

Abstract
Bacterial attachment to host cells is one of the earliest events during bacterial colonization of host tissues and thus a key step during infection. The biochemical and functional characterization of adhesins mediating these initial bacteria-host interactions is often compromised by the presence of other bacterial factors, such as cell wall components or secreted molecules, which interfere with the analysis. This protocol describes the production and use of biomimetic materials, consisting of pure recombinant adhesins chemically coupled to commercially available, functionalized polystyrene beads, which have been used successfully to dissect the biochemical and functional interactions between individual bacterial adhesins and host cell receptors. Protocols for different coupling chemistries, allowing directional immobilization of recombinant adhesins on polymer scaffolds, and for assessment of the coupling efficiency of the resulting “bacteriomimetic” materials are also discussed. We further describe how these materials can be used as a tool to inhibit pathogen mediated cytotoxicity and discuss scope, limitations and further applications of this approach in studying bacterial - host interactions.
Keywords: Infection, Issue 105, Host-pathogen interaction, bacterial adhesion, host cell attachment, adhesin, biomimicry, bioengineering, chemical biology
Introduction
Dissecting the interactions between bacterial adhesins and host surface receptors at the host-pathogen interface is an essential step towards our understanding of the underlying mechanisms driving bacterial adhesion. Ultimately, this will help us to identify strategies to interfere with these processes during infections. In conducting such studies, we often face a dilemma: Biochemical and biophysical analysis of the molecular mechanisms of adhesin binding requires their separation from other cell wall components, which may interfere with adhesion events. On the other hand, the use of recombinant soluble proteins over-simplifies the adhesion event, by disregarding protein anchoring in the bacterial cell wall and multivalency of binding achieved through bacterial surface display. Equally, from the host cell’s perspective, encountering and binding to single adhesin molecules does not always have the same impact in terms of plasma membrane organization, membrane fluidity and receptor clustering1 and this, ultimately, makes it difficult to evaluate the impact of adhesion on host cellular signaling and the outcome of bacteria-host interactions.
Recently a method was devised, whereby recombinant purified adhesins or adhesin fragments are directionally and covalently coupled to polymer particles similar in size to bacteria, thus mimicking bacterial surface display. This approach has been used on a range of different adhesins, from Gram-negative Multivalent Adhesion Molecules (MAMs)2, 3, to Gram-positive adhesins including Staphylococcus aureus fibronectin binding protein (FnBPA)4, 5 and Streptococcus pyogenes F1 6, 7. This method has allowed the dissection of adhesin fragments important for host cell binding, identification of host surface receptors and determination of their behavior on the host cell surface, while taking both binding affinity and avidity into consideration 1, 8. Additionally, this approach has been used to investigate the efficacy of immobilized adhesins and derivatives as inhibitors of host-pathogen interactions 8, 9. It has been demonstrated that surface coupled derivatives of the Vibrio parahaemolyticus Multivalent Adhesion Molecule (MAM) 7 can be used to attenuate a range of bacterial infections in vitro, including those caused by multidrug-resistant pathogens such as Acinetobacter baumannii and methicillin-resistant S. aureus (MRSA) 7, 9.
Herein, we describe different chemistries which can be used to immobilize adhesins to commercially available functionalized polystyrene particles. Due to the wide array of available surface functionalities, labels and particle sizes, these are a useful scaffold for the production of bacteriomimetic materials to investigate adhesin-host interactions. We further describe methods for the initial characterization of the coupling reaction, and for the calculation of important properties of the resulting materials. Finally, the use of bead-coupled adhesins as competitive inhibitors of in vitro bacterial infections is discussed as an example of their application, as well as the future scope and limitations of this approach.
Protocol
1. Chemical Coupling of Proteins to Polymer Beads
- Thiol-amine directional coupling Note: This protocol is suitable for coupling of cysteine containing proteins to amine-functionalized polymer beads, using Sulfosuccinimidyl 4-(p-maleimidophenyl)butyrate (Sulfo-SMPB) as cross-linking agent (Figure 1).
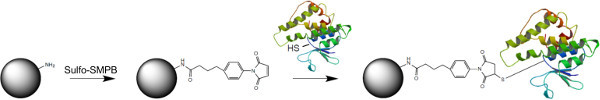 Figure 1. Cross-linking strategy used for directional thiol-amine coupling of proteins to polymer beads. Amine-modified polystyrene beads are activated with Sulfo-SMPB. The maleimide reacts with free cysteines to directionally couple proteins to beads. Please click here to view a larger version of this figure.
Figure 1. Cross-linking strategy used for directional thiol-amine coupling of proteins to polymer beads. Amine-modified polystyrene beads are activated with Sulfo-SMPB. The maleimide reacts with free cysteines to directionally couple proteins to beads. Please click here to view a larger version of this figure.
- Preparation of reagents:
- Prepare PBS (100 mM sodium phosphate, 150 mM NaCl, pH 7.0) and autoclave.
- Prepare a 100x stock (0.5 M or 287 mg/ml in PBS) of TCEP (tris(2-carboxyethyl)phosphine) immediately before use.
- Prepare a 5x stock (10 mM or 4.58 mg/ml in dH2O) of Sulfo-SMPB immediately prior to use.
- Prepare a 10x stock (500 mM or 88 mg/ml in PBS, pH 7.0) of cysteine immediately before use.
- Bead activation:
- Mix the bead suspension by gently inverting and transfer the required amount of bead suspension (e.g., 12 ml) into a sterile 1.5 ml tube containing 1 ml sterile PBS, pH 7.0.
- Gently pipette up and down to wash the beads and pellet by centrifugation in a microcentrifuge (2 min at 16,000 x g).
- Carefully remove the supernatant with a pipette and discard. Resuspend the bead pellet in 1 ml of fresh sterile PBS and repeat the washing step. Resuspend the bead pellet in 0.8 ml of PBS.
- Add 200 ml of freshly prepared 10 mM Sulfo-SMPB, to give a final concentration of 2 mM.
- Incubate the bead suspension for 1 hr at 25 °C on a rotating wheel.
- Protein reduction:
- During the incubation period of the activation step, prepare the protein for the following coupling step so it can be immediately added to the activated beads. Note: Although this reduction step is not always required for GST (Glutathione S-transferase) fusion proteins, it is recommended to ensure a high coupling efficiency.
- Check the protein concentration, and adjust it to the final concentration required for the coupling reaction. Note: 6 mM protein in PBS, and a volume of 1 ml are usually used.
- Add TCEP stock to give a final concentration of 5 mM. Incubate the solution for 30 min at RT. Note: The reaction mixture can be directly used for the following coupling reaction.
- Retain a small amount (a few ml) of the protein solution for determining the protein concentration and calculation of coupling efficiency (see section 2).
- Protein coupling step:
- Pellet the activated beads by centrifugation (2 min, 16,000 x g in a microcentrifuge), and wash the pellet once in 1 ml of fresh sterile PBS.
- Resuspend the pellet in the prepared protein solution (e.g., 1 ml), to give the desired protein concentration. Note: The protein concentration during the coupling step will depend on the average coupling efficiency (see section 2 for determination of the coupling efficiency) and desired coupling density (as calculated in 1.1.4.3). The efficiency is approximately 85% and the desired final concentration 5 mM in the 10x bead suspension, so protein concentration during the coupling step should be approximately 6 mM.
- Calculate the coupling density using the following formula:
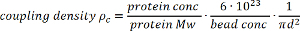 where ρc coupling density [number of protein molecules/nm2], protein conc protein concentration [mg/ml], protein Mw protein molecular weight [Da], bead conc bead concentration [number of beads/L], d bead diameter [nm2] Avogadro’s number
where ρc coupling density [number of protein molecules/nm2], protein conc protein concentration [mg/ml], protein Mw protein molecular weight [Da], bead conc bead concentration [number of beads/L], d bead diameter [nm2] Avogadro’s number - Use the coupling density to calculate the average ligand spacing:
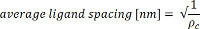
- Incubate the protein-bead suspension for 2 hr at 25 °C on a rotating wheel. Note: Some proteins may not be stable at RT. In these cases, the reaction can be carried out at 4 °C O/N.
- Deactivate remaining activated groups on the beads by adding cysteine stock to a final concentration of 50 mM and incubate the suspension for 30 min at 25 °C on a rotating wheel. Pellet the beads by centrifugation (2 min, 16,000 x g in a microcentrifuge).
- Keep the supernatant for determining the protein concentration and calculation of coupling efficiency (see section 2).
- Wash the bead pellet twice with 1 ml PBS and resuspend in 1 ml fresh PBS to give the final product. Note: The above protocol will typically give 1 ml of coupled protein, at a final concentration of 5 mM protein, which can be used as a 10x stock for subsequent experiments (see section 3).
- To proceed to section 3 of the protocol, work with 100 ml/ml of bead stock, or at a final concentration of 500 nM protein. Note: A good starting point for this procedure will be 2×1012 beads/ml, resulting in an average coupling density of 3×10-4 proteins/nm2 or an average spacing of 57 nm on a bead of 2 mm diameter.
- Thiol-carboxy directional coupling Note: This protocol is suitable for coupling cysteine containing proteins to carboxyl-functionalized polystyrene beads. The carboxyl moiety is first activated using 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC)/ N-hydroxysuccinimide (NHS), amine modified and then cross-linked using Sulfo-SMPB (Figure 2).
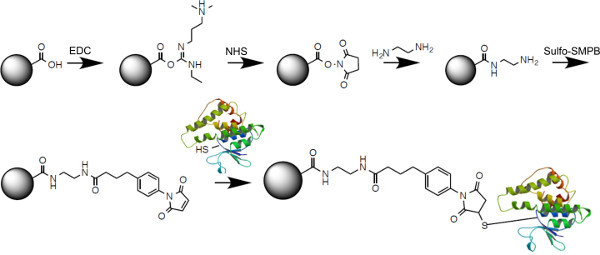 Figure 2. Cross-linking strategy used for directional thiol-carboxyl coupling of proteins to polymer beads. Carboxylated polystyrene beads are first activated with EDC and modified with NHS to form a semi-stable NHS-ester. Subsequent amine-coupling of ethylenediamine results in a free amine group, which reacts with Sulfo-SMPB. Maleimide activated beads can then react with free cysteines to directionally couple proteins to beads. Please click here to view a larger version of this figure.
Figure 2. Cross-linking strategy used for directional thiol-carboxyl coupling of proteins to polymer beads. Carboxylated polystyrene beads are first activated with EDC and modified with NHS to form a semi-stable NHS-ester. Subsequent amine-coupling of ethylenediamine results in a free amine group, which reacts with Sulfo-SMPB. Maleimide activated beads can then react with free cysteines to directionally couple proteins to beads. Please click here to view a larger version of this figure.
- Preparation of reagents:
- Prepare PBS (100 mM sodium phosphate, 150 mM NaCl, pH 7.0) and autoclave.
- Prepare a 100x stock (0.5 M or 287 mg/ml in PBS,) of TCEP immediately before use.
- Prepare a 10x stock (20 mM, or 4 mg/ml in PBS) of EDC (1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide) immediately before use.
- Prepare a 10x stock (50 mM, or 6 mg/ml in PBS) of NHS (N-hydroxysuccinimide) immediately prior to use.
- Prepare a 5x stock (10 mM or 4.58 mg/ml in dH2O) of Sulfo-SMPB immediately prior to use.
- Prepare a 10x stock (500 mM or 88 mg/ml in PBS, pH 7.0) of cysteine immediately before use.
- Bead activation:
- Mix bead suspension by gently inverting and transfer the required amount of bead suspension (e.g., 12 ml) into a sterile 1.5 ml tube containing 1 ml sterile PBS, pH 7.0.
- Gently pipette up and down to wash the beads and pellet by centrifugation in a microcentrifuge (2 min at 16,000 x g).
- Carefully remove the supernatant with a pipette and discard.
- Resuspend the bead pellet in 1 ml of fresh sterile PBS and repeat the washing step.
- Resuspend the bead pellet in 0.8 ml of PBS.
- Add 100 ml of 10x EDC stock (2 mM final concentration) and immediately 100 ml of 10x NHS stock solution (5 mM final concentration) to the bead suspension.
- Incubate the bead suspension for 30 min at 25 °C on a rotating wheel.
- Wash beads once in 1 ml of PBS and resuspend in 0.8 ml fresh sterile PBS.
- Add 200 ml ethylenediamine, and incubate the bead suspension for 1 hr at 25 °C on a rotating wheel.
- Wash beads once with 1 ml PBS and resuspend the beads in 0.8 ml of fresh PBS.
- Proceed from section 1.1.2.4 (bead activation with Sulfo-SMPB) as described under section 1.1.2 and follow the rest of the protocol described in section 1.1 (Thiol-amine directional coupling). Perform protein preparation and protein coupling steps in a manner identical to those described in section 1.1. Note: The typical coupling efficiency using this protocol is slightly lower (approx. 75%) compared to section 1.1, therefore adjust the initial protein concentration accordingly to achieve the same coupling density.
2. Determination of Coupling Efficiency
Note: To determine protein concentration, use Bradford Reagent 10 and colorimetric assays as follows:
Gently invert Bradford Reagent to ensure homogeneity of the reagent.
Using a 10 mg/ml BSA stock solution, prepare protein standards covering concentrations from 0.1 to 1.5 mg/ml BSA in buffer.
Add 250 ml of Bradford Reagent to wells of a 96-well plate. Prepare enough wells for all samples, protein standards and negative controls (buffer only).
Add 5 ml of protein sample (protein standards or buffer, for the negative control) to the reagent in the 96-well plate.
Incubate the plate on an orbital shaker at RT for 10 min.
Measure absorbance at 595 nm using a plate reader.
Generate a standard curve of BSA concentration versus A595 nm and use this to determine protein concentrations in initial and supernatant samples.
Calculate the concentration of coupled protein as follows:
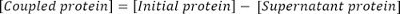
Calculate the coupling efficiency as:
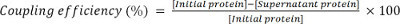
3. Use of Bead-coupled Adhesins in Competition Assays
- Preparation:
- Seed 1 ml per well of Hela cells at a concentration of 150,000 cells/ml into a 24-well plate the day before the competition assay, to allow cells to reach approximately 80% confluency prior to starting the experiment.
- Set up each experimental condition in triplicate. Include wells for negative controls (no bacteria added during competition assays), positive controls (control beads coupled to fusion-tag only added during competition assays) and lysis controls (for cytotoxicity experiments).
- Inoculate a 5 ml marine LB (MLB) culture with a fresh colony of V. parahaemolyticus and grow O/N at 30 °C, shaking.
- Prepare sufficient bead-coupled MAM, as described in section 1. (Coupling). Allow for 100 ml of 10x bead stock per well.
- Competition assay:
- On the day of the competition experiment, measure the OD600 of the bacterial culture.
- Prepare infection media by diluting bacterial cultures into colorless DMEM without additives, pre-warmed to 37 °C, containing 10% v/v bead suspension (either adhesin-coupled beads or control beads), to give an MOI of 10. Prepare 1 ml/well and 10-20% excess volume per sample. Note: For the above mentioned conditions (24-well plate, V. parahaemolyticus, MOI 10), the necessary volume of O/N culture to be added per well (ml/ml) is calculated as 3/OD600 of the culture. For example, 1 ml of infection medium will typically contain 100 ml bead suspension, a few ml of bacterial culture and be made up to 1 ml with colorless, unsupplemented DMEM.
- Remove old medium from wells and wash cultured Hela cells by adding 1 ml of sterile PBS pre-warmed to 37 °C to each well.
- Remove PBS and add 1 ml of infection medium per well. Also set up controls, by adding solutions containing control beads and bacteria (positive control) or adhesin beads and no bacteria (negative controls), or DMEM containing 0.1% Triton X-100 (lysis control, only necessary for cytotoxicity measurements).
- Incubate the plate in a tissue culture incubator at 37 °C for the desired amount of time (e.g. 4 hr for cytotoxicity measurements or 1 hr for adhesion measurements). Note: Both bacterial adhesion and cytotoxicity on host cells can be used as read-outs for the efficacy of inhibition. If the time points of cytotoxicity and adhesion measurements coincide, both assays may be performed using samples from the same well, since cytotoxicity is determined using the culture supernatant and attachment assays use samples derived from the remaining cell layer.
- Cytotoxicity measurements:
- At indicated time points (e.g., 4 hr post infection), remove three times 200 ml from each 24-well and transfer to a 96-well plate.
- Spin 96-well plates at 1,500 x g, 5 min and transfer 100 ml from each well into a fresh 96-well plate. Add 100 ml of the media used during infection experiments to fresh wells of the 96-well plate in triplicate (these will be used as blanks).
- Carry out the lactate dehydrogenase (LDH) release assay using a LDH cytotoxicity detection kit and following the manufacturer’s instructions.
- Briefly, calculate the amount of reagent needed in increments of 25 (e.g., if 62 samples are to be measured, make up enough reagent mix for 75 etc.).
- For example, for 100 samples, mix 11.25 ml of reagent A with 250 µl of reagent B. Invert, do not vortex to avoid foaming.
- Put the mixture in a reservoir to be able to pipet with a multi-channel pipette. Add 100 µl of reagent mix to each sample.
- Incubate plate at RT and read the absorbance at 490 nm on a plate reader at 10, 20, 30 min.
- Analyze the data set for which the absorbance of the lysis control sample is high but still within the linear range of the plate reader (typically, 2-3 absorbance units).
- Express results as % cytotoxicity, using the following formula for conversion:
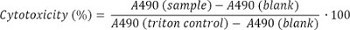
- Measurement of bacterial adhesion:
- At indicated time points (e.g., 1 hr post infection), remove media from the cell layer.
- Thoroughly wash the cell layer with sterile, pre-warmed PBS (at least 3-4 washes of 1 ml PBS each) to remove any un-attached cells.
- Lyse host cells by adding 1 ml of a sterile 1 % v/v Triton X-100 solution in PBS per well. Incubate the plate at 37 °C for 5 min.
- Pipette each sample up and down several times before transferring the contents of each well to separate 1.5 ml tubes. Prepare 10-fold serial dilutions of samples into sterile PBS (e.g., use 100 ml of sample and 900 ml of PBS).
- Plate 100 ml of each sample on MLB agar and spread using a cell spreader. Optimize which dilutions to plate depending on the bacterial strains and time point. Note: For the described experimental setup, 105 or 106 fold dilutions give a suitable number of CFUs.
- Incubate plates at 37 °C O/N and enumerate bacteria by colony counting.
Representative Results
The V. parahaemolyticus adhesin MAM7 contains seven tandem mammalian cell entry (MCE) domains involved in recognition of host surface receptors. We used polystyrene beads coupled to recombinant, purified fragments encompassing either all seven tandem MCE domains (MAM7) or only the first MCE domain (MAM1) to test the ability of these materials to compete with V. parahaemolyticus for host cell binding and the resulting efficacy of these materials as adhesion inhibitors. Hela cells were infected with V. parahaemolyticus strain POR1, and cytotoxicity resulting from in vitro infection was evaluated after 4 hr (Figure 3). Treatment of Hela cells with 0.1% Triton X-100 (positive lysis control) resulted in complete cell lysis, uninfected cells displayed very low levels of cytotoxicity. In vitro infection of untreated cells with POR1 resulted in very high levels of cell lysis, and this was inhibited by MAM7-coupled beads but not MAM1-coupled or GST-coupled control beads (Figure 3).
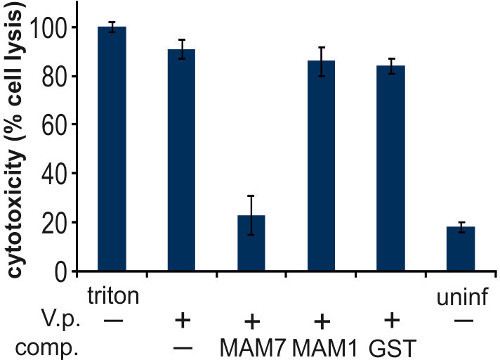 Figure 3. Characterization of Vibrio parahaemolyticus induced cytotoxicity in epithelial cells during competition assays. Cells were treated with V. parahaemolyticus (V.p), indicated with (+), or no bacteria (-) in the presence of different competing entities (comp.) as indicated. These included either no comp. (-), MAM7 beads (MAM7), MAM1 beads (MAM1) or GST control beads (GST). As controls, cells were treated either with Triton X-100 (triton, lysis control), or left uninfected (uninf). LDH release after 4 hr was measured as described in section 3, and results normalized to Triton controls (100 %) and blanks (0 %) as described above. Results are means ± s.e.m from triplicate experiments. Please click here to view a larger version of this figure.
Figure 3. Characterization of Vibrio parahaemolyticus induced cytotoxicity in epithelial cells during competition assays. Cells were treated with V. parahaemolyticus (V.p), indicated with (+), or no bacteria (-) in the presence of different competing entities (comp.) as indicated. These included either no comp. (-), MAM7 beads (MAM7), MAM1 beads (MAM1) or GST control beads (GST). As controls, cells were treated either with Triton X-100 (triton, lysis control), or left uninfected (uninf). LDH release after 4 hr was measured as described in section 3, and results normalized to Triton controls (100 %) and blanks (0 %) as described above. Results are means ± s.e.m from triplicate experiments. Please click here to view a larger version of this figure.
Enumeration of V. parahaemolyticus attached to either untreated Hela cells, or cells incubated with MAM7-, MAM1-, or GST control beads, revealed that MAM7-beads but not MAM1- or GST- control beads outcompete V. parahaemolyticus for attachment to host cell surface receptors (Figure 4).
 Figure 4. Characterization of bacterial attachment during competition experiments. Hela cells were infected with V. parahaemolyticus POR2 (V.p. +), in the absence (-) or presence of competing entities (comp.) as follows: MAM7 beads (MAM7), MAM1 beads (MAM1) or GST control beads (GST). Bacterial adhesion was measured after 1 hr, as described in section 3. Results are means ± s.e.m from triplicate experiments. Means (f.l.t.r. in CFU/ml) are 2.30106, 1.53105, 2.05106, 2.12106. Please click here to view a larger version of this figure.
Figure 4. Characterization of bacterial attachment during competition experiments. Hela cells were infected with V. parahaemolyticus POR2 (V.p. +), in the absence (-) or presence of competing entities (comp.) as follows: MAM7 beads (MAM7), MAM1 beads (MAM1) or GST control beads (GST). Bacterial adhesion was measured after 1 hr, as described in section 3. Results are means ± s.e.m from triplicate experiments. Means (f.l.t.r. in CFU/ml) are 2.30106, 1.53105, 2.05106, 2.12106. Please click here to view a larger version of this figure.
Adhesins coupled to fluorophore labeled beads result in the generation of materials that mimic bacterial adhesion to host cells and are powerful tools for cellular imaging (Figure 5). Using MAM7-coupled fluorescent blue beads, we characterized the process of MAM7-mediated attachment to epithelial cells. Attachment of MAM7 to host cells resulted in actin rearrangements and formation of stress fibers, which were co-visualized using rhodamine-phalloidin to stain for F-actin (Figure 5B).
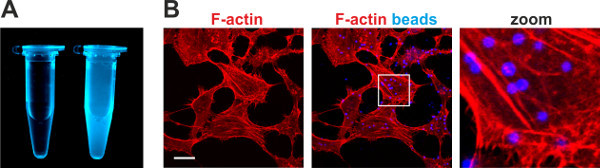 Figure 5. Preparation and use of fluorophore labeled biomimetic beads for imaging purposes.(A) Suspension of fluorescent blue biomimetic beads (right tube, 10x stock) and buffer control (left tube). (B) Attachment of MAM7-coupled beads to Hela cells results in actin rearrangements and stress fiber formation. Attachment of fluorescent blue beads and resulting actin stress fibers (red, stained with rhodamine-phalloidin) were imaged by microscopy. Bar, 10 μm. Images in panel B were adapted from Lim et al.1 and reproduced under the Creative Commons Attribution license. Please click here to view a larger version of this figure.
Figure 5. Preparation and use of fluorophore labeled biomimetic beads for imaging purposes.(A) Suspension of fluorescent blue biomimetic beads (right tube, 10x stock) and buffer control (left tube). (B) Attachment of MAM7-coupled beads to Hela cells results in actin rearrangements and stress fiber formation. Attachment of fluorescent blue beads and resulting actin stress fibers (red, stained with rhodamine-phalloidin) were imaged by microscopy. Bar, 10 μm. Images in panel B were adapted from Lim et al.1 and reproduced under the Creative Commons Attribution license. Please click here to view a larger version of this figure.
Discussion
Herein, we describe two protocols, which can be used to couple thiol-containing proteins to amine- or carboxylate-modified polystyrene beads, respectively. Due to ease of the procedure, thiol-amine coupling is preferable, but depending on the desired bead specification (diameter, fluorescence properties), use of amine-functionalized beads may not be possible and we have therefore included a protocol which will convert the carboxylate- into an amine moiety to give the researcher the greatest possible flexibility in choice of scaffold. Although both thiol-amine and thiol-carboxyl coupling work for any cysteine containing protein, directional coupling (i.e., immobilization of the protein per its N-terminus, mimicking surface display) requires a protein without cysteine residues, that is produced as a GST fusion protein, or that contains a single terminal cysteine residue introduced by site directed mutagenesis. If multiple reactive cysteines are contained within the protein, this will lead to random immobilization which may impede protein function. Many bacterial adhesins do not contain cysteines naturally. For others, these may be removed by site directed mutagenesis, although this would require extensive assays to ensure native structure and function are retained in the mutant. For GST fusion proteins, purified GST-tag coupled to beads can be used as a suitable negative control. Using uncoupled beads as a control should be avoided, as these often have a higher tendency to clump together or adhere to cells non-specifically. A simplified version of protocol 1.2., using only EDC, can be used to couple proteins to carboxyl-functionalized polymer beads, however in this case coupling takes place via primary amines in the protein and therefore does not guarantee directional coupling.
TCEP as a reducing agent must not be replaced with other commercially available and commonly used reducing agents, such as dithiothreitol (DTT) or 2-mercaptoethanol (BME), as the thiols contained within them will compete with protein coupling in the thiol-maleimide coupling step. PBS may be replaced with other buffers, but with the following considerations: Buffers may not contain primary amines (so Tris-containing buffers are not suitable). Use of buffers containing very low (< 10mM) salt concentrations leads to bead clumping and should also be avoided. Protein purity is also an important factor to consider during this procedure, and to achieve high quality data, pure proteins should be used. We routinely purify proteins in multiple steps, including at least an affinity purification and gel filtration step, but in some cases ion exchange chromatography is done as a third step. As a result the purity of proteins used for coupling is usually 90% or higher, as judged by SDS-PAGE.
It is recommended to determine protein concentrations both in the initial reaction mixture as well as of the supernatant after reaction completion. This will help to determine the apparent concentration of bead-coupled protein, and thus coupling density. Determination of both values will also allow calculation of the coupling efficiency. This can be taken into account when preparing the initial protein solution in subsequent reactions, to achieve the desired final concentration and coupling density. Bradford reagent is particularly suited for determination of protein concentration before and after the coupling reaction, as none of the substances in the reaction interferes with the dye complex formation at the concentrations used. If lower protein concentrations are to be used, this method may have to be replaced by a more sensitive detection method, however attention has to be paid to the fact it has to be compatible with the substances contained within the coupling reaction. It is also recommended to use freshly prepared reagents and handle powdered reagents with care (e.g., store in a sealed container and use silica beads, to avoid the reagents drawing moisture) since the quality of reagents will influence the coupling efficiency. If the coupling efficiency is lower than expected, possible remedies include increasing the initial bead and protein concentrations. If higher concentrations are being used, the concentration of coupling reagents has to be increased proportionally to ensure sufficient molar excess. Modifying bead/protein concentrations is usually a better step towards optimization rather than increasing reaction times. Since the protocols for bead coupling are lengthy, we commonly prepare a large batch of material. Aliquots of the suspension can be snap-frozen in liquid nitrogen and stored at -20 °C for several months. Thawed aliquots should not be refrozen and should be kept at 4 °C and used within 1-2 days. However, this will vary with the nature and stability of the protein used as should be tested on a small batch initially.
Bead-coupled adhesins can be used for many applications, as discussed below. This protocol described an assay that is commonly used to measure inhibition of bacterial binding and pathogen-mediated cytotoxicity on host cells. The assay is commonly performed to measure the capacity of bead-coupled MAMs to competitively inhibit infection of Hela epithelial cells with the sea-food borne pathogen Vibrio parahaemolyticus, using either a decrease in bacterial attachment to host cells or reduced cytotoxicity as a read-out. In both cases, preparation and competition assays follow the same protocol. Depending on the readout, different strains of V. parahaemolyticus are being used: the cytotoxic strain POR1 is used for cytotoxicity measurements, while the non-cytotoxic strain POR2 is used for measuring bacterial adhesion, since cell death and cell detachment compromises the procedure for quantifying attached bacteria.
Initially, competition experiments were set up as a step-wise protocol, where host cells were first pre-incubated with beads prior to the addition of bacteria. For V. parahaemolyticus and the bead specifications used (2 μm beads coupled to MAM7), both beads and bacteria can be added at the same time without changes in the resulting cytotoxicity. I.e., in this experimental setup, beads outcompete bacteria for host cell binding. Depending on the bacterial species and bead geometry used, there may be good reasons for maintaining the bead adhesion and bacterial infection as two separate steps. For example, to infect cells with non-motile bacteria, plates are commonly centrifuged after addition of the infection media. However, centrifugation of plates containing bead suspensions should be avoided, since this leads to a highly uneven distribution of beads on the cell layer. If smaller particle sizes are being used, beads will take longer to settle on the cell surface, in which case sufficient time should be allowed for bead attachment prior to the infection. When bacterial adhesion is used as a read out, samples should be taken at time points where host cells are not significantly damaged by the infecting strain, as cell detachment and lysis can compromise the quantification of attached bacteria.
Instead of enumerating bacterial adhesion by dilution plating, samples may alternatively be processed for imaging (Figure 5). In this case, tissue culture cells should be seeded onto glass cover slips, rather than directly into wells. Additionally, fluorescent beads and bacteria expressing a fluorescent protein may be used, along with infection-specific host cell markers. For example, competition experiments are commonly imaged using fluorescent red rhodamine-phalloidin to stain the host cells’ actin cytoskeleton and assess morphological changes resulting from infection, together with fluorescent blue beads and fluorescent green (GFP expressing or SYTO18 stained) bacteria.
A range of bead-coupled adhesins, including Staphylococcus aureus FnBPA, Streptococcus pyogenes F1 FUD and Vibrio parahaemolyticus MAM, have been used as biomimetic materials to study adhesion, adhesion inhibition and the contribution of adhesion to pathogen-mediated cytotoxicity 1, 7, 8. One of the advantages of using this approach is the ease of visualization of attachment events, since the polymer beads used as scaffolds are available in a wide range of colors (e.g., blue, fluorescent red, blue, green, orange). Thus, direct protein labeling, which may interfere with function, can be avoided. Additionally, surface coupling mimics the multivalent display of adhesins on the bacterial surface, thus reflecting a more physiologically relevant conformation compared to soluble proteins.
Compared to studies using intact bacteria or bacterial mutants, the bead approach circumvents problems associated with bacterial growth. For example, longer-term (e.g., O/N) studies of bacterial adhesion to host cells using intact bacteria are often compromised by phenomena accompanying bacterial growth – acidification of the growth medium and nutrient depletion negatively affect host cells, and bacterial replication eventually compromises imaging quality.
More recently, the use of bead-coupled adhesins has been extended to include their use as tools for affinity purifications of host cellular factors involved in signaling processes downstream of bacterial attachment. V. parahaemolyticus MAM7, via binding to phosphatidic acids in the host cell membrane, triggers RhoA activation and actin rearrangements 1, 3. MAM-coupled beads are being used to purify and identify proteins involved in the signaling platforms assembled as a consequence of MAM-host cell binding. Since beads can easily be separated from the supernatant by a short centrifugation step, and the protein of interest is covalently coupled, this is a good method to achieve separation from contaminant proteins and enrich relevant protein complexes, which can be used for downstream applications such as proteomics or Western Blotting.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors would like to thank members of the Krachler group for critical reading of the manuscript. DHS, DV and AMK were funded by the Biotechnology and Biological Sciences Research Council (BB/L007916/1), FA was funded by a Republic of Iraq Ministry of Higher Education and Scientific Research Scholarship and NPS was funded by a CONICYT Scholarship.
References
- Lim J, Stones DH, Hawley CA, Watson CA, Krachler AM. Multivalent Adhesion Molecule 7 clusters act as signaling platform for host cellular GTPase activation and facilitate epithelial barrier dysfunction. PLoS Pathog. 2014;10(9):e1004421. doi: 10.1371/journal.ppat.1004421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krachler AM, Ham H, Orth K. Outer membrane adhesion factor multivalent adhesion molecule 7 initiates host cell binding during infection by gram-negative pathogens. Proc Natl Acad Sci USA. 2011;108(28):11614–11619. doi: 10.1073/pnas.1102360108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krachler AM, Orth K. Functional characterization of the interaction between bacterial adhesin multivalent adhesion molecule 7 (MAM7) protein and its host cell ligands. J Biol Chem. 2011;286(45):38939–38947. doi: 10.1074/jbc.M111.291377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joh D, Speziale P, Gurusiddappa S, Manor J, Hook M. Multiple specificities of the staphylococcal and streptococcal fibronectin-binding microbial surface components recognizing adhesive matrix molecules. Eur J Biochem. 1998;258(2):897–905. doi: 10.1046/j.1432-1327.1998.2580897.x. [DOI] [PubMed] [Google Scholar]
- Bingham RJ, et al. Crystal structures of fibronectin-binding sites from Staphylococcus aureus FnBPA in complex with fibronectin domains. Proc Natl Acad Sci U.S.A. 2008;105(34):12254–12258. doi: 10.1073/pnas.0803556105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ensenberger MG, et al. Specific interactions between F1 adhesin of Streptococcus pyogenes and N-terminal modules of fibronectin. J Biol Chem. 2001;276(38):35606–35613. doi: 10.1074/jbc.M105417200. [DOI] [PubMed] [Google Scholar]
- Hawley CA, Watson CA, Orth K, Krachler AM. A MAM7 peptide-based inhibitor of Staphylococcus aureus adhesion does not interfere with in vitro host cell function. PLoS One. 2013;8(11):e81216. doi: 10.1371/journal.pone.0081216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krachler AM, Ham H, Orth K. Turnabout is fair play: use of the bacterial Multivalent Adhesion Molecule 7 as an antimicrobial agent. Virulence. 2012;3(1):68–71. doi: 10.4161/viru.3.1.18172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krachler AM, Mende K, Murray C, Orth K. In vitro characterization of multivalent adhesion molecule 7-based inhibition of multidrug-resistant bacteria isolated from wounded military personnel. Virulence. 2012;3(4):389–399. doi: 10.4161/viru.20816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]