

Abstract
Collection and processing of whole blood samples in a non-clinical setting offers a unique opportunity to evaluate community-dwelling individuals both with and without preexisting conditions. Rapid processing of these samples is essential to avoid degradation of key cellular components. Included here are methods for simultaneous peripheral blood mononuclear cell (PBMC), DNA, RNA and serum isolation from a single blood draw performed in the homes of consenting participants across a metropolitan area, with processing initiated within 2 hr of collection. We have used these techniques to process over 1,600 blood specimens yielding consistent, high quality material, which has subsequently been used in successful DNA methylation, genotyping, gene expression and flow cytometry analyses. Some of the methods employed are standard; however, when combined in the described manner, they enable efficient processing of samples from participants of population- and/or community-based studies who would not normally be evaluated in a clinical setting. Therefore, this protocol has the potential to obtain samples (and subsequently data) that are more representative of the general population.
Keywords: Medicine, Issue 105, PBMC isolation, whole blood, DNA isolation, RNA isolation, community-based study, non-clinical setting
Introduction
Multiple studies have characterized differences in gene expression, DNA methylation and cell subset in blood among individuals with and without mental (or other) illnesses1-4. These studies, however, have been obtained from clinical settings in which disease-associated differences may be magnified due to the generally more severe nature of the illnesses for which patients are seeking treatment. Due to advances in “omics” approaches, the past decade has seen an explosion of interest in obtaining biologic samples from community and/or epidemiologic settings5-7, in order to provide population-based estimates of disease prevalence and a broader picture of the environmental determinants of these mental and/or physical illnesses.
A key challenge in this regard is the requirement for rapid processing of the collected specimens. Degradation of mononuclear cells, key immune system components that are frequently used to assess the health of an individual, begins immediately upon blood draw with a significant decrease in recovery after 2 hr of collection8-10. To address this challenge, we present an optimized protocol in which multiple components of human whole blood are simultaneously isolated from samples obtained in homes of subjects living in a large metropolitan area. The protocol is based upon our compilation and modification of current techniques, including storage of all “extra” fractions in the event future techniques allow for further isolation/analyses. While alternative methods or kits may be employed in place of the individual methods described here, those outlined have proven to be a reliable and efficient means for processing samples in a high-throughput manner. High-quality fractions (PBMCs, DNA, serum, and RNA) of fresh blood can be produced within 2 hr of collection and all assay-ready specimens can be available within 2 days (Figure 1).
This protocol was developed to enable the efficient processing of samples collected from community-dwelling, adult residents of the city of Detroit for testing in the Detroit Neighborhood Health Study (DNHS; DA022720, RC1MH088283, DA022720-05-S1), a population-based study of the social and biological determinants of post-traumatic stress disorder (PTSD) and other mental illnesses. The prevalence of PTSD in Detroit is more than twice the national average11,12. Identifying biological determinants of PTSD in this population may help to develop appropriate pharmacologic and/or cognitive-behavioral interventions to aid those suffering from the disorder, both in this urban population, and in other high-risk populations (e.g., returning military veterans). Our laboratory, previously located at Wayne State University in Detroit, Michigan, was selected for processing based on our expertise in handling fresh tissue samples derived from a variety of sources, the necessity to begin processing the samples within 2 hr of collection, and our proximity to the collection sites. With this unique opportunity at hand, our goal was to optimize the processing for greatest yield of DNA, RNA, serum and peripheral blood mononuclear cells (PBMCs) from each specimen (a total of N=1,639 samples over 5 waves of specimen collection). The procedures outlined here can be performed simultaneously in a non-clinical setting, thus producing starting material (see Table 1 for average yields) for a multitude of downstream applications including microarray, epigenetic, real-time RT-PCR, and flow cytometry analyses.
 Figure 1. Overall work flow. The overall process depicted here includes the logistics of obtaining the blood specimens from identifying consenting participants to the blood draw itself. High-quality, fractions (peripheral blood mononuclear cells; PBMCs, DNA, serum, and RNA) of fresh whole blood can be produced within 2 hr of collection and all assay-ready specimens can be available within 2 days. Moreover, the fractions prepared through this method are suitable for long-term storage if samples are not to be tested immediately. The entire timeline outlined here could be completed in a single day (~5 hr total). However, such a day would be extremely labor intensive especially for a single technician with substantial experience with the techniques. Thus, we recommend dividing the procedures on Day 1 between at least two technicians and completing the RNA processing on Day 2. Please click here to view a larger version of this figure.
Figure 1. Overall work flow. The overall process depicted here includes the logistics of obtaining the blood specimens from identifying consenting participants to the blood draw itself. High-quality, fractions (peripheral blood mononuclear cells; PBMCs, DNA, serum, and RNA) of fresh whole blood can be produced within 2 hr of collection and all assay-ready specimens can be available within 2 days. Moreover, the fractions prepared through this method are suitable for long-term storage if samples are not to be tested immediately. The entire timeline outlined here could be completed in a single day (~5 hr total). However, such a day would be extremely labor intensive especially for a single technician with substantial experience with the techniques. Thus, we recommend dividing the procedures on Day 1 between at least two technicians and completing the RNA processing on Day 2. Please click here to view a larger version of this figure.
Protocol
The Detroit Neighborhood Health Study was reviewed and approved by the University of Michigan's Institutional Review Board. All participants provided informed consent prior to their participation in the study.
1. Overview
Properly document all stages from recruitment through analysis of endpoint data. Perform the protocols on Day 1 simultaneously, switching stages and overlapping as time permits. The sequence of stages is written to optimize performance efficiency. Figure 1 provides an overview of the entire process. Note: The term, “participant” is used throughout to signify the de-identified blood sample drawn from a consenting individual. The times in parentheses below indicate hands-on time. Examples of the tracking sheet and specimen log can be found in supplementary Tables S1 and S2, respectively. Samples are collected by phlebotomists in homes of individuals identified as consenting participants by the study coordinator and transported to the lab by a courier (see Supplementary Information (SI) 1 for further pre-processing details).
2. Day 1 Setup
Prepare 1x Phosphate-buffered saline (PBS).
Preheat a heat block to 56 ºC.
Prepare the protease (see SI 2) by aliquoting 20 µl into the appropriate number of microcentrifuge tubes for the day’s deliveries (2 per participant).
Ensure Buffers AW1 and AW2 (wash buffers provided in the DNA isolation kit that increase the purity of the DNA) are ready for use, adding 100% ethanol as indicated on the label, if necessary.
Cool down refrigerated centrifuge to 4 °C.
Replace the cap on the bottle of 1x PBS with the provided rubber septum cap and pierce the rubber septum with a transfer spike, retaining the screw cap on top of the transfer spike to prevent evaporation. Repeat with the bottle of RNA stabilization solution.
Following preparation of the buffers, centrifuge and heat block, document the time of vacutainer delivery on the sample tracking sheet (see Table SI).
3. Day 1: Processing Whole Blood Sample for DNA, PBMCs and Leukocyte RNA (2 hr)
- Overview
- Allot adequate time to minimize the delay between blood draw and processing start time. Begin processing of the Ficoll containing vacutainers within 2 hr of blood draw to avoid an increase in red blood cell contamination and a decrease in mononuclear cell recovery.
- Assume collection of 2 vacutainers containing Ficoll gel gradient, 2 K2 Ethylenediaminetetraacetic acid (EDTA) vacutainers, and 1 serum vacutainer per adult participant. Upon delivery of the vacutainers to the laboratory, have a technician sign the tracking sheet signifying acceptance of the samples. Begin processing immediately with the start time documented on a per vacutainer basis on the specimen log.
- Serum Isolation Stage 1 (1 min)
- Document the start time on the specimen log (see Table SI 2). Centrifuge (with brake and acceleration OFF) the 7.0 ml serum (red) vacutainer (1 per participant) using a swinging bucket rotor with aerosol caps (biosafety level 2; BSL2 certified) for 20 min, 1,300 x g, 4 ºC.
- DNA Isolation Stage 1 (15 min) Note: For further details on this isolation procedure, see manufacturer’s protocol13.
- Document the start time. In a BSL2 cell culture hood, invert the vacutainers containing the Ficoll gel 5 times then add 200 µl whole blood from the top of one of the vacutainers into each of two 20 µl aliquots of protease (400 µl total per participant). Leaving the protease + blood microcentrifuge tubes in the hood, continue to the centrifugation of the Ficoll containing vacutainers in step 3.4.1. Note: Use the vacutainer with the largest collection volume, keeping in mind the necessity of balancing the centrifuge (i.e., it may be necessary to remove 200 µl from each of the two vacutainers) when spinning the vacutainers in step 3.4.1. Additionally, to ensure proper separation during the processing of the blue vacutainers, the level of blood remaining in the vacutainer should not be less than 2.5 inches above the Ficoll layer.
- PBMC Isolation Stage 1 (1 min)
- Document the start time (must be within 2 hr of blood draw to avoid an increase in red blood cell contamination and a decrease in mononuclear cell recovery).Invert the Ficoll containing vacutainers 8-10 times. Centrifuge (with brake and acceleration OFF) the vacutainers (2 per participant) using a swinging bucket rotor with aerosol caps (BSL2 certified) for 30 min, 1,600 x g, 22 °C.
- DNA Isolation Stage 2 (20 min)
- Return to the hood and add 200 µl Buffer AL to each of the protease + blood microcentrifuge tubes (step 3.3.1). Cap, remove from hood, vortex 15 sec and flash spin.
- Incubate 56 ºC in a heat block, 10 min.
- Remove the tubes from the heat block. Flash spin. Return to the BSL2 hood. Add 200 µl 100% ethanol. Cap, remove from hood, vortex 15 sec and flash spin. The remaining steps can be completed outside of the hood.
- Apply lysate (within 30 min of step 3.5.3) to a labeled spin column (in a 2 ml collection tube). Close the cap to avoid cross contamination via aerosols. Centrifuge 6,000 x g, 1 min.
- Discard the collection tube containing the filtrate and place the spin column in a new 2 ml collection tube.
- Add 500 µl Buffer AW1 to the column without moistening the rim, close the cap, and centrifuge 6,000 x g, 1 min.
- Discard the collection tube containing the filtrate and place the spin column in a new 2 ml collection tube.
- Add 500 µl Buffer AW2 to the column without moistening the rim, close the cap, and centrifuge 20,000 x g, 3 min.
- Discard the collection tube containing the filtrate and place the spin column in a new 1.5 ml collection tube (not included in the kit), and centrifuge 20,000 x g, 1 min.
- Discard the collection tube containing the filtrate and place the spin column in a new 1.5 ml collection tube (not included in the kit).
- Add 200 µl Buffer AE or water to each column and incubate at RT for 5 min. Centrifuge 6,000 x g, 1 min.
- Repeat step 3.5.11 eluting into the same collection tube.
- Pool the eluted DNA from the 2 columns per participant. Total yield ~800 µl per participant.
- Quantify the samples using a spectrophotometer when time allows.
- Aliquot DNA as desired into 2 ml cryovials, documenting the concentration of each on the specimen log. Transfer cryovials to a cryobox and place at -80 °C for long term storage. Document the freezer start time.
- Leukocyte RNA Isolation Stage 1 (30 min) Note: Perform in a Biosafety Level 2 certified cell culture hood. For further details on this isolation see manufacturer’s instructions15.
- Pierce the rubber septum of the K2 EDTA vacutainer with a transfer spike. Retain the sheath and screw cap for use in step 3.6.11. Following BSL2 standard practices, take care to avoid exposure to bloodborne pathogens.
- Attach the white slip connector to the top of the transfer spike.
- Label the filter with the corresponding participant ID then connect the inlet (flared end) of the filter to the white slip connector.
- Attach a sheathed 25⅝ G needle to the outlet (tapered end) of the filter. Preparing the filter assembly prior to delivery expedites the process considerably and therefore, attaching the white slip connector to the transfer spike, adding the filter, then the sheathed needle and setting the assembly in a culture tube rack until use, is recommended.
- Following assembly of the K2 EDTA tube system, safely unsheathe the needle (use the end of a metal spatula). Stab the needle into an empty 10 ml evacuated blood collection tube (serum receiver tube) and invert the K2 EDTA vacutainer/filter/receiver tube assembly. Following BSL2 standard practices, take care to avoid exposure to bloodborne pathogens.
- Allow the blood to filter through until the wedge-shaped sections of the filter have cleared of blood. Filtration takes about 2 min and can be placed in a test tube rack during filtration.
- Remove the filter from the assembly. Leave the needle in the tube containing the filtrate and discard the entire assembly in a Sharps container.
- Attach a 5 ml syringe to the transfer spike on the bottle of 1x PBS, invert the bottle, and withdraw 3 ml.
- Attach the syringe with PBS to the inlet of the filter and flush the filter (3-5 drops per second). Collect the flow-through in a biological waste container. Detach the syringe from the filter without retracting the plunger.
- Following filtration and a PBS wash, withdraw 3 ml of the RNA stabilization agent using a new 5 ml syringe and the method described in step 3.6.8. Flush the filter as in step 3.6.9. The RNA stabilization agent should remain on the filter. Detach the syringe from the filter without retracting the plunger.
- Seal the filter inlet and outlet with the sheath and screw cap retained from the transfer spike leaving the filter saturated with RNA stabilization agent. The filter can be stored at this point. Store the filter at -80 ºC until time permits (~2 hr) to complete steps 5.3 to 5.4.7. Note: Filters stored at -80 ºC for up to a year after collection have been processed without a decrease in RNA quality.
- PBMC Isolation Stage 2 (30 min)
- Remove the Ficoll containing vacutainers from the centrifuge (step 3.4.1) and continue in a BSL2 hood. Vacutainers should display separation as in Figure 2. If not, see Table 2.
- Once the vacutainer is returned to the BSL2 hood, remove the stopper and withdraw 1.5 ml of the top, yellowish, plasma layer (Figure 2) using a serological pipette without getting close to the mononuclear (clear/white) layer. Transfer the plasma to a 5 ml cryovial — (Pool from 2 vacutainers — 1 participant). Log the volume collected. See step 3.7.6 for storage instructions. Note: The best separation and greatest PBMC yields come from participants who have fasted at least 12 hr prior to blood collection.
- Transfer the remaining plasma and the whitish, mononuclear layer (everything above the gel layer — Figure 2) using a serological pipette, to a 15 ml conical tube, pooling the mononuclear layer from each of the two Ficoll containing vacutainers per participant into one conical tube.
- Add 1x PBS to bring the total volume in the conical tube to 15 ml. Cap tube and invert 5 times. Centrifuge (with brake and acceleration OFF) 15 min, 300 x g, 22 °C.
- Return to the Ficoll containing vacutainers in the hood and collect the red blood cells (RBCs) by using a 5¾” Pasteur pipet to swirl around and loosen the outside of the Ficoll gel layer and remove it if possible. Use a serological pipet to collect and transfer the RBCs (~4.5 ml) to a 5 ml cryovial. Log the volume collected.
- Transfer both 5 ml cryovials (plasma in step 3.7.2 and RBCs in step 3.7.5) to a controlled rate freezing container and put at -80 ºC for at least 24 hr after which time they can be transferred to a cryobox and returned to -80 ºC for long term storage.
- Return the conical tube to the hood, when the centrifugation in step 3.7.4 is complete, and aspirate all but ~500 µl of the PBS without disturbing the pellet. PBMC yield is greater if ~200 µl of PBS is left above the pellet at this stage.
- Add fresh 1x PBS to bring the volume to 10 ml. Resuspend the pellet gently. Cap tube and invert 5 times. Centrifuge (with brake and acceleration OFF) 10 min, 300 x g, 22 °C.
- Serum Isolation Stage 2 (10 min) Note: Perform in a BSL2 hood.
- Aliquot the top serum layer from the serum vacutainer, following centrifugation (step 3.2.1), into 2 ml cryovials as desired. Typical yield is 2.5 ml (Table 1). Log the volume. For example, use 4 cryovials aliquoting 200 µl into cryovial 1, 1,000 µl into cryovial 2 and then divide the remaining into cryovials 3 and 4.
- Transfer the cryovials to a cryobox and place at -80 ºC for long-term storage. Document the freezer start time.
- PBMC Isolation Stage 3 (15 min)
- Return to the hood, after centrifugation (step 3.7.8), and aspirate as much supernatant/PBS as possible without disturbing the pellet. Resuspend pellet by pipetting in 2.5 ml PBMC Freezing Medium 1 (see SI 2).
- Add 2.5 ml PBMC Freezing Medium 2 (see SI 2) to the cell/medium solution in step 3.9.1. Vortex gently.
- Aliquot 10 µl of the cell solution into a 0.65 ml microcentrifuge tube (further dilution may be necessary). Add 10 µl of 0.4% trypan blue stain into the 0.65 ml microcentrifuge tube and mix by pipetting several times. For further details see the manufacturer's manual16.
- Pipet 10 µl of the mixture into a cell counting chamber slide and place slide into the cell counter within 3 min of mixing. Zoom in and focus the cells. Press the “Count Cells” to obtain PBMC count.
- Aliquot, if the viable PBMC number is above 3 million cells per milliliter (mc/ml), as desired into cryovials and continue to step 3.9.9. Store PBMCs in up to 5 cryovials at a concentration of at least 3 mc/ml each.
- If the viable PBMC number is below 3 mc/ml, calculate the total number of cells by multiplying the viable mc/ml by 5 ml. Divide that number by 4, 3, 2 or 1 ml so that there will be at least one tube with 3 mc/ml.
- Centrifuge the conical tube containing the cells/freezing medium solution for 5 min at 300 x g (brake and acceleration OFF). After centrifugation, aspirate the appropriate volume of freezing medium (supernatant) leaving the volume calculated above (4, 3, 2 or 1 ml).
- Resuspend the pellet in the remaining supernatant and aliquot at least 3 mc/ml into the appropriate number of cryovials (1-4) at 1 ml/cryovial. Final freezing medium is 10% Dimethyl Sulfoxide (DMSO)/20% Fetal Bovine Serum (FBS)/70% Roswell Park Memorial Institute (RPMI) 1640.
- Document the cell count per cryovial. Transfer the cryovials to a controlled rate freezing container and put at -80 ºC for at least 24 hr after which time the cryovials may be transferred to a cryobox and put in a liquid nitrogen tank (vapor phase) for long term storage. Document freezer start time.
4. Day 2 Setup
Heat an aliquot (220 µl per filter) of nuclease free 0.1 mM EDTA in a nuclease-free tube to 80 °C in a heat block.
Prepare wash solutions 1 and 2 (see SI 2).
5. Day 2: Long Term Sample Storage and Leukocyte Filter Processing (3 hr)
- Overview.
- Perform this procedure within 6 months of the application of the leukocytes to the filter. Note: Overall, Day 2 processing takes about 3 hr and while it is labeled “Day 2,” the key requirement is that the filter processing portion should be performed on a day when there is no Day 1 processing occurring in the lab.
- Long term sample storage (15 min)
- Perform this procedure every day, prior to the delivery of new vacutainers to the lab for Day 1 processing. Transfer the cryovials that were stored O/N in controlled rate freezing containers on Day 1 into appropriately labeled cryoboxes and return them to -80 ºC or liquid nitrogen (vapor phase) for long term storage. Organize the cryovials based on sample type (e.g., DNA, PBMC, RBCs, etc.) to expedite sample location tracking for endpoint assays.
- Leukocyte RNA Filters Processing (45 min). Note: For more information on this procedure see the manufacturer's instructions17.
- Bring the filter to RT (thawing about 5 min).
- Remove the sheath and screw cap from the filter. Retract the plunger of a 5 ml syringe and connect it to the inlet (flared end) of the filter, depress the plunger to expel the RNA stabilization agentfrom the filter ports into a biological waste container.
- Load a new 5 ml syringe with 4 ml of a phenol and guanidine isothiocyanate solution for RNA isolation and attach it to the inlet of the filter, depress the plunger to flush the solution through the filter, collecting the lysate in a labeled 15 ml conical tube (2 per participant).
- Disconnect the syringe from the filter, retract the plunger, re-attach it to the filter, and depress the plunger to expel residual sample trapped in the filter disk into the same 15 ml conical tube. Discard filter and syringe.
- Add 800 µl BCP to the conical tube, close the tube tightly and vigorously shake the prep for 30 sec.
- Incubate at RT for 5 min. Centrifuge for 10 min at 2,000 x g.
- Transfer the aqueous (top) phase to a freshly labeled 15 ml conical tube (~2.5 ml).
- Add 0.5 times the volume of the aqueous phase of nuclease-free water and mix well. Then add 1.25x the aqueous volume of 100% ethanol and mix again. Note: For a 2.5 ml aqueous volume, add 1.25 ml of nuclease free water, mix, then add 4.7 ml of 100% ethanol (2.5 ml x 0.5 = 1.25 ml nuclease free water THEN 2.5 ml + 1.25 ml = 3.75 ml new aqueous volume THEN 3.75 ml x 1.25 = 4.7 ml 100% ethanol). This step allows for isolation of total RNA that includes the small RNA fraction. A method to omit the small RNAs from the isolation can be found in the manual17.
- Remove the plunger from a 5 ml syringe and insert a spin cartridge in its place. Attach the cartridge/syringe assembly to a vacuum manifold. For a more secure connection to the vacuum manifold, place the cartridge/syringe assembly inside a 1.5 ml microcentrifuge tube with the bottom cut off.
- Apply the sample from step 5.3.8 to the spin cartridge slowly with the vacuum on, carefully adding more sample as it is pulled through the cartridge. Note: If no vacuum is present, see centrifugation method at http://www.ambion.com/techlib/misc/leuko_iso.pdf.
- Transfer the spin cartridge to a 1.5 ml microcentrifuge tube and add 750 µl of Wash 1. Centrifuge the spin cartridge/tube assembly 5 – 10 sec at 12,000 x g.
- Discard filtrate from the tube and return the spin cartridge to the same microcentrifuge tube.
- Add 750 µl of Wash 2 and centrifuge the spin cartridge/tube for 5 – 10 sec at 12,000 x g. Discard filtrate as in step 5.3.12. Return the spin cartridge to the same microcentrifuge tube.
- Repeat with another 750 µl of Wash 2 and centrifugation as in step 5.3.13. Discard filtrate. Return the spin cartridge to the same microcentrifuge tube. Centrifuge the spin cartridge/tube at maximum speed for 1 min to dry the filter.
- Transfer the spin cartridge to a fresh, labeled 1.5 µl microcentrifuge tube.
- Add 200 µl of nuclease-free 0.1 mM EDTA (preheated to 80 °C) to the center of the spin cartridge filter (2 per participant). Incubate at RT for 1 min.
- Centrifuge for 1 min at 12,000 x g to elute the RNA. Retain the filtrate. Discard spin cartridge.
- Split each 200 µl filtrate into two, 100 µl aliquots into fresh 1.5 ml microcentrifuge tubes, label and keep on ice. At this point, starting with 2 filters per participant will yield four 100 µl aliquots of RNA per participant.
- DNase treatment (1 hr) Note: For further details on this treatment see the manufacturer's protocol18.
- Create a master mix by combining 10 µl of DNase I Buffer and 2 µl rDNase I per aliquot + 1 (to account for pipetting error). Note: For four samples, 100 µl aliquots use 50 µl DNase I Buffer + 10 µl rDNase I.
- Aliquot 12 µl of the master mix in step 5.4.1 into each of the RNA aliquots from step 5.3.18 and mix gently. Incubate at 37 °C for 30 min in a heat block.
- Vortex the DNase Inactivation Reagent and add 11.2 µl to each aliquot, mix well. Incubate at RT for 2 min vortexing 2-3 times during incubation.
- Centrifuge at 10,000 x g for 1.5 min.
- Transfer the supernatant (RNA) to a fresh 1.5 ml tube. Aliquots from the same participant can be pooled here.
- Run each sample on a spectrophotometer to get the concentration. A quality analysis of the RNA is recommended using a Bioanalyzer (see step 5.5).
- Aliquot the RNA into cryovials as desired. If a Bioanalyzer analysis will not be performed immediately, aliquot 1.5 µl of sample into a microcentrifuge tube for later analysis. Document the concentrations and the freezer start time. Store samples at -80 °C.
- Bioanalyzer analysis (1 hr)
- Follow the manufacturer's protocol19. When the run is complete, the data is automatically stored, but save it again with a smaller, more recognizable file name. Expected results (Figure 3): the ladder should have 6 peaks, samples should have 3 peaks (2 ribosomal peaks at 44 sec and 50 sec, respectively and 1 early marker peak at 25 sec).
Representative Results
It is essential that the overall procedure produce high quality material for analysis in a multitude of downstream applications including gene expression via microarray and RT-PCR analysis, detection of epigenetic modifications, and cell subset variations. Table 1 indicates the average yield and quality of materials from each of the processes. Figure 3 provides an example of the quality output of the leukocyte RNA isolation and filter processing methods. The image on the upper left of Figure 3 is the gel image resulting from the capillary electrophoresis. Each lane should produce two distinct bands with minimal shadowing which would indicate degradation. The chromatograms below the gel provide an additional look at the level and type of degradation that can be determined based upon the location and size of the peaks. The RNA Integrity Number (RIN) is another quality measure that ranges from 1 (low; degraded) to 10 (high; pure, good quality RNA).
Such a high-throughput methodology lends itself to an occasional error, but there are several checkpoints throughout the various stages of processing to ensure quality control. Figure 2 displays the proper separation following centrifugation of the Ficoll containing vacutainer. There are several reasons a vacutainer may not display this separation including an error in centrifugation speed, low collection volume, or lack of a fasting participant as indicated in Table 2.
Subsets (n=100) of the specimens isolated using this procedure have been analyzed with success by HumanMethylation27 (HM27) DNA Analysis BeadChips including validation of those results by pyrosequencing11. Genotyping, targeted bisulfite pyrosequencing, and real-time RT-PCR has been successfully performed in the Wildman and Uddin laboratories20,21,22,23. Serum, isolated in parallel with the DNA isolation for both the Beadchip and genotyping analyses, was successfully analyzed for IL-6 and C-reactive protein activity24. T-cell subsets from the isolated PBMCs have been successfully analyzed by flow cytometry25-28. Additionally, RNA from the modified leukocyte procedure has been subjected to Genome Wide gene expression profiling 29.
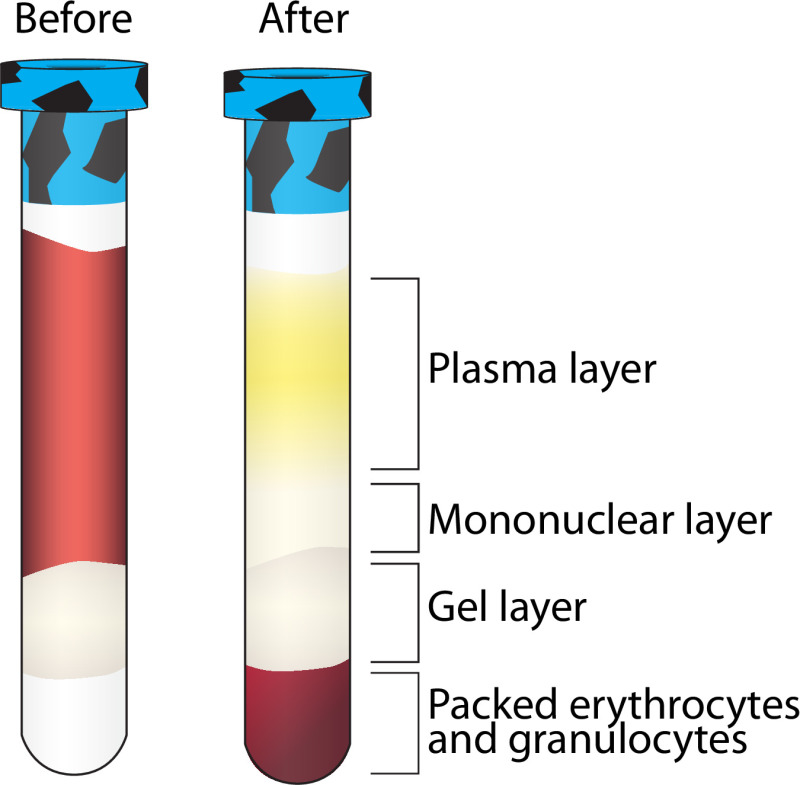 Figure 2. Layer separation following centrifugation of Ficoll containing vacutainer. A visualization of the separation of multiple blood components following centrifugation. The mononuclear layer is further purified while the other layers are stored at -80 °C. Please click here to view a larger version of this figure.
Figure 2. Layer separation following centrifugation of Ficoll containing vacutainer. A visualization of the separation of multiple blood components following centrifugation. The mononuclear layer is further purified while the other layers are stored at -80 °C. Please click here to view a larger version of this figure.
 Figure 3. Expected results from leukocyte RNA processing. Bioanalyzer results displaying two distinct bands representing 18S and 28S ribosomal RNA with RNA integrity numbers (RINs) above 8 isolated with the described leukocyte RNA isolation and filter processing methods (see protocol 3.6 and 5.3). Please click here to view a larger version of this figure.
Figure 3. Expected results from leukocyte RNA processing. Bioanalyzer results displaying two distinct bands representing 18S and 28S ribosomal RNA with RNA integrity numbers (RINs) above 8 isolated with the described leukocyte RNA isolation and filter processing methods (see protocol 3.6 and 5.3). Please click here to view a larger version of this figure.
| Total Average Yield (N≈500) | Average Quality | |
| Serum | 2.30 ml | N/A |
| DNA from whole blood | 39.97 µg | Absorbance at 260/280 = 1.74 |
| PBMC | 22.25 million viable cells | At least 95% viability of overall isolation |
| RNA from Leukocytes | 44.09 µg | Absorbance at 260/280 = 2.01 RNA Integrity Number (RIN) = 6.48 |
Table 1. Expected yields. Average quantity and quality of samples processed using the described methods.
| Problem | Potential Solution |
| No separation after Ficoll containing vacutainer centrifugation | Vacutainer is not filled to capacity — the minimum collection volume for adequate processing is 6 ml. |
| Check centrifuge settings, be sure the speed is in g-force. If incorrect, set to g-force and respin. | |
| Low PBMC count | Volume of blood draw too low. |
| Aspirated too close to the pellet in step 3.7.8 | |
| Non-fasting participant. | |
| No separation after serum vacutainer centrifugation | Check centrifuge settings, be sure the speed is in g-force. If incorrect, set to g-force and respin. |
| Unexpected concentration using the Nanodrop 1000 | Make sure the measurement is for the appropriate sample type (e.g., DNA or RNA). |
Table 2. Troubleshooting. Common problems encountered with the described methods and potential solutions.
 Supplementary Table 1. Tracking sheet. An example of tracking the vacutainers from blood collection to laboratory delivery. Please click here to view this table.
Supplementary Table 1. Tracking sheet. An example of tracking the vacutainers from blood collection to laboratory delivery. Please click here to view this table.
 Supplementary Table 2. Specimen log. An example of the document used to document the processing of the vacutainers upon delivery to the lab. Please click here to view this table.
Supplementary Table 2. Specimen log. An example of the document used to document the processing of the vacutainers upon delivery to the lab. Please click here to view this table.
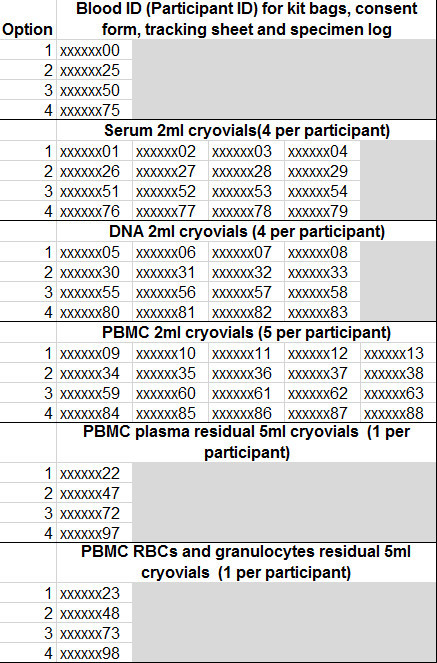 Supplementary Table 3. Cryovial Numbering System. An example of the numbering system used for each participant. Please click here to view this table.
Supplementary Table 3. Cryovial Numbering System. An example of the numbering system used for each participant. Please click here to view this table.
Supplementary Information 1. Preprocessing details. Details the duties of the Study Coordinator, Courier and Phlebotomist. Please click here to view supplementary information 1.
 Supplementary Information 2. Recipes. A list of recipes for the necessary reagents. Please click here to view supplementary information 2.
Supplementary Information 2. Recipes. A list of recipes for the necessary reagents. Please click here to view supplementary information 2.
Discussion
We have described a streamlined protocol that has been successfully applied to process more than 1,600 whole blood samples in the Detroit Neighborhood Health Study. Although many of these techniques are available in the existing literature, our step-by-step compilation, including precisely timed alterations between each step, reflects an optimized, efficient protocol that successfully produces a variety of biologic specimens with a wide range of downstream applications, including DNA methylation, mRNA expression and immunological analysis. These specimens have already been tested in a variety of experiments, results of which have been published in peer-reviewed literature and/or presented at national meetings11,20,21,23,24,29. This protocol should thus be of interest to other investigators seeking to collect biological specimens in population-based studies similar to the DNHS.
When processing samples in such a high-throughput manner, it is of utmost importance to maintain accurate records at all stages. We recommend developing a database to store all of the cryovial information at the outset. This database should include all aspects of the sample within each cryovial including the volume, concentration, quality, barcode, and storage location (storage box number and location within the box). We recommend preparing pre-labeled cryovials and storage boxes which contain a barcode. Further, we have found it convenient to have a “master” document containing the barcodes for each tube, specific to each participant (Table S3). This enables rapid input of the cryovial data into the database without excessive handling of the samples, introducing unnecessary freeze/thaw cycles to the samples and eliminates the potential for human error in entering in the barcodes manually. It is also essential that the labels adhere at extreme (e.g., vapor phase of liquid nitrogen -178 to -150 ºC) temperatures. The documentation can be time consuming, and with the necessity of efficient processing upon delivery, we have found that having at least two technicians divide the processes produces the greatest efficiency.
A limitation of this method is the proximity of the laboratory to the collection site. For PBMC isolation in particular, the samples must be processed within 2 hr of collection to avoid significant increases in red blood cell contamination and decreases in viable mononuclear cells. As such, the laboratory should be no more than 30 min from a collection site to account for any traffic related issues that may arise. In addition, any laboratory proposing to undertake the protocol outlined here will require two technicians on hand to ensure optimal sample processing. Furthermore, each laboratory must have access to the requisite equipment for each step outlined above. Thus, laboratories with fewer staff and/or limited equipment would likely be unable to undertake this protocol.
An advantage of this protocol is the ability to collect specimens directly in the homes of consenting individuals. This allows the study to reach individuals with mental or other health issues who would not typically seek medical help, perhaps because of a lack of insurance or transportation. It also the enables comparison of affected and unaffected individuals living within the same community who have experienced similar triggers, but differ in terms of their mental health symptoms. Obtaining specimens in this manner requires precise timing and coordination of phlebotomists in the field. Because our laboratory was located within the community we were studying, a phlebotomist could typically collect samples from two-three homes and deliver the samples to the laboratory within the 2 hr window of the first collection. Efficiency is key to our sample processing, and as such, we had multiple phlebotomists in the field, increasing the need for precise coordination. The laboratory received multiple blood deliveries with up to eight participants per delivery spaced 2 hr apart. The phlebotomists met at a designated location and combined their collections with only one phlebotomist delivering the specimens to the laboratory as a single batch. The methods described here can easily be adapted to study many other phenotypes and the biologic specimens collected can be used in a multitude of downstream assays.
Disclosures
The authors declare that they have no competing interests.
Acknowledgments
We would like to thank Henriette Mair-Meijers for invaluable attention to detail and hours devoted to processing the blood collections. We are grateful for the graphic design expertise of Natalie Jameson Kiesling. We also appreciate the approval of the manufacturers (Qiagen (Valencia, CA), BD Biosciences (San Jose, CA), Life Technologies (Grand Island, NY)) mentioned herein to publish the use of their products as described. Funding for this work was generously provided by the National Institutes of Health award numbers DA022720, RC1MH088283, and DA022720-05-S1.
References
- Hernandez ME, Martinez-Fong D, Perez-Tapia M, Estrada-Garcia I, Estrada-Parra S, Pavon L. Evaluation of the effect of selective serotonin-reuptake inhibitors on lymphocyte subsets in patients with a major depressive disorder. Eur Neuropsychopharmacol. 2010;20:88–95. doi: 10.1016/j.euroneuro.2009.11.005. [DOI] [PubMed] [Google Scholar]
- Weigelt K, et al. TREM-1 and DAP12 expression in monocytes of patients with severe psychiatric disorders EGR3, ATF3 and PU.1 as important transcription factors. Brain Behav Immun. 2011;25:1162–1169. doi: 10.1016/j.bbi.2011.03.006. [DOI] [PubMed] [Google Scholar]
- Robertson MJ, et al. Lymphocyte subset differences in patients with chronic fatigue syndrome, multiple sclerosis and major depression. Clin Exp Immunol. 2005;141:326–332. doi: 10.1111/j.1365-2249.2005.02833.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotter A, Asemann R, Decker A, Kornhuber J, Biermann T. Orexin expression and promoter-methylation in peripheral blood of patients suffering from major depressive disorder. J Affect Disord. 2011;131:186–192. doi: 10.1016/j.jad.2010.12.004. [DOI] [PubMed] [Google Scholar]
- Klengel T, et al. Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat Neurosci. 2013;16:33–41. doi: 10.1038/nn.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segman RH, Shefi N, Goltser-Dubner T, Friedman N, Kaminski N, Shalev AY. Peripheral blood mononuclear cell gene expression profiles identify emergent post-traumatic stress disorder among trauma survivors. Mol Psychiatry. 2005;10:500–513. doi: 10.1038/sj.mp.4001636. [DOI] [PubMed] [Google Scholar]
- Smith BH, et al. Cohort Profile: Generation Scotland: Scottish Family Health Study (GS:SFHS). The study, its participants and their potential for genetic research on health and illness. Int J Epidemiol. 2013;42:689–700. doi: 10.1093/ije/dys084. [DOI] [PubMed] [Google Scholar]
- Mallone R, et al. Isolation and preservation of peripheral blood mononuclear cells for analysis of islet antigen-reactive T cell responses: position statement of the T-Cell Workshop Committee of the Immunology of Diabetes Society. Clin Exp Immunol. 2011;163:33–49. doi: 10.1111/j.1365-2249.2010.04272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvigneau JC, Hartl RT, Teinfalt M, Gemeiner M. Delay in processing porcine whole blood affects cytokine expression. J Immunol Methods. 2003;272:11–21. doi: 10.1016/s0022-1759(02)00372-1. [DOI] [PubMed] [Google Scholar]
- Debey S, et al. Comparison of different isolation techniques prior gene expression profiling of blood derived cells: impact on physiological responses, on overall expression and the role of different cell types. Pharmacogenomics J. 2004;4:193–207. doi: 10.1038/sj.tpj.6500240. [DOI] [PubMed] [Google Scholar]
- Uddin M, et al. Epigenetic and immune function profiles associated with posttraumatic stress disorder. Proc Natl Acad Sci U S A. 2010;107:9470–9475. doi: 10.1073/pnas.0910794107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler RC, Wang PS. The descriptive epidemiology of commonly occurring mental disorders in the United States. Annu Rev Public Health. 2008;29:115–129. doi: 10.1146/annurev.publhealth.29.020907.090847. [DOI] [PubMed] [Google Scholar]
- Qiagen. QIAamp® DNA Mini and Blood Mini Handbook. Valencia, California, USA: 2010. Available from: https://www.qiagen.com/us/resources/resourcedetail?id=67893a91-946f-49b5-8033-394fa5d752ea. [Google Scholar]
- Thermo Scientific. NanoDrop 1000 Spectrophotometer. Waltham, Massachusetts, USA: 2010. Available from: http://www.nanodrop.com/Library/nd-1000-v3.8-users-manual-8. [Google Scholar]
- Life Technologies. LeukoLOCK™ Total RNA Isolation System. Grand Island, New York, USA: 2011. Available from: http://www.ambion.com/techlib/misc/leuko_iso.pdf. [Google Scholar]
- Life Technologies. Countess™ Automated Cell Counter. Grand Island, New York, USA: 2009. Available from: http://www.lifetechnologies.com/content/dam/LifeTech/migration/files/cell-analysis/pdfs.par.5257.file.dat/mp10227-countess. [Google Scholar]
- Life Technologies. Alternative Protocol for Extraction of RNA from Cells Captured on LeukoLOCK™ Filters Using TRI Reagent®. Grand Island, New York, USA: 2011. Available from: http://www.lifetechnologies.com/us/en/home/references/protocols/nucleic-acid-purification-and-analysis/rna-protocol/alternative-protocol-extraction-of-rna-from-cells-captured-on-leukolock-filters-using-tri-reagent.html. [Google Scholar]
- Life Technologies. DNA-free™ Kit. Grand Island, New York, USA: 2012. Available from: http://tools.lifetechnologies.com/content/sfs/manuals/cms_055739.pdf. [Google Scholar]
- Agilent Technologies. Agilent RNA 6000 Nano Kit Guide. Santa Clara, California, USA: 2006. Available from: http://www.genomics.agilent.com/files/Manual/G2938-90034_KitRNA6000Nano_ebook.pdf. [Google Scholar]
- Koenen KC, et al. SLC6A4 methylation modifies the effect of the number of traumatic events on risk for posttraumatic stress disorder. Depress Anxiety. 2011;28:639–647. doi: 10.1002/da.20825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh K, Uddin M, Soliven R, Wildman DE, Bradley B. Associations between the SS variant of 5-HTTLPR and PTSD among adults with histories of childhood emotional abuse: Results from two African American independent samples. J Affect Disord. 2014;161:91–96. doi: 10.1016/j.jad.2014.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustamante AC, et al. Childhood maltreatment is associated with epigenetic differences in hypothalamic-pituitary-adrenal (HPA) axis genes in the Detroit Neighborhood Health Study. American Association of Physical Anthropologists 82nd Annual Meeting; Knoxville, TN. Comprehensive Psychiatry; 2013. [Google Scholar]
- Sipahi L, et al. Longitudinal epigenetic variation of DNA methyltransferase genes is associated with vulnerability to post-traumatic stress disorder. Psychol Med. 2014;44:3165–3179. doi: 10.1017/S0033291714000968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddin M, Koenen KC, Aiello AE, Wildman D, de los Santos R, Galea S. Epigenetic and inflammatory marker profiles associated with depression in a community-based epidemiologic sample. Psychol Med. 2011;41:997–1007. doi: 10.1017/S0033291710001674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddin M, et al. Post-traumatic stress disorder is associated with immunosenescent T cell phenotypes in the Detroit Neighborhood Health Study. 3rd North American Congress of Epidemiology; Montreal, QC. 2011. [Google Scholar]
- Uddin M, et al. Post-traumatic stress disorder is associated with immunosenescent T cell phenotypes in the Detroit Neighborhood Health Study. 101st Annual Conference of the American Psychopathological Association; New York, NY. 2011. [Google Scholar]
- Uddin M. Biological signatures of post-traumatic stress disorder in the Detroit Neighborhood Health Study. 9th International Conference on Urban Health; New York Academy of Medicine. 2010. [Google Scholar]
- Aiello AE. Cytomegalovirus antibodies as a marker of immunosenescence in the Detroit Neighborhood Health Study. 9th International Conference on Urban Health; New York Academy of Medicine. 2010. [Google Scholar]
- Bustamante AC, et al. Distinct gene expression profiles characterize lifetime PTSD and childhood maltreatment in the Detroit Neighborhood Health Study. Philadelphia, PA: International Society for Traumatic Stress Studies; 2013. [Google Scholar]