

Abstract
DNA nanotechnology requires large amounts of highly pure DNA as an engineering material. Plasmid DNA could meet this need since it is replicated with high fidelity, is readily amplified through bacterial culture and can be stored indefinitely in the form of bacterial glycerol stocks. However, the double-stranded nature of plasmid DNA has so far hindered its efficient use for construction of DNA nanostructures or devices that typically contain single-stranded or branched domains. In recent work, it was found that nicked double stranded DNA (ndsDNA) strand displacement gates could be sourced from plasmid DNA. The following is a protocol that details how these ndsDNA gates can be efficiently encoded in plasmids and can be derived from the plasmids through a small number of enzymatic processing steps. Also given is a protocol for testing ndsDNA gates using fluorescence kinetics measurements. NdsDNA gates can be used to implement arbitrary chemical reaction networks (CRNs) and thus provide a pathway towards the use of the CRN formalism as a prescriptive molecular programming language. To demonstrate this technology, a multi-step reaction cascade with catalytic kinetics is constructed. Further it is shown that plasmid-derived components perform better than identical components assembled from synthetic DNA.
Keywords: Molecular Biology, Issue 105, DNA nanotechnology, DNA strand displacement, molecular programming, DNA computing, chemical reaction networks, plasmid DNA, reaction kinetics
Introduction
The predictability of Watson-Crick base pairing has allowed dynamic DNA nanotechnology to emerge as a programmable way to design molecular devices with dynamic properties1,2. In particular, DNA strand displacement — a programmable, competitive hybridization reaction — has proved to be a powerful mechanism for engineering dynamic DNA systems. In a DNA strand displacement reaction, an incoming oligonucleotide displaces a previously bound “output” strand from a complementary binding partner. Multiple such reactions can be chained together into multi-step reaction cascades with a high degree of control over the order and timing of individual reaction steps3. DNA strand displacement cascades have been used to create digital and analog molecular circuits4-7, switchable nanostructures8-10, autonomous molecular motors11-15, and non-covalent catalytic amplifiers13,16-21. Moreover, DNA devices using strand displacement reactions can be simulated and designed for diverse applications using computer-assisted design software22-24.
Currently, chemically synthesized DNA serves as the main material for DNA nanotechnology. However, errors in the DNA synthesis process, and the resulting imperfect oligonucleotides, are believed to limit performance of dynamic DNA devices by causing erroneous side reactions. For example, “leak” reactions can result in the release of an output oligonucleotide even in the absence of a reaction trigger. Such effects are most obvious in autocatalytic reaction cascades where even a minimal amount of initial leak will eventually result in the full activation of the cascade19,20. Conversely, reactions often fail to reach the expected level of activation because some components do not trigger even in the presence of the intended input7,25. To make the performance of DNA-based nanodevices comparable to their biological protein-based counterparts, such error modes need to be dramatically reduced.
Bacterial plasmids or other biological DNA could serve as a relatively cheap source of highly pure DNA for nanotechnology applications. Large amounts of DNA can be generated by replication in bacteria and the intrinsic proofreading abilities of living systems ensure the purity of the resulting DNA. In fact, several recent papers have recognized the potential utility of biological DNA for nanotechnology applications21,26-28. However, the fully double-stranded nature of plasmid DNA has so far prohibited its use as a material for making dynamic DNA devices, which typically consist of multiple oligonucleotides and contain both double-stranded and single-stranded domains. In a recent paper29 this issue was addressed and a new DNA gate architecture that consists primarily of nicked double-stranded DNA (ndsDNA) was introduced.
Importantly, systems of ndsDNA gates can be designed that realize the dynamics specified by any formal chemical reaction network (CRN)29. ndsDNA gates could thus be used, in principle, to create dynamical systems that exhibit oscillations and chaos, bistability and memory, Boolean logic or algorithmic behaviors30-38. For example, Ref. 29 demonstrated a three-reaction CRN that provided a molecular implementation of a “consensus” protocol, a type of distributed computing algorithm29,39,40. This work first demonstrated a novel use for the CRN formalism as a “programming language” for rapidly synthesizing functional molecular systems (Figure 1A).
Here, a detailed protocol for deriving ndsDNA gates from plasmid DNA is provided. First is a review of the sequence design process. Then follows an explanation of how synthetic oligonucleotides containing the gate sequences are cloned into plasmids and sequence verified and amplified via bacterial culture. Next, it is shown how ndsDNA gates can be derived from the plasmids by enzymatic processing (see Figure 2). Finally, a method for testing gate behavior using fluorescence kinetics assays is outlined.
Reaction mechanism As an example, the protocol focuses on the catalytic chemical reaction A+B->B+C. The species A, B, and C (“signals”, Figure 1B) all correspond to a different single-stranded DNA molecule. The sequences of these molecules are completely independent and the strands do not react with one another directly. The sequences of all signals have two different functional domains, i.e., subsequences that act together in strand displacement reactions: 1) a short toehold domain (labels ta, tb, tc) that is used for initiation of strand displacement reaction, and 2) a long domain (labels a, b, c) that determines the signal identity.
Interactions between signal strands are mediated by nicked double-stranded DNA (ndsDNA) gate complexes (called JoinAB and ForkBC) and auxiliary single-stranded species (<tr r>, <b tr>, <c tb> and <i tc>). The formal reaction A+B->B+C is executed through a series of strand displacement reaction steps, where each reaction step exposes a toehold for a subsequent reaction (Figure 1B). In this example, signals A and B are initially free in solution while signal C is bound to the fork gate. At the end of the reaction B and C are in solution. More generally, signals that are bound to a gate are inactive while signals that are free in solution are active, that is, they can participate in a strand displacement reaction as an input. The time course of the reaction is followed using a fluorescent reporter strategy (Figure 1C). In previous work29, it was demonstrated that this reaction mechanism not only realizes the correct stoichiometry but also the kinetics of the target reaction.
Protocol
1. Sequence Design
Note: Sequence design overview: In this section, the strategy for designing plasmid-derived DNA gates is described. Enzyme sites placed on either end of the gates to allow for the release of fully double stranded gates after digestion. Nicking sites are then placed such that enzymes create nicks on the top strand to create the final ndsDNA gates. Finally, the remaining sequences are chosen such that independent domains are orthogonal to one another and do not exhibit secondary structure.
Place the Nt.BstNBI nicking site four nucleotides away from the 3’ end of each long domain (a, b, c, r, and i). Place the Nb.BsrDI nicking site on the 5’ end of each long domain (a, b, c, and r. Note that the domain i does not have any Nb.BsrDI nicking site). Figure 2C shows the detailed sequence view of JoinAB and ForkBC gates.
Place the PvuII restriction site on both ends of ndsDNA gates so that PvuII digestion can release the gates from plasmids (see Figure 2C).
Design other unconstrained sequences by following two principles: (a) strands should not exhibit secondary structures (DNA structures can be predicted using Nupack41), and (b) all domains should be orthogonal to minimize crosstalk.
Place ndsDNA sequences in the center of a gate template. Place 30-40 bp random spacer sequences on both ends of the gate template, each spacer serves as a unique binding site for the following Polymerase Chain Reaction (PCR).
2. Cloning of NdsDNA Gates into Plasmids
Note: This section describes the Gibson cloning method for inserting 4 copies of the gate into a plasmid backbone.
Order ndsDNA gate templates as double-stranded genomic blocks from a DNA manufacturer (gate template sequences are shown in Table 1; strands occur in ndsDNA gates are shown in Table 2; domain level sequences are shown in Table 3).
After receiving the ordered DNA, spin the tubes containing genomic blocks at 10,000-14,000 x g for 1 min to ensure that all dried DNA is at the bottom of the tube.
Resuspend the dried genomic blocks in DNase-free water to achieve a final concentration of 10 ng/µl. Note: Alternatively, DNA can be resuspended using 1x Tris ethylenediaminetetraacetic acid (EDTA) buffer (TE buffer: 10 mM Tris and 1 mM EDTA, pH 8.0). However, EDTA is a chelating agent for divalent cations and could inhibit PCR.
Generate 4 gate fragments with different overlap regions through a standard PCR with a High-Fidelity DNA polymerase (see Figure 3A). The primer sequences are detailed in Table 4 (melting temperature of these primers is 62 °C).
Run a 2% agarose gel at 140 V for 30 min at RT (for a detailed agarose gel electrophoresis protocol see 42) and cut the bands corresponding to each PCR amplified fragment out of the gel. Then purify the gel slices using a gel extraction kit (please refer to the Materials) following the manufacturer’s instructions.
Digest a high copy number plasmid backbone (see materials) with PvuII-HF and PstI-HF at 37 °C for 1 hr (see Table 5) according to the manufacturer’s protocol. PvuII-HF and PstI-HF are high fidelity restriction enzymes, which dramatically reduce unspecific cuts.
Run a 1.5% agarose gel and cut the linearized backbone (typically run the gel at 140 V for 30-40 min at RT). Then extract the DNA from the gel slice using gel extraction kit following the manufacturer’s instructions.
Perform Gibson assembly43 with the linearized vector and purified PCR fragments (see Table 6 and Figure 3B) at 50 °C for 1 hr.
Transform the Gibson assembly product from step 2.8 into Escherichia coli (E. coli) and plate on a Lysogeny Broth (LB) agar plate containing Ampicillin antibiotics (at a concentration of 100 µg/ml). Perform transformation through electroporation or a heat shock method 44,45, and use the appropriate E. coli strain. For example, use E. coli strain JM109 for heat shock transformation, and use DH5α electrocompetent E. coli cells for electroporation. Note: The plasmid backbone used contains an Ampicillin resistance cassette. If using a different selection marker, use the appropriate antibiotics instead of Ampicillin.
3. Bacterial Culture Amplification and Quality Control
Note: This section describes the mass production and isolation of plasmids containing the DNA gates after quality control.
Pick a single colony from the Ampicillin selective plate from step 2.9 and incubate a culture of 3 ml enriched medium containing Ampicillin antibiotics (at a concentration of 100 µg/ml). Mark the colony such that it can be utilized again in subsequent experimental steps. Grow the culture at 37 °C O/N with vigorous shaking (200-300 rpm). Typically, incubate for 16-24 hr.
Extract the plasmid DNA from the bacterial culture using a Mini-prep kit following the manufacturer’s instructions.
Measure the purified plasmid DNA using a spectrophotometer following the manufacturer’s instructions. Typical yield ranges from 50-1,000 ng/µl.
Get the extracted plasmid DNA sequenced by sending the sample to a DNA sequencing company. Sequencing primers should be located about 100 nucleotides upstream and downstream of the region to be sequenced; the sequencing primer for the plasmid (see materials for plasmid) has the following sequence: ATTACCGCCTTTGAGTGAGC. Note: If there is sequence error or recombination in the inserted ndsDNA gates, select a different colony from the plate from step 2.9. Follow steps 3.1-3.4 to verify that the sequences of the inserted gates are correct.
After verifying that the sequences are correct, pick the corresponding colony from the Ampicillin selective plate (from step 2.9), and incubate a culture of 800 ml Terrific Broth (TB) containing Ampicillin antibiotics (at a concentration of 100 µg/ml). Grow the culture at 37 °C for 16-24 hr with vigorous shaking (200-300 rpm). TB particularly is well suited for high yield plasmid production. Note: Alternatively, LB could also be used to grow bacteria although the plasmid yield can be an issue.
Purify the DNA using a Maxi-prep kit following the manufacturer’s instructions.
Follow step 3.3-3.4 to check whether the sequences are correct. If any recombination occurred, see the following note. Otherwise, move on to step 4. Note: One possible issue here is that multiple copies of inserted gates in the plasmid may recombine due to DNA repair. To address this problem, use an E. coli strain lacking the recA protein (a protein related to DNA repair) such as JM109 or DH5α to transform a previously sequence-verified plasmid (i.e., without any sequence errors and recombination). Then pick one colony from this plate and verify the plasmid sequence by sending the sample to a DNA sequencing company.
4. Enzymatic Processing
Note: This section describes the process for digesting the plasmids such that they are cut and nicked in the correct locations and ready to be used for kinetics experiments.
Digest the purified plasmid DNA from step 3.7 with restriction enzyme PvuII-HF for 1 hr at 37 °C (see Table 7). Typically digest the plasmid with 4 units of PvuII-HF per 1 mg of plasmid. High fidelity restriction enzymes are recommended for use because they dramatically reduce unspecific cuts.
- Perform ethanol precipitation on the sample.
- Add 2 equivalent volumes of ice-cold absolute ethanol to the sample.
- Incubate the mixture at -80 °C for at least 1 hr (this mixture can also sit at -80 °C for O/N).
- Centrifuge at 10,000-14,000 x g at 0 °C for 30 min.
- Remove the supernatant.
- Add 1,000 µl of RT 95% ethanol to the sample, and invert 10-15 times.
- Centrifuge at 10,000-14,000 x g at 4 °C for 10 min.
- Remove the supernatant and air dry on bench for 10-20 min.
- Resuspend the DNA pellets in an appropriate volume of Nuclease free H2O (typically 100-200 µl). Adding more than 200 µl will generally make the sample too dilute for use in kinetics experiments.
Measure the resuspended DNA using a spectrophotometer following the manufacturer’s instructions.
Digest join gates with nicking enzyme Nb.BsrDI at 65 °C for 1 hr using 4 units of enzyme per 1 µg of plasmid (see Table 8); digest fork gates with nicking enzyme Nt.BstNBI at 55 °C for 1 hr using 8 units of enzyme per 1 µg of plasmid (see Table 9). Note: Step 4.2 removes enzyme digestion buffer and helps concentrate the gates for kinetics experiments. Step 4.2 can be skipped for join gates because both restriction enzyme PvuII-HF and nicking enzyme Nb.BsrDI share the same digestion buffer. In step 4.2.8, Nuclease free H2O is used instead of TE because EDTA is a chelating agent for divalent cations and can inhibit restriction enzymes that need these ions to function. Note: The addition of excess amounts of enzymes may lead to high amounts of initial circuit leakage (Figure 4), which is most likely caused by over-digestion46. This issue can be addressed by optimizing the enzyme amounts (see Figure 4). Typical range of enzymes is from 1-10 units/1 µg plasmid.
5. Preparation of Single-stranded Oligonucleotides
Note: This section describes the protocol for resuspending and quantitating the chemically synthetized single-stranded DNA (ssDNA) that will be used for signal strands and auxiliary strands. For strand sequences see Table 10. Note that the following protocol is an example of preparing 10 µM ssDNA. Other concentrations of ssDNA can be prepared similarly.
After receiving oligos from DNA manufacturer, spin the tubes containing DNA at 10,000-14,000 x g for 1 min to ensure that all dried DNA is at the bottom of the tube.
Resuspend the DNA using 1x Tris ethylenediaminetetraacetic acid (EDTA) buffer (TE buffer: 10 mM Tris and 1mM EDTA, pH 8.0) to achieve a final concentration of 100 µM. For example, resuspend 8 nmol of DNA in 80 µl of TE buffer.
Mix 10 µl of the DNA at 100 µM with 90 µl of molecular water in a microcentrifuge tube, which should achieve a final concentration of 10 µM.
- Measure the exact concentration of the DNA sample using a spectrophotometer following the manufacturer’s instructions. The following protocol gives an example of how DNA concentration can be measured.
- Blank the spectrophotometer with 2 µl of molecular water.
- Measure the absorbance at 260 nm (A260) of the DNA sample. Use the following equation to calculate the stock concentration. Note: The sample concentration is M = A260/extinction coefficient. The extinction coefficient can be found on the specification datasheet by the DNA manufacturer.
6. Preparation of Fluorescent Reporters
Note: This section describes the protocol for preparing ReporterC, Other fluorescent reporters can be assembled similarly.
Order high-performance liquid chromatography (HPLC) purified oligonucleotides ROX-<c* tc*> (the top strand of ReporterC) and <c>-RQ (the bottom strand of ReporterC) from DNA manufacturer (see Table 10 for sequences).
After receiving the synthesized oligonucleotides, resuspend and quantitate samples as explained in Step 5.
Mix the reporter top and bottom strands (i.e., ROX-<c* tc*> and <c>-RQ) in 1x Tris-Acetate-EDTA (TAE) with 12.5 mM Mg2+ (see the Table 11 for the detailed recipe). Note that here 30% excess quencher labeled strand <c>-RQ is added to assemble the reporter, which ensures that all fluorophore-labeled strands are quenched even with imperfect stoichiometry.
Anneal the ReporterC complex using a thermal cycler, cooling from 95 °C to 20 °C at a rate of 1 °C/min. The samples can be stored at 4 °C.
7. Fluorescence Measurements
Note: The section describes a general protocol for fluorescence kinetics measurements (see Figure 5 for the experimental procedure), and this protocol will be used in steps 8, 9, and 10. Also note that this protocol is for the use of a spectrofluorimeter. Alternatively, these experiments could also be performed in a plate reader although sensitivity, well-to-well variations and lack of temperature control in long-term experiments can be an issue.
Set the temperature controller to 25 °C, and wait for the temperature to stabilize. Using a temperature controller can reduce variability in the signal that can result from temperature variation.
- Set proper parameters for kinetics measurements in the data acquisition software of the spectrofluorimeter. Detailed example settings are as follows:
- Set the slit width to 2.73 nm for both excitation and emission monochromators.
- Set the integration time to 10 sec for every 60 sec time-point. Set the total measurement time to 24 hr.
- Set the excitation/emission wavelengths to match the fluorophores used in the experiment. Example wavelengths are as follows: ROX (588 nm / 608 nm), and TAMRA (559 nm / 583 nm).
Add Nuclease free H2O and 10x Tris-acetate-EDTA buffer containing 125 mM Mg2+ (10x TAE/Mg2+) to a synthetic quartz cell. See Tables 12, 13, and 14 for example volumes to use.
Add polyT strands to achieve a final concentration of ~1 µM (see Table 12, 13, and 14 for volumes), and then vortex the synthetic quartz cells for 10-15 sec. Generally, pipette tips will non-specifically bind DNA. Adding high concentrations of polyT strands can reduce this non-specific binding error.
Add reporters and auxiliary strands. See Table 12, 13, and 14 for example volumes to use. Note that for reporter calibration, no auxiliary strands are needed.
Add 10% sodium dodecyl sulfate (SDS) to achieve a final concentration of 0.15% SDS. Note: SDS is used to dissociate enzymes from the plasmid-derived gates because enzymes may interfere with the strand displacement reaction (see Figure 6). SDS is recommended here instead of heat denaturation of enzymes to avoid the dissociation and incorrect recombination of gate strands, which may adversely affect the circuit function.
- [Skip this step for reporter calibration.]
- Add join and fork gates (see Table 13, and 14 for volumes) to the synthetic quartz cell and mix the solution by pipetting it up and down for at least 20 times (do not vortex the cuvette because vortexing solutions with SDS can result in bubbles which will affect fluorescence kinetics measurements).
- Also, move to the following measurement steps as soon as possible because the leak reaction initiates immediately after the addition of join and fork gates to the synthetic quartz cell.
Place the synthetic quartz cells into the chamber of a spectrofluorimeter.
Start the kinetics measurement.
After 5 min of measurement, add input strands (see Table 12, 13, and 14 for volumes) to the synthetic quartz cell and mix the reaction by pipetting it up and down for at least 20 times. Note that the sample should be mixed gently to avoid bubbles. Perform this step while the data acquisition program is paused to avoid measuring signals triggered by external light.
Record the reaction kinetics until it reaches steady state. The reaction kinetics are displayed on the computer.
8. Calibrate Fluorescent Reporters
Note: This section describes the protocol for making calibration curves of fluorescent reporters. Calibration curves will be used to convert arbitrary fluorescence units to molar signal concentration.
Calibrate fluorescent reporters following the protocol described in Step 7. Use the volumes of reactants and buffers as summarized in Table 12. The standard concentration for this example is 50 nM (1x); reporters are at 3x; input is 1x. For cases where the input <tc c> are at 0.25x, 0.5x, 0.75x, adjust the volume of Nuclease free H2O correspondingly to keep the final volume of each reaction to be 600 µl. An example data are shown in Figure 7A.
Make a calibration curve of the ReporterC by a linear fit of the final fluorescence values against the initial concentration of signal C (An example calibration curve is shown in Figure 7B). This calibration curve can be used to convert arbitrary fluorescence units to its corresponding signal concentration.
9. Quantify the Concentration of Plasmid-derived ndsDNA Gates
Note: Each independently processed batch of plasmid-derived ndsDNA gates results in a different yield of functional gates, and this section describes a protocol for quantifying the concentration of plasmid-derived ndsDNA gates.
Quantify the concentration of plasmid-derived ndsDNA gates following the protocol described in Step 7. Use the volumes of reagents as summarized in Table 13. Note: Table 13 describes an example recipe for ForkBC quantification. JoinAB and other gates can be performed similarly but using different input strands, auxiliary strands and reporters.
Convert the final fluorescence value measured in this experiment to a concentration of signal C using the calibration curve from step 8.2. Then back-calculate the ndsDNA gate concentration. For example, a final fluorescence value for the gate quantification experiment of corresponds to 25 nM signal C (0.5x) based on the calibration curve in Figure 7B. Since the stock of ForkBC is diluted 40 times in this reaction, the stock concentration of the ForkBC gate is 1 µM.
10. Kinetics Measurements for the Reaction A+B->B+C
Note: This section describes a protocol for testing the DNA realization of a formal chemical reaction using fluorescence kinetics measurements.
Perform kinetics measurement by following the protocol described in Step 7. Use volumes of reagents and buffers as summarized in Table 14.
Representative Results
For a functional test, a DNA implementation of the bimolecular catalytic reaction (i.e., A+B->B+C) was created. The performance of plasmid-derived gates was compared to gates assembled from synthetic DNA. Catalytic reactions are a good test for gate purity because a faulty gate can irreversibly trap a catalyst, causing a disproportionate effect on the amount of product produced18,19. At the same time, a small leak reaction resulting in the untriggered release of the catalytic signal will be linearly amplified, leading to a disproportionate error signal. Experimental data for plasmid-derived and synthesized gates are shown in Figure 8B and 8C, respectively. In the experiments, the concentration of signal strand A is fixed while the amount of the catalytic signal B is varied. Signal C is used to read out the progress of the reaction without interrupting the catalytic cycle. Catalysis can be observed in the data since reactions approach completion even with amounts of catalyst B much smaller than the amount of A. Since SDS was not added to experiments done with the synthesized system, reaction speed (that could be affected by the addition of SDS) is not compared and the analytical focus is instead on catalytic turnover (detailed as follows).
Further analysis of the catalytic turnover of this reaction was conducted. Turnover is defined as the amount signal C produced for each catalyst B at a given time. Specifically, turnover was calculated from our experimental data by dividing the leak subtracted signal C by the initial amount of catalyst B added. For an ideal catalytic system, this turnover number should linearly increase with time and to be independent of the amount of catalyst as long as the substrate is not limiting. In a real system, faulty gates can disable catalysts, and the turnover will reach a maximum value even if not all available substrate is converted to product. The maximum turnover value indicates how many substrates (signal A) a catalyst (signal B) can convert before becoming inactivated. Here, it is observed that the synthesized system deviates from the ideal linear increase of turnover much earlier than the plasmid-derived system does, indicating sequestration of the catalyst through an undesirable side reaction (Figure 8D). The turnover comparison is only shown for low concentrations because at high concentrations of catalysts, all gates will be triggered and release signal C. The circuit leakage is also compared, and it is observed that the ratio of leak signal using plasmid-derived gates is about 8% less than that using synthesized gates after 10 hr of reaction (Figure 8E).
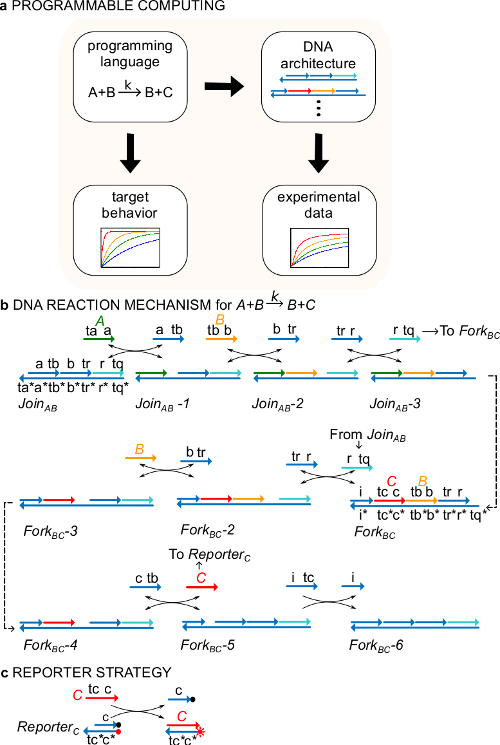 Figure 1. (A) CRNs serve as a prescriptive programming language. DNA reaction networks can be engineered to approximate the dynamics of a formal CRN. (B) DNA implementation of an example chemical instruction: A+B->B+C. DNA strands are drawn as lines with arrows at the 3’ end and * indicates complementarity. All signal strands A (<ta a>, green), B (<tb b>, orange), and C (<tc c>, red) are consisted of one toehold domain (labeled as ta, tb, and tc) and one identity domain (labeled as a, b, and c). The bimolecular reaction A+B->B+C requires two multi-stranded complexes JoinAB and ForkBC, and four auxiliary strands <tr r>, <b tr>, <c tb>, and <i tc>. The reaction proceeds through seven steps of strand displacement, where each step starts with toehold binding. (C) Reporter strategy. The reaction is followed using a reporter in which the bottom strand is labeled with a fluorophore (red dot) and the top strand is attached to a quencher (black dot). Because of the co-localization of the fluorophore and quencher, reporter fluorescence is quenched in the intact reporter. The signal C can replace the top strand of the reporter, leading to an increase of fluorescence. (This figure has been modified from Ref29.) Please click here to view a larger version of this figure.
Figure 1. (A) CRNs serve as a prescriptive programming language. DNA reaction networks can be engineered to approximate the dynamics of a formal CRN. (B) DNA implementation of an example chemical instruction: A+B->B+C. DNA strands are drawn as lines with arrows at the 3’ end and * indicates complementarity. All signal strands A (<ta a>, green), B (<tb b>, orange), and C (<tc c>, red) are consisted of one toehold domain (labeled as ta, tb, and tc) and one identity domain (labeled as a, b, and c). The bimolecular reaction A+B->B+C requires two multi-stranded complexes JoinAB and ForkBC, and four auxiliary strands <tr r>, <b tr>, <c tb>, and <i tc>. The reaction proceeds through seven steps of strand displacement, where each step starts with toehold binding. (C) Reporter strategy. The reaction is followed using a reporter in which the bottom strand is labeled with a fluorophore (red dot) and the top strand is attached to a quencher (black dot). Because of the co-localization of the fluorophore and quencher, reporter fluorescence is quenched in the intact reporter. The signal C can replace the top strand of the reporter, leading to an increase of fluorescence. (This figure has been modified from Ref29.) Please click here to view a larger version of this figure.
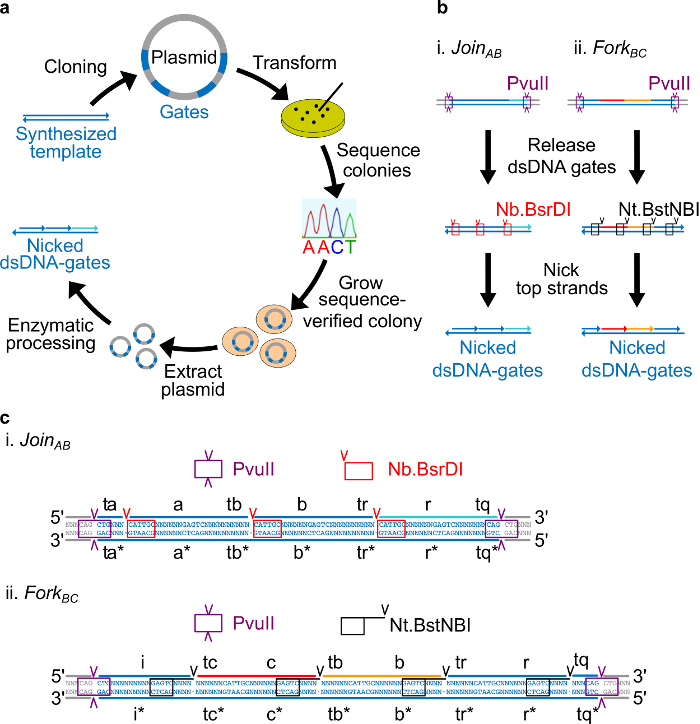 Figure 2. (A) NdsDNA gates made from bacterial plasmid DNA. Several copies of the double stranded ndsDNA gate template are cloned into a plasmid. The cloned plasmids are then transformed into E. coli cells and colonies on the plate are sequence verified. Once the sequence is confirmed, plasmid DNA is amplified and extracted. Finally, the double stranded plasmid is processed into the desired ndsDNA gates through enzymatic processing. (B) Enzymatic processing of ndsDNA gates. The restriction enzyme PvuII is used to release the gate from the plasmid. The released gates are further processed using nicking enzymes: Nb.BsrDI is used to generate nicks for JoinAB (Panel i); Nt.BstNBI is used to generate nicks for ForkBC (Panel ii). Restriction and nicking sites are indicated as color-coded boxes. (C) Sequence view of the gate template of JoinAB (Panel i) and ForkBC (Panel ii). The PvuII restriction site (highlighted in purple box) is at both ends of the ndsDNA gates. The Nb.BsrDI and Nt.BstNBI nicking sites are highlighted in red and black boxes, respectively. The locations of cut are marked with arrowheads. Sequence N is any nucleotide. (This figure has been modified with permission from Ref29.) Please click here to view a larger version of this figure.
Figure 2. (A) NdsDNA gates made from bacterial plasmid DNA. Several copies of the double stranded ndsDNA gate template are cloned into a plasmid. The cloned plasmids are then transformed into E. coli cells and colonies on the plate are sequence verified. Once the sequence is confirmed, plasmid DNA is amplified and extracted. Finally, the double stranded plasmid is processed into the desired ndsDNA gates through enzymatic processing. (B) Enzymatic processing of ndsDNA gates. The restriction enzyme PvuII is used to release the gate from the plasmid. The released gates are further processed using nicking enzymes: Nb.BsrDI is used to generate nicks for JoinAB (Panel i); Nt.BstNBI is used to generate nicks for ForkBC (Panel ii). Restriction and nicking sites are indicated as color-coded boxes. (C) Sequence view of the gate template of JoinAB (Panel i) and ForkBC (Panel ii). The PvuII restriction site (highlighted in purple box) is at both ends of the ndsDNA gates. The Nb.BsrDI and Nt.BstNBI nicking sites are highlighted in red and black boxes, respectively. The locations of cut are marked with arrowheads. Sequence N is any nucleotide. (This figure has been modified with permission from Ref29.) Please click here to view a larger version of this figure.
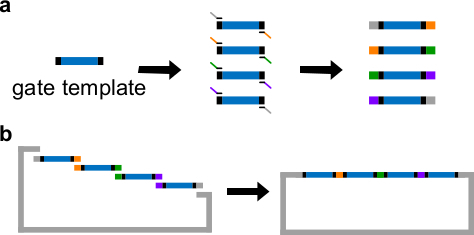 Figure 3. (A) PCR of a DNA gate template. A DNA gate template contains the ndsDNA gate sequences in the center (a blue region), and spacer sequences on both ends (black regions; these two end sequences are orthogonal). Primers can bind to the spacer sequences of the gate template, and generate four overlapping DNA fragments through PCR (overlapping sequences are color-coded in the figure). (B) Gibson assembly. The four amplified DNA fragments are then assembled into a linearized plasmid backbone through Gibson assembly method43. (This figure has been modified with permission from Ref29.) Please click here to view a larger version of this figure.
Figure 3. (A) PCR of a DNA gate template. A DNA gate template contains the ndsDNA gate sequences in the center (a blue region), and spacer sequences on both ends (black regions; these two end sequences are orthogonal). Primers can bind to the spacer sequences of the gate template, and generate four overlapping DNA fragments through PCR (overlapping sequences are color-coded in the figure). (B) Gibson assembly. The four amplified DNA fragments are then assembled into a linearized plasmid backbone through Gibson assembly method43. (This figure has been modified with permission from Ref29.) Please click here to view a larger version of this figure.
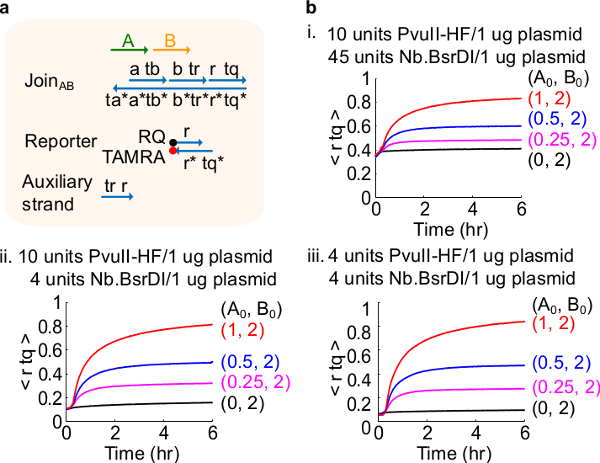 Figure 4.Circuit performance with different enzyme amounts. (A) A simplified representation of the gate, reporter, auxiliary strands, and signal strands used for the corresponding experiments. (B) Kinetics experiments with plasmid-derived JoinAB processed with different enzyme amounts. i. 10 units of PvuII-HF and 45 units of Nb.BsrDI per 1 µg of plasmid; ii. 10 units of PvuII-HF and 4 units of Nb.BsrDI per 1 µg of plasmid; iii. 4 units of PvuII-HF and 4 units of Nb.BsrDI per 1 µg of plasmid. All auxiliary strands were at 2x (1x = 10nM). The gate complex was 1.5x, and the experiments were performed at 35 °C in 1x TAE/Mg2+. (This figure has been modified with permission from Ref29.) Please click here to view a larger version of this figure.
Figure 4.Circuit performance with different enzyme amounts. (A) A simplified representation of the gate, reporter, auxiliary strands, and signal strands used for the corresponding experiments. (B) Kinetics experiments with plasmid-derived JoinAB processed with different enzyme amounts. i. 10 units of PvuII-HF and 45 units of Nb.BsrDI per 1 µg of plasmid; ii. 10 units of PvuII-HF and 4 units of Nb.BsrDI per 1 µg of plasmid; iii. 4 units of PvuII-HF and 4 units of Nb.BsrDI per 1 µg of plasmid. All auxiliary strands were at 2x (1x = 10nM). The gate complex was 1.5x, and the experiments were performed at 35 °C in 1x TAE/Mg2+. (This figure has been modified with permission from Ref29.) Please click here to view a larger version of this figure.
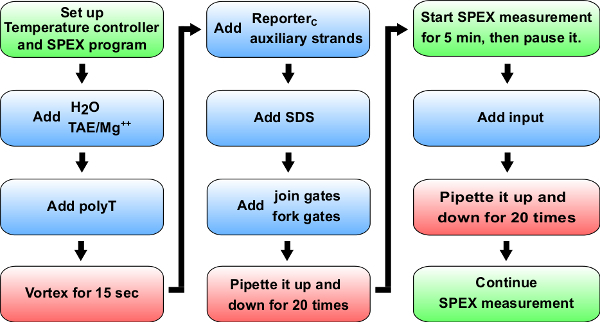 Figure 5. Flow chart of kinetics experiments. Blue: Materials to add to cuvette (0.875 ml synthetic quartz cell). Reference Table 14 for specific volumes to add for kinetic experiment of A+B -> B+C. Green: Instructions of a spectrofluorimeter (labeled as SPEX). Red: Mixing instructions. Please click here to view a larger version of this figure.
Figure 5. Flow chart of kinetics experiments. Blue: Materials to add to cuvette (0.875 ml synthetic quartz cell). Reference Table 14 for specific volumes to add for kinetic experiment of A+B -> B+C. Green: Instructions of a spectrofluorimeter (labeled as SPEX). Red: Mixing instructions. Please click here to view a larger version of this figure.
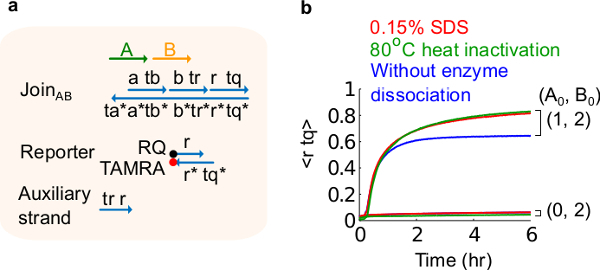 Figure 6.Enzyme dissociation and circuit behavior. (A) A simplified representation of the gate, reporter, auxiliary strands, and signal strands used for the corresponding experiments. (B) Kinetics experiments of the plasmid-derived JoinAB using 80 °C heat inactivation (green traces), 0.15% sodium dodecyl sulfate (SDS) (red), and a control without heat inactivation or addition of SDS (blue). The standard concentration was 1x = 10 nM, and all auxiliary strands and input B were at 2x. The gate complex was 1.5x, and the experiments were performed at 35 °C in 1x Tris-acetate-EDTA buffer containing 12.5 mM Mg2+ (1x TAE/Mg2+). (This figure has been modified from Ref29.) Please click here to view a larger version of this figure.
Figure 6.Enzyme dissociation and circuit behavior. (A) A simplified representation of the gate, reporter, auxiliary strands, and signal strands used for the corresponding experiments. (B) Kinetics experiments of the plasmid-derived JoinAB using 80 °C heat inactivation (green traces), 0.15% sodium dodecyl sulfate (SDS) (red), and a control without heat inactivation or addition of SDS (blue). The standard concentration was 1x = 10 nM, and all auxiliary strands and input B were at 2x. The gate complex was 1.5x, and the experiments were performed at 35 °C in 1x Tris-acetate-EDTA buffer containing 12.5 mM Mg2+ (1x TAE/Mg2+). (This figure has been modified from Ref29.) Please click here to view a larger version of this figure.
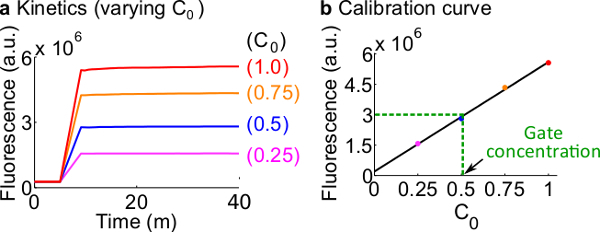 Figure 7.Reporter calibration. (A)ReporterCkinetics. The reporter concentration was at 3x (1x = 50 nM), and the initial concentration of signal C is indicated in the figure. (B) The fluorescence levels of signal C at the measurement end point (40 min) shows a linear relationship with the initial concentration of signal C. In a quantification example of ForkBC gate (green dashed line), the fluorescence value of ForkBC was measured as 3 x 106 (a.u.), which corresponds to 25 nM (0.5x) based on the calibration curve. Please click here to view a larger version of this figure.
Figure 7.Reporter calibration. (A)ReporterCkinetics. The reporter concentration was at 3x (1x = 50 nM), and the initial concentration of signal C is indicated in the figure. (B) The fluorescence levels of signal C at the measurement end point (40 min) shows a linear relationship with the initial concentration of signal C. In a quantification example of ForkBC gate (green dashed line), the fluorescence value of ForkBC was measured as 3 x 106 (a.u.), which corresponds to 25 nM (0.5x) based on the calibration curve. Please click here to view a larger version of this figure.
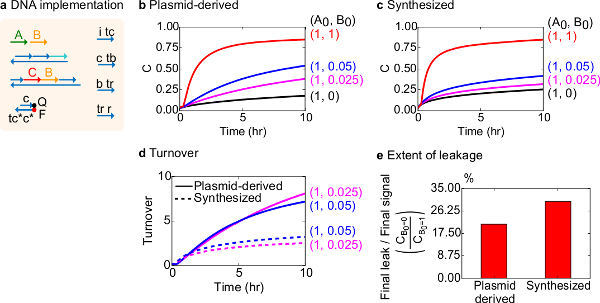 Figure 8. Bimolecular catalytic reaction kinetics (A+B->B+C). (A) A simplified representation of the gate, reporter, auxiliary strands, and signal strands used for the corresponding experiments. Experiments were run in 1x Tris-acetate-EDTA buffer containing 12.5 mM Mg2+ (1x TAE/Mg2+). All gate complexes were at 75 nM concentration (1.5x), and auxiliary strands were at 100 nM concentration (2x). Kinetics data for plasmid-derived gates and data for synthesized gates are shown in (B) and (C), respectively. Signal was at 50 nM (1x). Different amounts of signal (catalyst) were introduced in the system, and the reaction was tested at 35 °C. (D) Plasmid-derived gates exhibited higher turnover than synthesized DNA gates when low amounts of input were added. (E) Extent of leakage. The bar chart shows the ratio of the final leakage to the final signal (CB0=0/CB0=1) at the end points (10 hr). (This figure has been modified from Ref29.) Please click here to view a larger version of this figure.
Figure 8. Bimolecular catalytic reaction kinetics (A+B->B+C). (A) A simplified representation of the gate, reporter, auxiliary strands, and signal strands used for the corresponding experiments. Experiments were run in 1x Tris-acetate-EDTA buffer containing 12.5 mM Mg2+ (1x TAE/Mg2+). All gate complexes were at 75 nM concentration (1.5x), and auxiliary strands were at 100 nM concentration (2x). Kinetics data for plasmid-derived gates and data for synthesized gates are shown in (B) and (C), respectively. Signal was at 50 nM (1x). Different amounts of signal (catalyst) were introduced in the system, and the reaction was tested at 35 °C. (D) Plasmid-derived gates exhibited higher turnover than synthesized DNA gates when low amounts of input were added. (E) Extent of leakage. The bar chart shows the ratio of the final leakage to the final signal (CB0=0/CB0=1) at the end points (10 hr). (This figure has been modified from Ref29.) Please click here to view a larger version of this figure.
| Gate Templates | Sequences | Length (nt) |
| JoinAB | TCTAGTTCGATCAGAGCGTTATTACCAGTAGTCGATTGCTCAGCTGCTACATTGCTTCTACGAGTCATCCTTCCACCATTGCACCTTAGAGTCCGAATCCTACCATTGCTTAACCGAGTCTCACAACCAGCTGTCATTATGGACTTGACACACAGATTACACGGGAAAGTTGC | 173 |
| FORKBC | TCTAGTTCGATCAGAGCGTTATTACCAGTAGTCGATTGCTCAGCTGCCATCATAAGAGTCACCATACCCACATTGCCACATCGAGTCCCTTTTCCACCATTGCACCTTAGAGTCCGAATCCTACCATTGCTTAACCGAGTCTCACAACCAGCTGTCATTATGGACTTGACACACAGATTACACGGGAAAGTTGC | 194 |
Table 1. Sequences of ndsDNA gate templates.
| Gate | Strand | Length of bottom strand (nt) |
| JoinAB | JoinAB-Bottom, <a tb>, <b tr>, <r tq> | 87 |
| ForkBC | ForkBC-Bottom, <i>, <tc c>, <tb b>, <tr r> | 108 |
Table 2. Strands comprising JoinAB and ForkBC. (This table has been modified from Ref29.)
| Domain | Sequence | Length (nt) |
| ta | CTGCTA | 6 |
| tb | TTCCAC | 6 |
| tc | TACCCA | 6 |
| tr | TCCTAC | 6 |
| tq | AACCAG | 6 |
| a | CATTGCTTCTACGAGTCATCC | 21 |
| b | CATTGCACCTTAGAGTCCGAA | 21 |
| c | CATTGCCACATCGAGTCCCTT | 21 |
| r | CATTGCTTAACCGAGTCTCAC | 21 |
| i | CTGCCATCATAAGAGTCACCA | 21 |
Table 3. Domain level sequences for implementing the chemical reaction A+B -> B+C. (This table has been modified from Ref29.)
| Primer strand | Sequences | Length (nt) |
| Forward primer-1 | AAGAGAGACCACATGGTCCTTCTTGAGTTTGTAACAG CGTTATTACCAGTAGTCGATTGC | 60 |
| Reverse primer-1 | ACTACTATTTACTAATCCCATTGCGTGTTCTTATT TAATCTGTGTGTCAAGTCCATAATG | 60 |
| Forward primer-2 | AATAAGAACACGCAATGGGATTAGTAAATAGTAGT CGTTATTACCAGTAGTCGATTGC | 58 |
| Reverse primer-2 | GCGAAACTAGCTTGTGGTGATATTGTCTCGTGTGT TAATCTGTGTGTCAAGTCCATAATG | 60 |
| Forward primer-3 | ACACACGAGACAATATCACCACAAGCTAGTTTCGC CGTTATTACCAGTAGTCGATTGC | 58 |
| Reverse primer-3 | ACATTGTACGCCTAAATCATCAAGAATAATTGTTG TAATCTGTGTGTCAAGTCCATAATG | 60 |
| Forward primer-4 | CAACAATTATTCTTGATGATTTAGGCGTACAATGT CGTTATTACCAGTAGTCGATTGC | 58 |
| Reverse primer-4 | GAGCGCAGCGAGTCAGTGAGCGAGGAAGCCTGCAG TAATCTGTGTGTCAAGTCCATAATG | 60 |
Table 4. Primer sequences for the PCR of ndsDNA gate templates.
| Reagent | Volume for 1x reaction (µl) |
| High-copy plasmid backbone (~300 ng/µl) | 10 |
| PvuII-HF (20,000 units/ml) | 2 |
| PstI-HF (20,000 units/ml) | 2 |
| 10x Cut smart buffer | 2 |
| H2O | 4 |
| Total volume | 20 |
Table 5. Protocol for plasmid backbone digest.
| Reagent | Volume for 1x reaction (µl) |
| DNA vector (~50 ng/µl) | 1 |
| PCR amplified fragment-1 (~50 ng/µl) | 1 |
| PCR amplified fragment-2 (~50 ng/µl) | 1 |
| PCR amplified fragment-3 (~50 ng/µl) | 1 |
| PCR amplified fragment-4 (~50 ng/µl) | 1 |
| 2x Gibson Assembly master Mix | 5 |
| Total volume | 10 |
Table 6. Protocol for Gibson assembly.
| Reagent | Volume for 1x reaction (µl) |
| Plasmid DNA (~1 µg/µl concentration) | 1,000 |
| PvuII-HF (20,000 units/ml) | 200 |
| 10x Cut smart buffer | 133.3 |
| Total volume | 1333.3 |
Table 7. Protocol for ndsDNA gates inserted plasmid digest with Restriction enzyme PvuII-HF.
| Reagent | Volume (µl) |
| Join gates (~5 µg/µl concentration) | 150 |
| Nb.BsrDI (10,000 units/ml) | 300 |
| 10x Cut smart buffer | 50 |
| Total volume | 500 |
Table 8. Protocol for join gates digest with nicking enzyme Nb.BsrDI.
| Reagent | Volume (µl) |
| Fork gates (~5 µg/µl concentration) | 150 |
| Nt.BstNBI (10,000 units/ml) | 600 |
| 10x NEB buffer 3.1 | 83.3 |
| Total volume | 833.3 |
Table 9. Protocol for fork gates digest with nicking enzyme Nt.BstNBI.
| Strand | Domain | Sequence | Length (nt) |
| JoinAB-Bottom | tq* r* tr* b* tb* a* ta* | CTGGTT GTGAGACTCGGTTAAGCAATG GTAGGA TTCGGACTCTAAGGTGCAATG GTGGAA GGATGACTCGTAGAAGCAATG TAGCAG | 87 |
| FORKBC-Bottom | tq* r* tr* b* tb* c* tc* i* | CTGGTT GTGAGACTCGGTTAAGCAATG GTAGGA TTCGGACTCTAAGGTGCAATG GTGGAA AAGGGACTCGATGTGGCAATG TGGGTA TGGTGACTCTTATGATGGCAG | 108 |
| <ta a> | CTGCTA CATTGCTTCTACGAGTCATCC | 27 | |
| <tb b> | ta a | TTCCAC CATTGCACCTTAGAGTCCGAA | 27 |
| <tc c> | tb b | TACCCA CATTGCCACATCGAGTCCCTT | 27 |
| <a tb> | tc c | CATTGCTTCTACGAGTCATCC TTCCAC | 27 |
| <b tr> | a tb | CATTGCACCTTAGAGTCCGAA TCCTAC | 27 |
| <r tq> | b tr | CATTGCTTAACCGAGTCTCAC AACCAG | 27 |
| <i> | r tq | CTGCCATCATAAGAGTCACCA | 21 |
| <tr r> | i | TCCTAC CATTGCTTAACCGAGTCTCAC | 27 |
| <i tc> | tr r | CTGCCATCATAAGAGTCACCA TACCCA | 27 |
| <c tr> | i tc | CATTGCCACATCGAGTCCCTT TCCTAC | 27 |
| <c tb> | c tr | CATTGCCACATCGAGTCCCTT TTCCAC | 27 |
| <b tr> | c tb | CATTGCACCTTAGAGTCCGAA TCCTAC | 27 |
| <i tb> | b tr | CTGCCATCATAAGAGTCACCA TTCCAC | 27 |
| <b tb> | i tb | CATTGCACCTTAGAGTCCGAA TTCCAC | 27 |
| <b tc> | b tb | CATTGCACCTTAGAGTCCGAA TACCCA | 27 |
| <c tr> | b tc | CATTGCCACATCGAGTCCCTT TCCTAC | 27 |
| <b tr> | c tr | CATTGCACCTTAGAGTCCGAA TCCTAC | 27 |
| ROX-<c* tc*> | b tr | /56-ROXN/ AAGGGACTCGATGTGGCAATG TGGGTA | 27 |
| <c>-RQ | c* tc* | CATTGCCACATCGAGTCCCTT /3IAbRQSp/ | 21 |
| <tq* r*>-TAMRA | c | CTGGTT GTGAGACTCGGTTAAGCAATG /36-TAMTSp/ | 27 |
| RQ-<r> | tq* r* | /5IAbRQ/ CATTGCTTAACCGAGTCTCAC | 21 |
| r |
Table 10. Strand sequences for implementing the chemical reaction A+B -> B+C. (This table has been modified from Ref29.)
| Reagent | Volume (µl) | Final concentration |
| ROX-<c* tc*> at 100 µM | 10 | 10 µM (1x) |
| <c>-RQ at 100 µM | 13 | 13 µM (1.3x) |
| 10x TAE with 125 mM Mg2+ | 10 | 1x TAE with 12.5 mM Mg2+ |
| H2O | 67 | -- |
| Total volume | 100 | 10 µM (1x) |
Table 11. Protocol for assembling ReporterC.
| Reagent | Volume (µl) | Final concentration |
| H2O | 514 | - |
| 10x TAE with 125 mM Mg2+ | 60 | 1x TAE with 12.5 mM Mg2+ |
| PolyT at 300 µM | 2 | 1 µM |
| ReporterC at 10 µM | 9 | 150 nM (3x) |
| 10% SDS | 9 | 0.15% |
| <tc c> at 5 µM | 6 | 50 nM (1x) |
| Total volume | 600 | - |
Table 12. Protocol for the calibration of ReporterC. The volumes provided here is for a total reaction volume of 600 µl (corresponding to the use of a 0.875 ml synthetic quartz cell), but can be adjusted to work with different size cells.
| Reagent | Volume (µl) | Final concentration |
| H2O | 493 | - |
| 10x TAE with 125 mM Mg2+ | 60 | 1x TAE with 12.5 mM Mg2+ |
| polyT at 300 µM | 2 | 1 µM |
| ReporterC at 10 µM | 9 | 150 nM (3x) |
| <i tc> at 100 µM | 3 | 10x |
| <c tb> at 100 µM | 3 | 10x |
| <b tr> at 100 µM | 3 | 10x |
| 10% SDS | 9 | 0.15% |
| ForkBC at ~1 µM (concentration unknown) | 15 | ~0.5x |
| <r tq> at 100 µM | 3 | 10x |
| Total volume | 600 | - |
Table 13. Protocol for the calibration of ForkBC. The volumes provided here is for a total reaction volume of 600 µl, but can be adjusted to work with different size cells.
| Reagent | Volume (µl) | Final concentration | |
| H2O | 407.2 | - | |
| 10x TAE with 125 mM Mg2+ | 52.8 | 12.5 mM Mg2+ | |
| polyT at 300 µM | 2 | 1 µM | |
| ReporterC at 10 µM | 9 | 150 nM (3x) | |
| <i tc> at 10 µM | 6 | 100 nM (2x) | |
| <c tb> at 10 µM | 6 | 100 nM (2x) | |
| <b tr> at 10 µM | 6 | 100 nM (2x) | |
| <tr r> at 10 µM | 6 | 100 nM (2x) | |
| 10% SDS | 9 | 0.15% | |
| JoinAB at 1 µM | 45 | 75 nM (1.5x) | |
| ForkBC at 1 µM | 45 | 75 nM (1.5x) | |
| <ta a> at 10 µM | 3 | 50 nM (1x) | |
| <tb b> at 10 µM | 3 | 50 nM (1x) | |
| Total volume | 600 | - |
Table 14. Protocol for a chemical reaction A+B->B+C. The volumes provided here is for a total reaction volume of 600 µl, but can be adjusted to work with different size cells.
| Synthesized gates | Plasmid-derived gates | ||||
| Description | Cost | Join gates | Fork gates | ||
| PAGE purified long strand (100 nt; served as the bottom strands of a gate) | ~$75 | Description | Cost | Description | Cost |
| PAGE purified short strand (~30 nt, served as top strands of a gate) | ~$185 | Gate template | ~$100 | Gate template | ~$100 |
| Total | ~$260 | Plasmid extraction kit | ~$26 | Plasmid extraction kit | ~$26 |
| Restriction enzyme (PvuII-HF) | ~$11 | Restriction enzyme (PvuII-HF) | ~$11 | ||
| Nicking enzyme (Nt.BsrDI, Join gates) | ~$29 | Nicking enzyme (Nt.BstNBI, Fork gates) | ~$62 | ||
| Total | ~$166 | Total | ~$199 |
Table 15. Cost comparison between plasmid-derived gates and synthesized gates. (This table has been modified from Ref29.)
| Synthesized gates | Plasmid-derived gates | ||
| Processing | Processing time | Processing | Processing time |
| Annealing | 1 hr | Cloning | 5 hr |
| PAGE purification | 2 hr | Plasmid extraction | 2 hr |
| Total | 3 hr | Two steps of enzyme digestion | 0.5 hr |
| Ethanol precipitation | 1 hr | ||
| Total | 8.5 hr |
Table 16. Processing time comparison between plasmid-derived gates and synthetic gates. (This table has been modified from Ref29.)
Discussion
This paper describes a method for deriving ndsDNA gates from highly pure plasmid DNA. Moreover, a protocol is presented for characterizing gate performance using a fluorescence kinetics assay. Experimental data shows that the plasmid-derived system outperforms its synthetic counterpart even if the synthetic system is assembled from strands purified using polyacrylamide gel electrophoresis (PAGE). Likely, the improved performance of plasmid-derived gates is primarily due to the very high purity of the biological DNA. Synthetic DNA contains a variety of errors, in particular deletions that result in oligonucleotides of length n-1, and such side products are typically not completely removed in PAGE or high-performance liquid chromatography (HPLC) purification procedures. Similar improvements to the ones reported here were also observed in a previous study of a catalyzed hairpin amplifier that used DNA derived from biological sources21.
However, even the use of plasmid-derived gates cannot completely eliminate errors in the gate performance, for which there are at least two reasons: first over-digestion or lack of cut precision can lead to gates with too many nicks or nicks in the wrong positions. In either case, gates are more likely to participate in undesired reactions. Such problems may be eased by optimizing the amount of enzyme used (see Figure 4). Second, in these experiments, most inputs and auxiliary strands were synthetic DNA and thus contained deletions and mutations. In principle, all single-stranded input and auxiliary strands could also be obtained from phagemid DNA through a nicking enzyme digestion of the pre-encoded m13 viral genome26. Perhaps the circuit performance can be further improved using ssDNA derived from the bacterial genome.
While the use of plasmid-derived gates was found to improve circuit performance, an analysis of the cost and processing times revealed that while the production of plasmid-derived gates is slightly cheaper (Table 15), it takes 2-3 times longer processing time compared to assembly and purification of gates from commercially synthesized oligos (Table 16). The primary costs of plasmid-derived gates are gene synthesis and the use of restriction enzymes. For 300 pmole of gates (enough for 15 reactions at 30 nM), the estimated cost for Join gates is approximately $170 and $200 for Fork gates, the cost difference being due to the use of different nicking enzymes. In contrast, the chemical synthesis of the strands for the same gate costs around $260 including a PAGE purification fee. The primary time cost for plasmid-derived gates is in the cloning procedure, which, just like DNA synthesis, can be outsourced to a gene synthesis company. However, once assembled, plasmid-derived gates have the advantage that the host plasmids can easily be replicated and can be stored in the form of bacterial glycerol stocks. This makes it possible to reuse the gates many times over.
Looking forward, the improved performance of plasmid-derived gates could enable a much larger range of dynamics behaviors than have been experimentally demonstrated so far with DNA CRNs. For example, recent theoretical work47,48 suggested that self-organized spatial patterns at the macro-scale can be realized with DNA CRNs through a reaction diffusion mechanism. The method presented here provides a viable path for constructing the underlying molecular components for such self-patterning DNA materials. Though challenging, developing macro-scale morphologies in a programmable way would have significant implications in areas ranging from biomaterials research to regenerative medicine.
Disclosures
The authors declare no competing financial interests.
Acknowledgments
Figures 1, 2, 3, 4, 6, 8 and Tables 2, 3, 10, 15, 16 are modified from Ref29. This work was supported by the National Science Foundation (grant NSF-CCF 1117143 and NSF-CCF 1162141 to G.S.). Y.-J. C. was supported by Taiwanese Government Fellowships. S.D.R was supported by the National Science Foundation Graduate Research Fellowship Program (GRFP).
References
- Zhang DY, Seelig G. Dynamic DNA nanotechnology using strand-displacement reactions. Nat. Chem. 2011;3:103–113. doi: 10.1038/nchem.957. [DOI] [PubMed] [Google Scholar]
- Krishnan Y, Simmel FC. Nucleic acid based molecular devices. Angew. Chem. Int. Ed. Engl. 2011;50:3124–3156. doi: 10.1002/anie.200907223. [DOI] [PubMed] [Google Scholar]
- Zhang DY, Winfree E. Control of DNA strand displacement kinetics using toehold exchange. J. Am. Chem. Soc. 2009;131:17303–17314. doi: 10.1021/ja906987s. [DOI] [PubMed] [Google Scholar]
- Qian L, Winfree E, Bruck J. Neural network computation with DNA strand displacement cascades. Nature. 2011;475:368–372. doi: 10.1038/nature10262. [DOI] [PubMed] [Google Scholar]
- Qian L, Winfree E. Scaling up digital circuit computation with DNA strand displacement cascades. Science. 2011;332:1196–1201. doi: 10.1126/science.1200520. [DOI] [PubMed] [Google Scholar]
- Zadegan RM, Jepsen MD, Hildebrandt LL, Birkedal V, Kjems J. Construction of a fuzzy and boolean logic gates based on DNA. Small. 2015;11:1811–1817. doi: 10.1002/smll.201402755. [DOI] [PubMed] [Google Scholar]
- Seelig G, Soloveichik D, Zhang DY, Winfree E. Enzyme-free nucleic acid logic circuits. Science. 2006;314:1585–1588. doi: 10.1126/science.1132493. [DOI] [PubMed] [Google Scholar]
- Zadegan RM, et al. Construction of a 4 zeptoliters switchable 3D DNA box origami. ACS Nano. 2012;6:10050–10053. doi: 10.1021/nn303767b. [DOI] [PubMed] [Google Scholar]
- Andersen ES, et al. Self-assembly of a nanoscale DNA box with a controllable lid. Nature. 2009;459:73–76. doi: 10.1038/nature07971. [DOI] [PubMed] [Google Scholar]
- Zhang DY, Hariadi RF, Choi HM, Winfree E. Integrating DNA strand-displacement circuitry with DNA tile self-assembly. Nat. Commun. 1965;4 doi: 10.1038/ncomms2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurke B, Turberfield AJ, Mills AP, Simmel FC, Neumann JL. A DNA-fuelled molecular machine made of DNA. Nature. 2000;406:605–608. doi: 10.1038/35020524. [DOI] [PubMed] [Google Scholar]
- Green SJ, Lubrich D, Turberfield AJ. DNA hairpins: fuel for autonomous DNA devices. Biophys. J. 2006;91:2966–2975. doi: 10.1529/biophysj.106.084681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataraman S, Dirks RM, Rothemund PW, Winfree E, Pierce NA. An autonomous polymerization motor powered by DNA hybridization. Nat. Nanotechnol. 2007;2:490–494. doi: 10.1038/nnano.2007.225. [DOI] [PubMed] [Google Scholar]
- Green SJ, Bath J, Turberfield AJ. Coordinated chemomechanical cycles: a mechanism for autonomous molecular motion. Phys. Rev. Lett. 2008;101:238101. doi: 10.1103/PhysRevLett.101.238101. [DOI] [PubMed] [Google Scholar]
- Omabegho T, Sha R, Seeman NC. A bipedal DNA Brownian motor with coordinated legs. Science. 2009;324:67–71. doi: 10.1126/science.1170336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turberfield AJ, et al. DNA fuel for free-running nanomachines. Phys. Rev. Lett. 2003;90:118102. doi: 10.1103/PhysRevLett.90.118102. [DOI] [PubMed] [Google Scholar]
- Dirks RM, Pierce NA. Triggered amplification by hybridization chain reaction. Proc. Natl. Acad. Sci. U. S. A. 2004;101:15275–15278. doi: 10.1073/pnas.0407024101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelig G, Yurke B, Winfree E. Catalyzed relaxation of a metastable DNA fuel. J. Am. Chem. Soc. 2006;128:12211–12220. doi: 10.1021/ja0635635. [DOI] [PubMed] [Google Scholar]
- Zhang DY, Turberfield AJ, Yurke B, Winfree E. Engineering entropy-driven reactions and networks catalyzed by DNA. Science. 2007;318:1121–1125. doi: 10.1126/science.1148532. [DOI] [PubMed] [Google Scholar]
- Yin P, Choi HM, Calvert CR, Pierce NA. Programming biomolecular self-assembly pathways. Nature. 2008;451:318–322. doi: 10.1038/nature06451. [DOI] [PubMed] [Google Scholar]
- Chen X, Briggs N, McLain JR, Ellington AD. Stacking nonenzymatic circuits for high signal gain. Proc. Natl. Acad. Sci. U. S. A. 2013;110:5386–5391. doi: 10.1073/pnas.1222807110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips A, Cardelli L. A programming language for composable DNA circuits. J. R. Soc. Interface. 2009;6(Suppl 4):S419–S436. doi: 10.1098/rsif.2009.0072.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakin MR, Youssef S, Polo F, Emmott S, Phillips A. Visual DSD: a design and analysis tool for DNA strand displacement systems. Bioinformatics. 2011;27:3211–3213. doi: 10.1093/bioinformatics/btr543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakin MR, Youssef S, Cardelli L, Phillips A. Abstractions for DNA circuit design. J. R. Soc. Interface. 2012;9:470–486. doi: 10.1098/rsif.2011.0343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DY, Winfree E. Robustness and modularity properties of a non-covalent DNA catalytic reaction. Nucleic Acids Res. 2010;38:4182–4197. doi: 10.1093/nar/gkq088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducani C, Kaul C, Moche M, Shih WM, Hogberg B. Enzymatic production of 'monoclonal stoichiometric' single-stranded DNA oligonucleotides. Nat. Methods. 2013;10:647–652. doi: 10.1038/nmeth.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, et al. In vivo cloning of artificial DNA nanostructures. Proc. Natl. Acad. Sci. U. S. A. 2008;105:17626–17631. doi: 10.1073/pnas.0805416105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia D, et al. Icosahedral DNA nanocapsules by modular assembly. Angew. Chem. Int. Ed. Engl. 2009;48:4134–4137. doi: 10.1002/anie.200806000. [DOI] [PubMed] [Google Scholar]
- Chen YJ, et al. Programmable chemical controllers made from DNA. Nat. Nanotechnol. 2013;8:755–762. doi: 10.1038/nnano.2013.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkin A, Ross J. Computational functions in biochemical reaction networks. Biophys. J. 1994;67:560–578. doi: 10.1016/S0006-3495(94)80516-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Érdi P, Tóth J. Mathematical models of chemical reactions: theory and applications of deterministic and stochastic models. Manchester University Press; 1989. [Google Scholar]
- Magnasco MO. Chemical kinetics is Turing universal. Phys. Rev. Lett. 1997;78:1190. [Google Scholar]
- Oishi K, Klavins E. Biomolecular implementation of linear I/O systems. IET Syst. Biol. 2011;5:252–260. doi: 10.1049/iet-syb.2010.0056. [DOI] [PubMed] [Google Scholar]
- Senum P, Riedel M. Rate-independent constructs for chemical computation. PLoS One. 2011;6 doi: 10.1371/journal.pone.0021414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soloveichik D, Cook M, Winfree E, Bruck J. Computation with finite stochastic chemical reaction networks. Natural Computing. 2008;7:615–633. [Google Scholar]
- Soloveichik D, Seelig G, Winfree E. DNA as a universal substrate for chemical kinetics. Proc. Natl. Acad. Sci. U. S. A. 2010;107:5393–5398. doi: 10.1073/pnas.0909380107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson JJ, Chen KC, Novak B. Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signaling pathways in the cell. Curr. Opin. Cell. Biol. 2003;15:221–231. doi: 10.1016/s0955-0674(03)00017-6. [DOI] [PubMed] [Google Scholar]
- Cardelli L. Two-domain DNA strand displacement. Math. Struct. Comput. Sci. 2013;23:247–271. [Google Scholar]
- Angluin D, Aspnes J, Eisenstat D. A simple population protocol for fast robust approximate majority. Distrib. Comput. 2008;21:87–102. [Google Scholar]
- Cardelli L, Csikasz-Nagy A. The cell cycle switch computes approximate majority. Sci. Rep. 2012;2:656. doi: 10.1038/srep00656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zadeh JN, et al. NUPACK: Analysis and design of nucleic acid systems. J. Comput. Chem. 2011;32:170–173. doi: 10.1002/jcc.21596. [DOI] [PubMed] [Google Scholar]
- Lee PY, Costumbrado J, Hsu CY, Kim YH. Agarose gel electrophoresis for the separation of DNA fragments. J. Vis. Exp. 2012. [DOI] [PMC free article] [PubMed]
- Gibson DG, et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- Froger A, Hall JE. Transformation of plasmid DNA into E. coli using the heat shock method. J. Vis. Exp. 2007. p. e253. [DOI] [PMC free article] [PubMed]
- Lessard JC. Transformation of E. coli via electroporation. Methods Enzymol. 2013;529:321–327. doi: 10.1016/B978-0-12-418687-3.00027-6. [DOI] [PubMed] [Google Scholar]
- Nasri M, Thomas D. Alteration of the specificity of PvuII restriction endonuclease. Nucleic Acids Res. 1987;15:7677–7687. doi: 10.1093/nar/15.19.7677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalchau N, Seelig G, Phillips A. Computational design of reaction-diffusion patterns using DNA-based chemical reaction networks. DNA Computing and Molecular Programming. 2014. pp. 84–99.
- Scalise D, Schulman R. Designing modular reaction-diffusion programs for complex pattern formation. Technology. 2014;2:55–66. [Google Scholar]