

Abstract
Specific metabolic pathways are increasingly being recognized as critical hallmarks of macrophage subsets. While LPS-induced classically activated M1 or M(LPS) macrophages are pro-inflammatory, IL-4 induces alternative macrophage activation and these so-called M2 or M(IL-4) support resolution of inflammation and wound healing. Recent evidence shows the crucial role of metabolic reprogramming in the regulation of M1 and M2 macrophage polarization.
In this manuscript, an extracellular flux analyzer is applied to assess the metabolic characteristics of naive, M1 and M2 polarized mouse bone marrow-derived macrophages. This instrument uses pH and oxygen sensors to measure the extracellular acidification rate (ECAR) and oxygen consumption rate (OCR), which can be related to glycolytic and mitochondrial oxidative metabolism. As such, both glycolysis and mitochondrial oxidative metabolism can be measured in real-time in one single assay.
Using this technique, we demonstrate here that inflammatory M1 macrophages display enhanced glycolytic metabolism and reduced mitochondrial activity. Conversely, anti-inflammatory M2 macrophages show high mitochondrial oxidative phosphorylation (OXPHOS) and are characterized by an enhanced spare respiratory capacity (SRC).
The presented functional assay serves as a framework to investigate how particular cytokines, pharmacological compounds, gene knock outs or other interventions affect the macrophage’s metabolic phenotype and inflammatory status.
Keywords: Immunology, Issue 105, M1 M2 macrophage activation/polarization, Seahorse, Extracellular flux analysis, immunometabolism, Glycolysis, Oxidative phosphorylation (OXPHOS), Mitochondrial (dys)function, Inflammation, Macrophage activation, Innate Immunity, Macrophage function, interleukin-4 (IL-4), cytokine
Introduction
While macrophages play a central role in virtually all diseases, the molecular mechanisms that regulate their phenotype are still not fully unraveled. Macrophages display high heterogeneity and in response to the microenvironment adopt different phenotypes1. LPS(+IFNγ)-induced classically activated M1 or M(LPS) macrophages promote inflammation and protect the host against different types of microbial threats2. Others use IFNγ alone or in combination with TNF as stimuli to elicit M1 polarization. IL-4 and/or IL-13 induces alternative macrophage activation and these M2 or M(IL-4) cells are potent suppressors and controllers of ongoing immune responses3. Polarized macrophages display a distinct regulation of cellular metabolism with LPS-activated M1 macrophages undergoing a metabolic switch to enhanced glycolysis4-6. Conversely, enhanced fatty acid oxidation (FAO) and mitochondrial oxidative phosphorylation (OXPHOS) provides sustained energy in IL-4-induced M2 macrophages7-9. Thus, altered metabolism is not only a characteristic of polarized macrophage subsets, it is also a prerequisite for proper polarization and inflammatory regulation. Importantly, the inhibition of glycolysis or OXPHOS/FAO has been demonstrated to impair M1 or M2 activation, respectively8,10. As such, identifying metabolic changes in macrophages could be applied as a tool to assess their polarization status and inflammatory potential.
A consistent assay that measures macrophage metabolism could therefore be used to predict whether a drug, gene knock down or other treatment affects macrophage polarization and function. In this video, an extracellular flux analyzer is used to characterize the bioenergetics profiles of naïve, M1 and M2 macrophages.
This manuscript details an optimized protocol that allows the measurement of all relevant glycolysis parameters (glycolysis, maximal glycolysis and glycolytic reserve) and mitochondrial function characteristics (total respiration, basal mitochondrial respiration, ATP production, proton leak, maximal respiration and spare respiratory capacity) in one single assay. Using this experimental setup, bioenergetics can be compared between control and ‘altered’ (e.g. gene knock down, transgenic overexpression or pharmacological treatment) cells.
Protocol
1. Culturing and Polarizing Bone Marrow-derived Macrophages
Day 0: Isolate femur and tibia bones from 6-10 week old mice of interest (C57Bl/6 in this assay).
Remove muscle tissue from the bones and place the bones in a 9 cm petri-dish filled with ice-cold PBS.
Transfer the bones to a 9 cm Petri dish filled with 70% ethanol and gently shake the plate for 30 sec.
Transfer the bones to a fresh 9 cm Petri dish filled with ice-cold sterile PBS.
Cut the femur and tibia at both ends with sterile scissors.
Flush the bones with 10 ml ice-cold sterile PBS using a 10 ml syringe and a 25 G needle. Collect the bone marrow in a 50 ml tube.
Transfer the bone marrow cells to a new 50 ml tube using a 23 G needle.
Repeat this step with a 25 G needle. Note: These steps are needed to remove cell clumps and bone residues.
Centrifuge at 300 x g for 5 min at 4 °C.
Discard the supernatant and resuspend the cells in 40 ml cell culture medium (RPMI-1640 plus 2 mM L-glutamine, 10% FCS, penicillin (100 U/ml), streptomycin (100 µg/ml) and 15% filtered L-929 cell (ATCC,CCL-1)-conditioned medium containing M-CSF (or alternatively 10 ng/ml recombinant M-CSF instead of L-929 cell-conditioned medium). Note: The generation of L-929 cell-conditioned medium was detailed earlier in this journal in 2013.11
Culture the cells from one mouse in two 15 cm Petri dishes (do not use cell culture-treated plates since macrophage will not detach from this plastic) in 20 ml culture medium per plate in a 37 °C, 5% CO2 incubator.
Add 10 ml fresh cell culture medium on day 3, without removing the 20 ml older cell culture medium.
Replace the old medium of each plate with 20 ml fresh culture medium on day 6. Doing this, non-adherent cells will also be removed.
- Day 8:
- Detach cells by incubating them 5 min at 37 °C in 10 ml citrate saline (135 mM potassium chloride plus 15 mM sodium citrate in H2O, autoclaved) and wash with 10 ml PBS. Count mature bone marrow-derived macrophages (BMDM) on day 8 using a cell counter.
- Verify the formation of mature (CD11b+ F4/80+) BMDM by visual inspection or by flow cytometry. Block 105 cells with 1/100 anti-CD16/CD32 in PBS for 20 min in 100 µl PBS, incubate with 1/200 anti-CD11b-FITC plus 1/200 anti-F4/80-APC-Cy7 at RT, wash and analyze by flow cytometry. Note: Culturing mouse bone marrow-derived macrophages has been published in detail earlier in this journal by Ying et al.11.
Seed 50,000 cells (optimal cell counts need to be optimized) per well in a cell culture microplate in 100 µl culture medium. Do not seed cells in background correction wells (A1, A12, H1, H12) and put medium only (no cells) in these wells. Tip: Hold the pipette at an angle about halfway down the side of the wells for most homogeneous cell layer.
Allow the cells to settle at RT in a cell culture hood for 1 hr. This will promote even cell distribution and reduce edge effects. Monitor adherence using a microscope and culture in a 37 °C, 5% CO2 incubator.
Three hr after plating, stimulate cells for 24 hr with 10 ng/ml LPS, or 20 ng/ml recombinant mouse IL-4 to generate M1 or M2 macrophages, respectively. Include untreated naïve (M0) macrophages as a control. Assess proper M1 and M2 macrophage polarization by measuring LPS-induced secretion of IL-6, IL-12, TNF (ELISA) and IL-4-induced arginase-1 activity and/or MMR (CD206) and MGL (CD301) surface expression (flow cytometry) as described earlier3,11. Note: At this point, cells can be (pre)-treated with pharmacological compounds of interest or macrophages can be stimulated with other factors. Since, the variation between separate wells might be substantial, it is advisable to use at least 4 and ideally 6 wells per condition.
2. Preparation of the Extracellular Flux Assay
- Hydrate the sensor cartridge one day before running the assay:
- Place the sensor cartridge upside down next to the utility plate.
- Fill each well of the utility plate with 200 µl of calibrant solution and lower the sensor cartridge onto the utility plate submerging the sensors in the calibrant solution.
- Verify the calibrant solution level is high enough to keep the sensors submerged and place in a non-CO2 37 °C incubator O/N.
- Prepare assay medium on the day of the assay as follows:
- Warm 50 ml XF base medium to 37 °C.
- Add 500 µl 200 mM L-glutamine to get a 2 mM final concentration.
- Adjust pH to 7.4 using 1 N NaOH and sterilize with a 0.2 µM filter and keep the assay medium at 37 °C.
Gently remove the cell culture medium from the polarized macrophages and store it at -20 °C for future use (e.g. ELISA to examine proper macrophage polarization). Wash the cells with 100 µl assay medium, add 180 µl assay medium per well and verify under the microscope that cells were not washed away. Place the cell culture plate in a 37 °C incubator without CO2 for 1 hr prior to the assay run.
- Prepare 10 x injection compound mixtures A till D as described in Table 1, warm to 37 °C, adjust the pH to 7.4 and filter sterilize.
- Make all compounds in these mixtures at 10x the final concentration in the cell culture wells and optimize these concentrations for each cell type.
Load the sensor cartridge using the provided loading guides with indicated volumes (Table 1) of the prepared 10x injection compound mixtures in ports A, B, C and D, respectively.
| Mixture/Injection | Compounds | Volume added to get 10x mixture (µl) | Volume Assay Medium (ml) | Volume injected during run (µl) | Final concentration in the assay |
| A | 2.5 M (45%) glucose | 300 | 2.7 | 20 | 25 mM |
| B | 5 mM oligomycin A (OM) | 9 | 3.0 | 22 | 1.5 µM |
| C | 5 mM FCCP | 9 | 2.7 | 25 | 1.5 µM |
| 100 mM Sodium Pyruvate solution | 300 | 1 mM | |||
| D | 5 mM antimycin A (AA) | 15 | 3.0 | 28 | 2.5 µM |
| 5 mM rotenone (Rot) | 7.5 | 1.25 µM |
Table 1. Injection mixtures.
3. Bioenergetic Characterization of Polarized M1 and M2 BMDM using a Extracellular Flux Analyzer
Create an assay template with the assay wizard with 2 minute MIX and 3 minute MEASURE times and (at least) 3 mix and rate measurements loops before each of the 4 injections (Table 1) and after the last injection. Note: These concentrations are valid for bone marrow-derived macrophages and Raw264.7 macrophage cell lines, but should be optimized for each cell type. Adding pyruvate in the FCCP mixture is needed to fuel maximal respiration.
Start the run by pushing the start bottom and follow the instructions of the apparatus. First load the cartridge plate with the hydrated probes to allow calibration by apparatus and next load the cell plate.
- After the assay, carefully discard all assay medium and store the analyzed cell culture microplate at -20 °C for future cell normalization using the cell proliferation assay kit as described in detail by the supplier.
- Briefly, prepare a 1x cell-lysis buffer by diluting component B 20-fold in distilled water and dilute compound A 400-fold in the 1x cell-lysis buffer. Add 200 µl per well, incubate 5 min at RT and measure fluorescence with ~180 nm excitation and ~520 nm emission maxima. Note: When working with non-adherent cells or if poor adherence is a concern, the direct cell proliferation kit can be used as an alternative since this protocol does not require discarding all assay medium. Yet, this direct protocol is slightly less sensitive compared to the protocol used in this lab.
After fluorescence measurement, normalize the cell count in each well as a ratio in which the average cell count of all naïve macrophage wells is set at 1. Note: Protein quantification (e.g. using a BCA assay) could be used as an alternative way to normalize cells counts. Yet, in our hands this assay was less sensitive compared to the cell proliferation assay kit.
Representative Results
After finishing the extracellular flux assay and cell quantification, data can be normalized for cell counts and analysed. This will typically yield ECAR and OCR plots as shown in Figure 1A and Figure 1B.
After the injection of glucose (A), the increase in ECAR represents the glycolysis rate. The additional increase in ECAR after ATP synthase inhibition with oligomycin (B) provides information about the glycolytic reserve and capacity (Figure 1A). When analysing the OCR values, oligomycin injection (B) allows calculation of the oxygen consumption used for mitochondrial ATP synthesis. FCCP (C) uncouples mitochondrial respiration and the corresponding OCR measurements yield data about the maximal and spare respiratory capacity. Finally, injection of rotenone (Rot) and antimycin A (AA) block mitochondrial complex I and III and the residual OCR represents the non-mitochondrial oxygen consumption (Figure 1B).
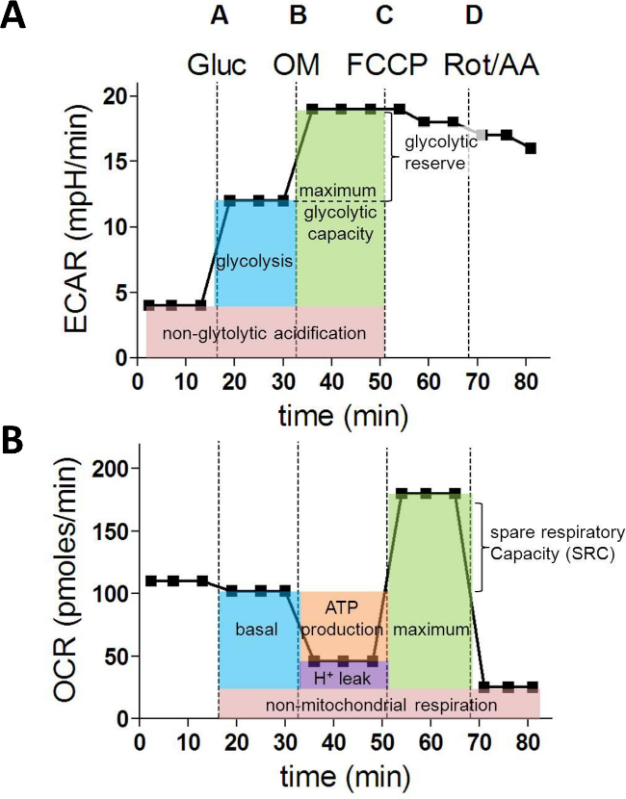 Figure 1: Metabolic parameters derived from a XF Extracellular flux assay. (A) Following parameters of cellular glycolysis are calculated from the ECAR values (in mpH/min): glycolysis, maximum glycolytic capacity and glycolytic reserve. (B) OCR measurements (in pMoles/min) are used to calculate the next fundamental parameters of mitochondrial function: basal respiration, ATP production, proton leak, maximum respiration and spare respiratory capacity. Gluc = glucose; OM = oligomycin A; FCCP = Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone; AA = antimycin A; Rot = Rotenone Please click here to view a larger version of this figure.
Figure 1: Metabolic parameters derived from a XF Extracellular flux assay. (A) Following parameters of cellular glycolysis are calculated from the ECAR values (in mpH/min): glycolysis, maximum glycolytic capacity and glycolytic reserve. (B) OCR measurements (in pMoles/min) are used to calculate the next fundamental parameters of mitochondrial function: basal respiration, ATP production, proton leak, maximum respiration and spare respiratory capacity. Gluc = glucose; OM = oligomycin A; FCCP = Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone; AA = antimycin A; Rot = Rotenone Please click here to view a larger version of this figure.
After the run, ECAR and OCR measurements 1 till 15 for each well can be exported to Excel and the following glycolytic parameters can be calculated from the ECAR measurements as follows:
Non-glycolytic acidification = avg. ECAR(1,2,3)
Glycolysis = avg. ECAR(4,5,6)– avg. ECAR(1,2,3)
Maximum glycolytic capacity = avg. ECAR(7,8,9)– avg. ECAR(1,2,3)
Glycolytic reserve= avg. ECAR(7,8,9) - avg. ECAR(4,5,6)
From the OCR rates, the next metabolic characteristics can be determined:
Non-mitochondrial respiration = avg. OCR(13,14,15)
Basal respiration = avg. OCR(4,5,6) - avg. OCR(13,14,15)
ATP production = avg. OCR(4,5,6) - avg. OCR(7,8,9)
Proton leak = avg. OCR(7,8,9) - avg. OCR(13,14,15)
Maximum respiration = avg. OCR(10,11,12) - avg. OCR(13,14,15)
Spare respiratory capacity (SRC) = avg. OCR(10,11,12) - avg. OCR(4,5,6)
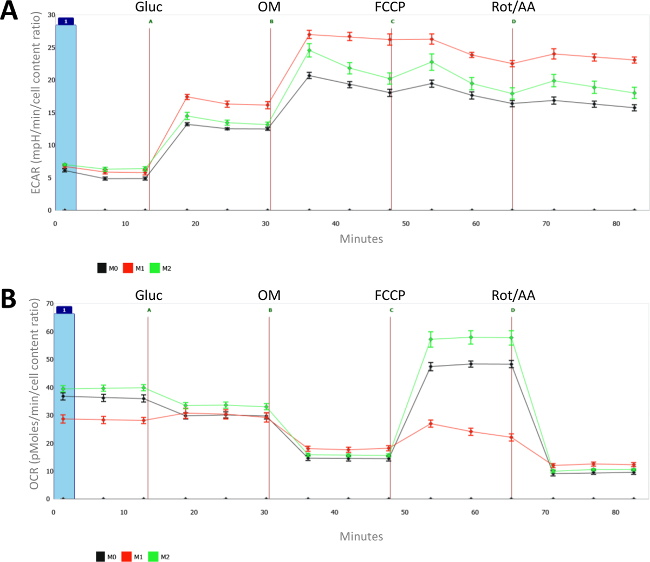 Figure 2: Metabolic characteristics of naïve (M0), LPS- (M1) and IL-4- (M2) polarized macrophages. For each measurement, the mean and standard error of the mean (SEM) of 8 individual wells is presented. (A) Extracellular acidification rates (ECAR, in mpH/min) and (B) oxygen consumption rates (OCR, in pMoles/min) are measured for differentially polarized (M1 and M2) and naïve macrophages (M0). All measurements were normalized for cell count (as measured with CyQUANT) and the average cell count of the naïve macrophages was set at 1. Injection A = glucose; B = oligomycin; C = FCCP; D= antimycin A + Rotenone. Please click here to view a larger version of this figure.
Figure 2: Metabolic characteristics of naïve (M0), LPS- (M1) and IL-4- (M2) polarized macrophages. For each measurement, the mean and standard error of the mean (SEM) of 8 individual wells is presented. (A) Extracellular acidification rates (ECAR, in mpH/min) and (B) oxygen consumption rates (OCR, in pMoles/min) are measured for differentially polarized (M1 and M2) and naïve macrophages (M0). All measurements were normalized for cell count (as measured with CyQUANT) and the average cell count of the naïve macrophages was set at 1. Injection A = glucose; B = oligomycin; C = FCCP; D= antimycin A + Rotenone. Please click here to view a larger version of this figure.
As shown in Figure 2A, macrophage activation with LPS induces increased glycolytic metabolism. The differences between LPS and IL-4-treated macrophages are even more apparent when looking at oxygen consumption rates (OCR) in Figure 2B. Indeed, while the maximal oxidative metabolism is highly suppressed in M(LPS), IL-4 induces basal and especially maximal respiration in M2 macrophages. One should note that increased glycolytic is rather unique to M(LPS) macrophages and is not necessarily a characteristic of M1 macrophages since M(IFNγ) macrophages do not display enhance glycolysis.
Overall, this extracellular flux analysis demonstrates that IL-4-induced M2 macrophages are characterized by enhanced oxidative metabolism and especially a high spare respiratory capacity (SRC), whereas LPS-activated macrophages display enhanced glycolytic metabolism and do not possess any spare respiratory capacity.
Discussion
The aim of our laboratory is to understand how macrophage polarization and function can be influenced with the ultimate goal to improve outcome of atherosclerosis and other inflammatory conditions12. To assess macrophage polarization, one typically measures the LPS(+IFNγ)-induced and IL-4-induced gene expression of M1 and M2 marker genes (qPCR), determines IL-6, IL-12, TNF and NO secretion (by ELISA and Griess reaction) and examines IL-4-induced CD71, CD206, CD273 and CD301 surface expression (by flow cytometry) and arginase activity3,13. Additionally, apoptosis assays (by Annexin-V/PI staining), phagocytosis assays (with fluorescent latex beads and/or pHrodo E. coli) and foam cell formation assays (by DiI-oxLDL uptake, LipidTOX neutral lipid staining) can be performed to evaluate macrophage functionality14. Additionally, extracellular flux analysis can be performed to measure bioenergetics profiles of polarized macrophages as a new and alternative functional readout.
This manuscript shows that macrophage polarization induces metabolic reprogramming with LPS-induced macrophages switching to increased glycolysis as their energy source. Conversely, IL-4-induced M2 macrophages use mitochondrial oxidative phosphorylation as the main source of ATP. Importantly, metabolic reprogramming not only provides energy for the particular requirements of M1 and M2 polarized macrophages. Indeed, metabolic changes affect metabolite concentrations that are direct regulators of the macrophage phenotype 10,15,16. Hence, macrophage metabolism is not only a characteristic (as shown here in Figure 2) but also a driver of distinct macrophage polarization states and this new knowledge supported the rapid growth of the immunometabolism research area during the last couple of years.
However, robust measurements of metabolic profiles in macrophages remained difficult and had their limitations. In this study, an extracellular flux analyser is applied to robustly measure glycolysis, glycolytic reserve, maximal glycolytic capacity, non-glycolytic acidification, basal and maximal respiration, ATP production, spare respiratory capacity, proton leak and non-mitochondrial respiration in real-time in a minimal amount of macrophages.
Therefore, we modified and improved the manufacturer’s protocols as follows. The standard Mito Stress Test (# 103015-100) suggests using medium with glucose and pyruvate and a protocol with 3 injections (OM, FCCP, AA/Rot). We opt to start the assay with glucose/pyruvate-free medium, add glucose as a first injection in port A (like in the Glycolysis Stress Test # 103020-100) and add pyruvate together with the FCCP uncoupler in port C. In this way, all relevant glycolysis parameters are derived from the ECAR measurements 1-9 and all relevant information about mitochondrial function from OCR measurements 4-15.
Note that it is critical to inject pyruvate together with FCCP to fuel maximal respiration upon uncoupling. Before using this combined protocol, one should also verify that 2-Deoxy-D-Glucose (2-DG) lowers ECAR levels after the assay to the basal values (1-3) and that the recorded ECAR rates 4-6 are indeed due to glycolysis. Additionally, one should comprehend that the compound concentrations and cell numbers used in this manuscript are valid for bone marrow-derived macrophages and Raw264.7 macrophage cell lines and should be optimized for other cell types. Moreover, one should note that M-CSF which is used to generate bone marrow-derived macrophages promotes an M2-like anti-inflammatory phenotype. The presented results from bone marrow-derived macrophages may therefore be not fully comparable to measurements with primary tissue resident macrophages or macrophages cell lines that do not require M-CSF during cell culturing.
Compared to existing methods, this protocol requires minimal amounts of macrophages and quickly provides virtually all fundamental metabolic cell characteristics. This results in enough surplus cells to perform other immunological assays and even the cells used for bioenergetics profiling could still be used for other purposed after the assay. Moreover, the particular 96 well plate format allows assessing multiple conditions in real time at the same time. Yet, the variation between individual wells can be considerable and therefore the use of at least 4 wells per condition is often desired. Taken this limitation into account, the described extracellular flux analysis still allows to run more than 10 experimental conditions (n = 6) at once, which is much more than traditional techniques.
Overall, this article provides an easy and reproducible functional assay that allows measuring all relevant glycolysis and mitochondrial parameters in polarized macrophage subsets. This technique can serve as a future application to assess macrophage function and to evaluate the effect of distinct manipulations (e.g. gene knock down, new pharmaceuticals, etc.) on the macrophage’s (metabolic) phenotype. As such, extracellular flux data can be used in conjunction with other assays to allow in-depth characterisation of the macrophage activation status.
Disclosures
Free access to this article is sponsored by Seahorse Bioscience.
Acknowledgments
Jan Van den Bossche received a Junior Postdoc grant from the Netherlands Heart Foundation (2013T003) and a VENI grant from ZonMW (91615052). Menno de Winther is an Established Investigator of the Netherlands Heart Foundation (2007T067), is supported by a Netherlands Heart Foundation grant (2010B022) and holds an AMC-fellowship. We also acknowledge the support from the Netherlands Cardiovascular Research Initiative, Dutch Federation of University Medical Centers, the Netherlands Organization for Health Research and Development and the Royal Netherlands Academy of Sciences for the GENIUS project “Generating the best evidence-based pharmaceutical targets for atherosclerosis” (CVON2011-19). We acknowledge Seahorse Bioscience for making open access publication possible. The authors thank Riekelt Houtkooper and Vincent de Boer (Laboratory Genetic Metabolic Diseases, Academic Medical Center, Amsterdam, The Netherlands) for all previous assistance with the Seahorse analyzer.
References
- Murray PJ, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeksema MA, et al. IFN gamma Priming of Macrophages Represses a Part of the Inflammatory Program and Attenuates Neutrophil Recruitment. J Immunol. 2015. [DOI] [PubMed]
- Vanden Bossche J, et al. Pivotal Advance Arginase 1 independent polyamine production stimulates the expression of IL 4 induced alternatively activated macrophage markers while inhibiting LPS-induced expression of inflammatory genes. Journal of leukocyte biology. 2012;91:685–699. doi: 10.1189/jlb.0911453. [DOI] [PubMed] [Google Scholar]
- Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38:633–643. doi: 10.1016/j.immuni.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, et al. PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nature communications. 2014;5:4436. doi: 10.1038/ncomms5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freemerman AJ, et al. Metabolic reprogramming of macrophages glucose transporter 1 (GLUT1) mediated glucose metabolism drives a proinflammatory phenotype. The Journal of biological chemistry. 2014;289:7884–7896. doi: 10.1074/jbc.M113.522037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavakoli S, Zamora D, Ullevig S, Asmis R. Bioenergetic profiles diverge during macrophage polarization implications for the interpretation of 18F FDG PET imaging of atherosclerosis. J Nucl Med. 2013;54:1661–1667. doi: 10.2967/jnumed.112.119099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vats D, et al. Oxidative metabolism and PGC 1beta attenuate macrophage mediated inflammation. Cell metabolism. 2006;4:13–24. doi: 10.1016/j.cmet.2006.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SC, et al. Cell intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nature immunology. 2014;15:846–855. doi: 10.1038/ni.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannahill GM, et al. Succinate is an inflammatory signal that induces IL 1beta through HIF 1alpha. Nature. 2013;496:238–242. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying W, Cheruku PS, Bazer FW, Safe SH, Zhou B. Investigation of macrophage polarization using bone marrow derived macrophages. J Vis Exp. 2013. [DOI] [PMC free article] [PubMed]
- Hoeksema MA, et al. Targeting macrophage Histone deacetylase 3 stabilizes atherosclerotic lesions. EMBO Mol Med. 2014;6:1124–1132. doi: 10.15252/emmm.201404170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Bossche J, et al. Alternatively activated macrophages engage in homotypic and heterotypic interactions through IL 4 and polyamine induced E cadherin catenin complexes. Blood. 2009;114:4664–4674. doi: 10.1182/blood-2009-05-221598. [DOI] [PubMed] [Google Scholar]
- Van den Bossche J, et al. Inhibiting epigenetic enzymes to improve atherogenic macrophage functions. Biochem Biophys Res Commun. 2014;455:396–402. doi: 10.1016/j.bbrc.2014.11.029. [DOI] [PubMed] [Google Scholar]
- Galvan Pena S, O Neill LA. Metabolic reprograming in macrophage polarization. Frontiers in immunology. 2014;5:420. doi: 10.3389/fimmu.2014.00420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Zhao Q, Yang T, Ding W, Zhao Y. Cellular metabolism and macrophage functional polarization. International reviews of immunology. 2015;34:82–100. doi: 10.3109/08830185.2014.969421. [DOI] [PubMed] [Google Scholar]