conditions for mutant allele amplification bias.
| Temperature | Time | Cycles | Ramp rate | Acquisition mode | Analysis mode | |
| Denature | 95 °C | 30 sec | 1 | 20 | None | None |
| Cycle | 95 °C | 2 sec | 55 | 20 | None | Quantification |
| 56 °C* | 15 sec | 20 | Single | |||
| Melt | 40 °C | 0 sec | 0.3 | None | None | |
| 45 °C* | 0 sec | Continous | Melt | |||
| 90 °C* | 0 sec |
* this temperature is amplicon-dependent and will be reduced when performing MAAB
Discussion
Continous HRM is a post-PCR analysis step; therefore, sample and assay setup is similar to standard PCR protocols, with the additional incorporation of a fluorescent intercalating dye used to track the transition from double-stranded to single-stranded DNA during the high-resolution melting step. The single most important factor for successful HRM analysis is a robust PCR product. Assay design is key, and several tools optimized for HRM assay design are available commercially and online13. Amplicon lengths of 80-150 base pairs with accompanying ~20 base pair blocked probes centered over the SNP or SNPs of interest work best to differentiate SNPs as well as haplotypes of multiple SNPs within the probe region. Probes can be blocked using either a 3’ C3 spacer or a 2 base pair mismatch at the 3’ end, which prevent extension during amplification. Probes can be designed to match the Watson or Crick template strand; the choice depends on empirical testing to determine which probe design produces acceptable melting peaks. Excess forward or reverse primers are used to produce single-stranded amplification product that anneals to the blocked probes. If the probe contains the forward strand sequence, then reverse primer is used in excess, typically in a 1:5 ratio, and vice-versa for probes that match the reverse strand.
Similarly, HRM works best with sufficient and robust PCR product. PCR reaction optimization requires gradient PCR and agarose gels or Bioanalyzer analysis to determine optimal annealing temperatures. Template concentration can be increased using methods such as pre-amplification, biased multiplexed amplification of all targets using low primer concentration and cycle numbers to increase the template concentration13.
Only a handful of instruments are compatible with MAAB. The thermal properties of the glass capillaries tubes used in these systems facilitate this method. The small interior diameter of the capillaries as well as the rapid heat transfer through glass offer more stringent temperature control than standard PCR plasticware, which has slower transition rates between annealing and denaturation cycles due to the thermal insulating properties of the plastics. With this method, detection sensitivities can be reduced from 2-5% to less than 1% of a mutant allele in a mixture of wild-type and mutant alleles10.
HRM is a facile, efficient, and economical tool for SNP genotyping in a variety of settings and numerous applications, from infectious disease surveillance to scanning for cancer gene variants. Several other instruments, such as the Roche Nano and LightCycler systems (480 and 96) as well as the Eco Real-time PCR system offer HRM in addition to standard amplification, real-time, and copy-number functionality. Using higher-throughput machines, HRM barcoding costs less than $0.50 per assay in our hands, including all reagents and consumables.
In the context described here, field-based applications allow real-time population surveillance for changes associated with malaria control efforts, including reduced population diversity, detection of imported parasite types, and the appearance and spread of SNPs associated with reduced drug sensitivity. Refinements to the method offer additional sensitivity useful for informing progress towards malaria elimination and eradication.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors thank the Bill and Melinda Gates Foundation for its support of technology development and training.
References
- World Health Organization report 2013. 2013. pp. 1–286.
- Hayton K, Su X-Z. Drug resistance and genetic mapping in Plasmodium falciparum. Current genetics. 2008;54(5) doi: 10.1007/s00294-008-0214-x. [DOI] [PubMed] [Google Scholar]
- Neafsey DE, et al. Genome-wide SNP genotyping highlights the role of natural selection in Plasmodium falciparum population divergence. Genome biology. 2008;9(12):R171. doi: 10.1186/gb-2008-9-12-r171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkman SK, et al. A genome-wide map of diversity in Plasmodium falciparum. Nature. 2007;39(1):113–119. doi: 10.1038/ng1930. [DOI] [PubMed] [Google Scholar]
- Volkman SK, Neafsey DE, Schaffner SF, Park DJ, Wirth DF. Harnessing genomics and genome biology to understand malaria biology. Nature Reviews Genetics. 2012;13(5):315–328. doi: 10.1038/nrg3187. [DOI] [PubMed] [Google Scholar]
- Manske M, et al. Analysis of Plasmodium falciparum diversity in natural infections by deep sequencing. Nature. 2012;487(7407):375–379. doi: 10.1038/nature11174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auburn S, et al. Characterization of within-host Plasmodium falciparum diversity using next-generation sequence data. PLoS ONE. 2012;7(2):e32891. doi: 10.1371/journal.pone.0032891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels R, et al. A general SNP-based molecular barcode for Plasmodium falciparum identification and tracking. Malaria Journal. 2008;7:223. doi: 10.1186/1475-2875-7-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels R, et al. Genetic surveillance detects both clonal and epidemic transmission of malaria following enhanced intervention in Senegal. PLoS ONE. 2013;8(4):e60780. doi: 10.1371/journal.pone.0060780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels RR, et al. field-deployable method for genotyping and discovery of single-nucleotide polymorphisms associated with drug resistance in Plasmodium falciparum. Antimicrobial agents and chemotherapy. 2012;56(6):2976–2986. doi: 10.1128/AAC.05737-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olbadia N, et al. Clonal outbreak of Plasmodium falciparum in eastern Panama. The Journal of infectious diseases. 2014. [DOI] [PMC free article] [PubMed]
- Montgomery J, Wittwer CT, Palais R, Zhou L. Simultaneous mutation scanning and genotyping by high-resolution DNA melting analysis. Nature protocols. 2007;2(1):59–66. doi: 10.1038/nprot.2007.10. [DOI] [PubMed] [Google Scholar]
- Mharakurwa S, Daniels R, Scott A, Wirth DF, Thuma P, Volkman SK. Pre-amplification methods for tracking low-grade Plasmodium falciparum populations during scaled-up interventions in Southern Zambia. Malaria Journal. 2014;13:89. doi: 10.1186/1475-2875-13-89. [DOI] [PMC free article] [PubMed] [Google Scholar]