

Abstract
A plant’s cell surface is its interface for perceiving environmental cues; it responds with cell biological changes such as membrane trafficking and cytoskeletal rearrangement. Real-time and high-resolution image analysis of such intracellular events will increase the understanding of plant cell biology at the molecular level. Variable angle epifluorescence microscopy (VAEM) is an emerging technique that provides high-quality, time-lapse images of fluorescently-labeled proteins on the plant cell surface. In this article, practical procedures are described for VAEM specimen preparation, adjustment of the VAEM optical system, movie capturing and image analysis. As an example of VAEM observation, representative results are presented on the dynamics of PATROL1. This is a protein essential for stomatal movement, thought to be involved in proton pump delivery to plasma membranes in the stomatal complex of Arabidopsis thaliana. VAEM real-time observation of guard cells and subsidiary cells in A. thaliana cotyledons showed that fluorescently-tagged PATROL1 appeared as dot-like structures on plasma membranes for several seconds and then disappeared. Kymograph analysis of VAEM movie data determined the time distribution of the presence (termed ‘residence time’) of the dot-like structures. The use of VAEM is discussed in the context of this example.
Keywords: Plant Biology, Issue 106, Arabidopsis thaliana, Cell surface, Exocytosis, Fluorescent proteins, Image analysis, Kymograph, Membrane trafficking, Plant cell biology, Real-time imaging, Stomata, Total internal reflection microscopy, Variable-angle epifluorescence microscopy
Introduction
The plant cell surface, including the plasma membrane and its immediately adjacent cytoplasm, is the main region of a plant cell’s perception and integration of biotic and abiotic cues from the extracellular environment. In response to these cues, cell surface components including plasma membrane proteins and the cortical cytoskeleton undergo dynamic changes, on a time scale of seconds to minutes1-4. Thus, real-time and high-resolution imaging of fluorescent proteins on the cell surface can illuminate a plant’s responses to environmental cues at the molecular level.
Confocal laser scanning microscopy is a powerful tool for determination of fluorescently-tagged protein localization3, however, it is often difficult to monitor the real-time protein dynamics because of its relatively long capturing times. An emerging technique for real-time monitoring of proteins in the plant cell is variable angle epifluorescence microscopy (VAEM), which is an adaptation of equipment usually used for total internal reflection fluorescence (TIRF) microscopy. In TIRF microscopy, the fluorescence-excitation light source is an evanescent light field that is generated when the entry angle of the laser is shallow enough to totally internally reflect light at the glass–water interface. The penetration depth of the evanescent light field is around 100 nm. TIRF microscopy is an outstanding tool for single molecule imaging, such as the detection of exocytosis in animal cells5. However, evanescent light cannot reach plasma membranes or the cortical cytoplasm in plant cells, because they have thick cell walls. Recently, TIRF microscopy equipment has been adapted by plant cell biologists, observing that a laser, if angled slightly more deeply than when being used to induce total internal reflection phenomena, could excite the surface of plant cell samples, resulting in high-quality plant cell imaging6,7. The excitation illumination depth is varied by adjusting the entry angle of the laser; therefore, this technique is described as VAEM. This optical system is also called variable angle TIRF microscopy (VA-TIRFM) because there is a possibility that total reflection may take place at the cell wall-periplasm interface7, however, the term VAEM is used in this article, as per the first report in plants6.
The goal of this protocol is to demonstrate practical procedures for using VAEM to visualize fluorescently-tagged protein dynamics on plant cell surfaces. Additionally, an image analysis protocol to quantify the residence time (duration of presence) of molecules is described for VAEM movie analysis. GFP-PATROL1 dot blinking on stomatal complex cells in Arabidopsis thaliana cotyledons is used as an example. PATROL1 was identified by forward genetic approaches as a causal gene of a stomatal response defect mutant in A. thaliana8. PATROL1 is a plant homolog of MUNC-13, which is a priming factor in synapse vesicle exocytosis8. In response to environmental cues, such as light or humidity, it is thought that PATROL1 reversibly regulates the delivery of a proton pump to plasma membranes in the stomatal complex. Stomatal complexes each comprise a pair of guard cells8 and subsidiary cells9, and they require a proton pump for stomatal movement. In these cells, GFP-tagged PATROL1 localizes to dot-like structures that remain on the plasma membrane for less than 1 min9.
Protocol
1. Preparation of Seedlings
- Sterilize the seeds.
- Prepare the sterilization solution by adding 500 µl NaClO (available chlorine: 5.0%) and 1 µl 10% Triton X-100 to 500 µl sterile water.
- Place ca. 10 transgenic A. thaliana seeds carrying GFP-PATROL18 into a 1.5-ml tube.
- Add 1 ml 70% ethanol solution, and mix well by inverting five times. Leave for 1 min.
- Observe the seeds sink to the bottom of the tube. In a clean laminar flow cabinet, gently remove the 70% ethanol using a micropipette, and add 1 ml sterilization solution. Mix well by inverting five times and leave for 5 min.
- Wash the seeds. Still working under aseptic conditions on a clean bench, gently remove the solution using a micropipette, and add 1 ml sterile water. Repeat this five times. The sterilized seeds may be stored in sterile water at 4 °C for 2 days.
Sow the sterilized seeds on a 0.5% gellan gum-solidified 1/2 Murashige and Skoog medium plate (pH 5.8)9. Tape the lid onto the plate using two layers of surgical tape.
Incubate the plate in the dark at 4 °C with a cold storage chamber, O/N.
Transfer the plate into a growth chamber set at 23.5 °C with a 12-hr/12-hr light-dark cycle using 100 µmol m−2 sec−1 white lights, and incubate for 7 days. Seedlings with cotyledons of around 1 mm long can then be harvested.
2. Sky Drop Mounting of Cotyledon Specimens
NOTE: An important factor in specimen preparation for VAEM observation is avoiding the inclusion of air bubbles between the specimen and the cover glass. Bubbles greatly reduce the image quality of VAEM by causing differences in the refractive index. A simple method, which we have called ‘sky drop’ mounting, can be used to avoid bubbles between the A. thaliana cotyledons and the cover glass. This should be done immediately prior to observation.
Place 30 µl of basal buffer [5 mM 2-(N-morpholino)-ethanesulfonic acid-Tris (MES-Tris) at pH 6.5, 50 mM KCl, 100 µM CaCl2] on the center of a glass slide (size: 76×26 mm, thickness: 1.0–1.2 mm).
Remove a cotyledon from a 7-day-old seedling using dissecting scissors. Float the cotyledon with the observation side facing up on the basal buffer drop (Figure 1, step 1).
Place 30 µl of basal buffer on the center of a cover glass (size: 18×18 mm, thickness: 0.12–0.17 mm) (Figure 1, step 2). Turn the cover glass upside down gently. Surface tension will prevent the buffer drop from falling off. Hold the cover glass with tweezers on one edge. Place the opposite edge on the glass slide so that the buffer drop is approximately above the cotyledon.
With the edge of the cover glass still on the slide, and still holding the opposite edge with tweezers, adjust the position of the buffer drop under the cover glass so that it is directly above the cotyledon sample (Figure 1, step 3). Let go of the cover glass. Mount a drop on the sample resulting in a preparation without air bubbles.
Wipe off excess buffer using lint-free tissues. Observe the preparation immediately (see step 3). NOTE: There is no need to seal the samples.
3. VAEM Observation and Movie Acquisition
NOTE : The TIRF microscope system9 used in the present study is described as follows: An inverted microscope is equipped with a TIRF unit and a TIRF objective lens with a numerical aperture of 1.49. For computerized control of the laser entry angle, a control box is used. Green fluorescent protein (GFP) is excited with a 488 nm optically pumped semiconductor laser, and the fluorescence is detected through a 510–550 nm band-pass filter to prevent autofluorescence from chloroplasts. The measured maximum value of fiber output power is 13.0–13.5 mW. For detection, an electron multiplying charge-coupled device (EM-CCD) camera head system and a C-mount camera magnification change unit are used.
- Calibrate the laser centering and focusing according to the manufacturer’s instructions. NOTE: This step is crucial for precise VAEM observations. It is strongly recommended that periodic checks of the laser path adjustment procedures, appropriate to your microscope, are carried out.
- Determine the central position illuminated with an objective lens-free light path on the ceiling of the microscopy room. To mark the central position, put a colored circle seal on the ceiling (Figure 2, step 1, 2).
- Illuminate the ceiling with an objective lens (Figure 2, step 3, 4). Move the illuminated region to the center position (Figure 2, step 5). Focus the laser (Figure 2, step 6). Fine-tune the position of the focused laser to the center (Figure 2, step 7).
Set a specimen on the stage of the microscope and select cells for observation using bright field illumination.
Check that the fluorescent protein can be observed in the cells, and set the z-axis position at the cell surface using epifluorescence illumination (Figure 3A).
- Perform VAEM observations.
- Incline the entry angle of the laser beam gradually with the controller box. At the same time, monitor the live image carefully. Initially, the image will be blurred (Figure 3B).
- As the laser angle increases, the VAEM image will become less blurred, eventually producing a clear image (Figure 3C and D). At this point, stop increasing the laser angle. If the fluorescence signal is lost, decrease to a shallower angle.
- Fine-tune the laser entry angle to obtain a better image. Fine-tuning of the z-axis position may also improve the image. If necessary, adjust the optical parameters (laser power, wavelength and filter set) and image sensor parameters [image size, exposure time, gain, digitizer and electron-multiplying (EM) gain]. NOTE: In the case of the TIRF microscope equipment described above, representative parameters are as follows. Magnification of lens just in front of the camera: 2X; Laser output: 1.0 mW; Image size: 512 × 512 pixels; Exposure time: 100 msec; Gain: 5×; Digitizer: 11 MHz; and EM gain: 100.
Acquire a movie as a multi-page TIFF image file using the commercial microscope software. Here, the movie is of GFP-labeled dots blinking on the surface of stomatal cells. NOTE: Representative image acquisition conditions are as follows. Acquisition Mode: Stream to RAM; Number of frames: 600; and multi-page TIFF file size: ~302 MB.
4. Kymograph Analysis for Quantification of GFP-labeled Dot Residence Time Using Fiji Software
Install Fiji (‘Fiji is Just ImageJ’) software10 using the authors’ instructions (http://fiji.sc/Fiji).
Run Fiji and open the acquired multi-page TIFF file using the Fiji menu “File-Open”.
Place a line on the site of interest, using the Fiji tool bar menu “Straight Line Selection Tool”. Optionally, use the “Segmented Line Selection Tool” or “Freehand Line Selection Tool”. In the results presented here, a straight line of 100 pixels (corresponding to 8.0 µm) was placed on a subsidiary cell (Figure 4A).
Make a kymograph image using the Fiji menu “Image-Stacks-Dynamic Reslice” (http://fiji.sc/Dynamic_Reslice) (Figure 4B). Optionally, "Flip Vertically” and “Rotate 90 Degrees” in a check box window are also available (not used in the results presented here). The x- and y-axis in the kymograph image indicate the line position (100 pixels corresponding to 8.0 µm) and observation time (600 pixels corresponding to 60 sec), respectively. If the kymograph image is not satisfactory, the position and type (straight, segmented and freehand) of the line on the multi-page image may be changed. As the line changes, the kymograph image dynamically changes. Save a kymograph image as a TIFF file using the Fiji menu “File-Save As-Tiff”.
- Measure the length of the blob in the orientation of time axis.
- To perform noise reduction, apply a Gaussian filter to the kymograph images using the Fiji menu “Process-Filters-Gaussian Blur”. The “Sigma (Radius)” parameter is tunable (1 pixel radius is used for the presented results).
- Segment the signal regions by thresholding, using the Fiji menu “Image-Adjust-Threshold”. NOTE: There are several thresholding algorithms to choose from. In presented results, the “Yen” algorithm was chosen (Figure 4C).
- Prepare to measure the time (y)-axis length of the blob using the menu “Analyze-Set Measurements”. Check the “Bounding rectangle” in the “Set Measurements” window. Ideally the area set should be as small as possible, to exclude noise. Here, the minimum area was set at 50 pixels. Measure the time (y)-axis length of the blob using the menu “Analyze-Analyze Particles”. To obtain the result values and prove credibility of the measurement regions, check the “Display results” and “Add to Manager” boxes.
- Values in the “Height” column are the time (y)-axis lengths for the measured blob (Figure 4D). Save the values using the Results table menu “File-Save As” and calculate and process the dataset of blob image durations using a statistical package and/or spreadsheet software (Figure 4E).
Representative Results
In this video article, the protocols for VAEM observation of GFP-PATROL1 in A. thaliana cotyledon stomatal complex cells are provided. Sky drop mounting is a simple preparation method that may help reduce the occurrence of air bubbles in VAEM preparations of A. thaliana cotyledons (Figure 1). Overtilting of the entry laser and/or z-positioning of specimens for VAEM will provide an unclear image. If that happens, it is recommended to start again from a position immediately above the sample, as judged by epifluorescence illumination. After a few days’ experience of VAEM observation, it should be possible to acquire VAEM movies successfully and routinely, such as the one shown here of GFP-PATROL1 dot blinking. Using the kymograph analysis presented in Figure 4, the residence times of 185 GFP-PATROL1 dots from 12 independent subsidiary cells in A. thaliana cotyledons were measured. The fitted density function revealed that most GFP-PATROL1 dots were resident in a subsidiary cell for between 2 and 10 sec, with a peak of around 3.5 sec (Figure 4E), suggesting fast exocytosis/endocytosis of proton pumps on the subsidiary cell surface.
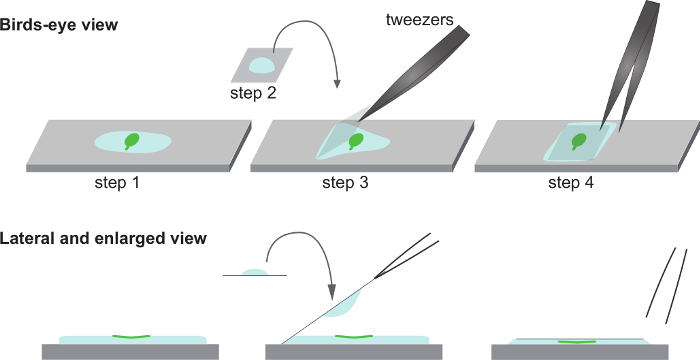 Figure 1. Schematic of the sky drop mounting method. Step 1: Specimen Arabidopsis thaliana cotyledons are floated on a drop of basal buffer on a glass slide. Step 2: A drop of basal buffer drop is placed on a cover glass. Step 3: The cover glass with the drop is turned upside down and the drop is placed above the specimen on the glass slide. Step 4: The cover glass is released and the drop falls on to the specimen. (Top) Birds-eye view. (Bottom) Lateral and enlarged view. Please click here to view a larger version of this figure.
Figure 1. Schematic of the sky drop mounting method. Step 1: Specimen Arabidopsis thaliana cotyledons are floated on a drop of basal buffer on a glass slide. Step 2: A drop of basal buffer drop is placed on a cover glass. Step 3: The cover glass with the drop is turned upside down and the drop is placed above the specimen on the glass slide. Step 4: The cover glass is released and the drop falls on to the specimen. (Top) Birds-eye view. (Bottom) Lateral and enlarged view. Please click here to view a larger version of this figure.
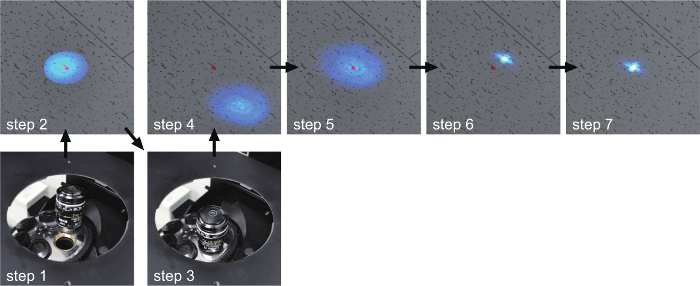 Figure 2. Calibration of the laser centering and focusing. Step 1: The empty position of the microscope revolver is selected to send laser light onto the ceiling above the microscope. Step 2: The central position is marked using a colored circle seal on the ceiling. Step 3: An objective is selected. Step 4: Check the position illuminated by the laser light through the objective lens. Step 5: The illumination is adjusted to the marked central position. Step 6: The laser light is focused. Step 7: The focused laser light is moved to the marked central position. Please click here to view a larger version of this figure.
Figure 2. Calibration of the laser centering and focusing. Step 1: The empty position of the microscope revolver is selected to send laser light onto the ceiling above the microscope. Step 2: The central position is marked using a colored circle seal on the ceiling. Step 3: An objective is selected. Step 4: Check the position illuminated by the laser light through the objective lens. Step 5: The illumination is adjusted to the marked central position. Step 6: The laser light is focused. Step 7: The focused laser light is moved to the marked central position. Please click here to view a larger version of this figure.
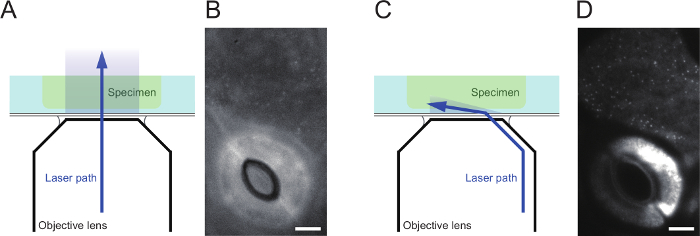 Figure 3. Comparison of epifluorescence microscopy and variable angle epifluorescence microscopy (VAEM). (A) Schematic of the light path of the laser in epifluorescence microscopy. (B) Representative epifluorescence image of GFP-PATROL1 at cotyledon surfaces in 7-day-old seedlings of Arabidopsis thaliana. Scale bar indicates 5 µm. (C) Schematic of the light path of the laser in VAEM. Note that the laser’s entry is tilted, and it illuminates the specimen’s surface with variable depth. (D) VAEM images of the same region as shown in (B). Scale bar indicates 5 µm. Please click here to view a larger version of this figure.
Figure 3. Comparison of epifluorescence microscopy and variable angle epifluorescence microscopy (VAEM). (A) Schematic of the light path of the laser in epifluorescence microscopy. (B) Representative epifluorescence image of GFP-PATROL1 at cotyledon surfaces in 7-day-old seedlings of Arabidopsis thaliana. Scale bar indicates 5 µm. (C) Schematic of the light path of the laser in VAEM. Note that the laser’s entry is tilted, and it illuminates the specimen’s surface with variable depth. (D) VAEM images of the same region as shown in (B). Scale bar indicates 5 µm. Please click here to view a larger version of this figure.
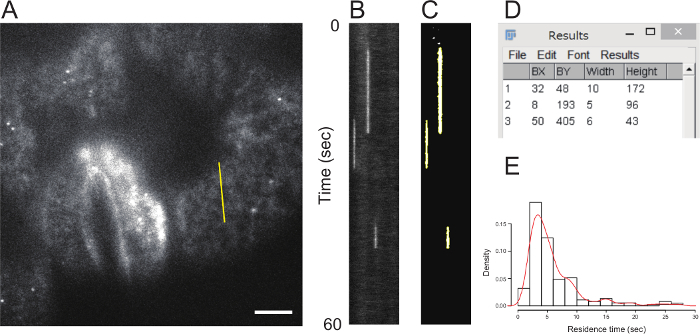 Figure 4. Kymograph analysis to quantify a GFP-PATROL1 dot’s residence time. (A) VAEM image with a straight line placed to make a kymograph. Scale bar indicates 5 µm. (B) A kymograph image along the line shown in (A). (C) A binary image of (B) based on Yen’s algorithm, generated in Fiji software. Yellow lines show the automatically-detected region of GFP-PATROL1 signal. (D) Screenshot of results table of the Fiji “Analyze Particles” function. (E) A histogram of GFP-PATROL1 dot’s residence time in subsidiary cells. The red line shows the density function. Please click here to view a larger version of this figure.
Figure 4. Kymograph analysis to quantify a GFP-PATROL1 dot’s residence time. (A) VAEM image with a straight line placed to make a kymograph. Scale bar indicates 5 µm. (B) A kymograph image along the line shown in (A). (C) A binary image of (B) based on Yen’s algorithm, generated in Fiji software. Yellow lines show the automatically-detected region of GFP-PATROL1 signal. (D) Screenshot of results table of the Fiji “Analyze Particles” function. (E) A histogram of GFP-PATROL1 dot’s residence time in subsidiary cells. The red line shows the density function. Please click here to view a larger version of this figure.
Discussion
In this video article, protocols are given for monitoring and measuring the dynamic behavior of GFP-PATROL1 dots on the stomatal complex of Arabidopsis thaliana. As shown here, VAEM observation is a powerful tool for live imaging of plant cell surfaces. Under the experimental conditions used here for GFP-PATROL1 monitoring, there was very little fluorescence photobleaching in the sample used for 1 min of video capture, because the highly sensitive EM-CCD permits the use of a relatively weak excitation laser in VAEM optics. Laser centering and focusing, before the beginning of each experiment, are important for successful VAEM observation. Users should be trained by professional staff from the chosen microscope company before using VAEM.
VAEM reveals that GFP-PATROL1 has a dynamic localization with an immobile dot-like compartment blinking on the plasma membrane. The fluorescently-labeled plasma membrane proton pump AHA1 mis-localizes in patrol1 mutant guard cells and subsidiary cells, as previously reported8,9. Thus, the VAEM-visualized GFP-PATROL1 dot dynamics may represent the delivery of the proton pump into the plasma membrane. This dynamic behavior of GFP-PATROL1 is difficult to visualize using conventional galvanometer mirror-type confocal laser scanning microscopy, indicating the usefulness of VAEM in uncovering real-time protein dynamics on plant cell surfaces. GFP-PATROL1 observation is just one example of VAEM application in plant cell biology. VAEM would be applicable to the monitoring of numerous events in the thin cortical layer of cytoplasm in plant cells (e.g., enzyme secretion in cell wall synthesis or modification11,12, plasma membrane protein dynamics7,13,14, cortical microtubule dynamics4, rearrangement of actin microfilaments4,15 and organelle movement6). A wide range of plant cell scientists should benefit from VAEM techniques.
Quantitative evaluation of biological image data has recently become important, driven by advances in microscope equipment and computing. VAEM movie data containing 512(x)×512(y)×600(t) pixels can be easily obtained in only a few minutes. Image analysis helps not only in measuring the lifetime or movement of a fluorescently-labeled protein, but also in mining biological information from large, multidimensional image datasets. A kymograph is a basic, but valuable, approach to determining target protein movement from VAEM movie data, as shown by the sequential recruitment of proteins onto plasma membranes2. The kymograph permitted the visualization of the GFP-PATROL1 dots’ residence times, and their low motility on plasma membranes, as shown in Figure 4. Conversely, a kymograph has limited effectiveness for evaluating more complicated movements, such as cytoplasmic streaming. For more complicated movement, other image analysis approaches such as optical flow16 would be helpful.
Disclosures
The author has nothing to disclose.
Acknowledgments
I am grateful to Dr. Masaru Fujimoto for his technical suggestions for VAEM. I am also grateful to Prof. Koh Iba and Dr. Mimi Hashimoto-Sugimoto for providing GFP-PATROL1 transgenic plants, and discussions about PATROL1. I thank Prof. Seiichiro Hasezawa for his continuing support of my work. This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI grant number 25711017.
References
- Konopka CA, Bednarek SY. Comparison of the dynamics and functional redundancy of the Arabidopsis dynamin-related isoforms DRP1A and DRP1C during plant development. Plant Physiol. 2008;147(4):1590–1602. doi: 10.1104/pp.108.116863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto M, et al. Arabidopsis dynamin-related proteins DRP2B and DRP1A participate together in clathrin-coated vesicle formation during endocytosis. Proc. Natl. Acad. Sci. USA. 2010;107(13):6094–6099. doi: 10.1073/pnas.0913562107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higaki T, Sano T, Hasezawa S. Actin microfilament dynamics and actin side-binding proteins in plants. Curr. Opin. Plant Biol. 2007;10(6):549–556. doi: 10.1016/j.pbi.2007.08.012. [DOI] [PubMed] [Google Scholar]
- Rosero A, Žársky V, Cvrčková F. AtFH1 formin mutation affects actin filament and microtubule dynamics in Arabidopsis thaliana. J. Exp. Bot. 2013;64(2):585–597. doi: 10.1093/jxb/ers351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelrod D. Total internal reflection fluorescence microscopy in cell biology. Traffic. 2001;2(11):764–774. doi: 10.1034/j.1600-0854.2001.21104.x. [DOI] [PubMed] [Google Scholar]
- Konopka CA, Bednarek SY. Variable-angle epifluorescence microscopy: a new way to look at protein dynamics in the plant cell cortex. Plant J. 2008;53(1):186–196. doi: 10.1111/j.1365-313X.2007.03306.x. [DOI] [PubMed] [Google Scholar]
- Wan Y, Ash WM, Fan L, Hao H, Kim MK, Lin J. Variable-angle total internal reflection fluorescence microscopy of intact cells of Arabidopsis thaliana. Plant Methods. 2011;7(27) doi: 10.1186/1746-4811-7-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto-Sugimoto M, et al. A Munc13-like protein in Arabidopsis mediates H+-ATPase translocation that is essential for stomatal responses. Nat. Commun. 2013;4:2215. doi: 10.1038/ncomms3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higaki T, Hashimoto-Sugimoto M, Akita K, Iba K, Hasezawa S. Dynamics and environmental responses of PATROL1 in Arabidopsis subsidiary cells. Plant Cell Physiol. 2014;55(4) doi: 10.1093/pcp/pct151. [DOI] [PubMed] [Google Scholar]
- Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9(7):676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chebli Y, Kaneda M, Zerzour R, Geitmann A. The cell wall of the Arabidopsis pollen tube-spatial distribution, recycling, and network formation of polysaccharides. Plant Physiol. 2012;160(4):1940–1955. doi: 10.1104/pp.112.199729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto M, Suda Y, Vernhettes S, Nakano A, Ueda T. Phosphatidylinositol 3-kinase and 4-kinase have distinct roles in intracellular trafficking of cellulose synthase complexes in Arabidopsis thaliana. Plant Cell Physiol. 2015;56(2):287–298. doi: 10.1093/pcp/pcu195. [DOI] [PubMed] [Google Scholar]
- Li X, Wang X, Yang Y, Li R, He Q, Fang X, Luu DT, Maurel C, Lin J. Single-molecule analysis of PIP2; 1 dynamics and partitioning reveals multiple modes of Arabidopsis plasma membrane aquaporin regulation. Plant Cell. 2011;23(10):3780–3797. doi: 10.1105/tpc.111.091454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Liu P, Wan Y, Chen T, Wang Q, Mettbach U, Baluska F, Samaj J, Fang X, Lucas WJ, Lin J. A membrane microdomain-associated protein, Arabidopsis Flot1, is involved in a clathrin-independent endocytic pathway and is required for seedling development. Plant Cell. 2012;24(5):2105–2122. doi: 10.1105/tpc.112.095695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staiger CJ, Sheahan MB, Khurana P, Wang X, McCurdy DW, Blanchoin L. Actin filament dynamics are dominated by rapid growth and severing activity in the Arabidopsis cortical array. J. Cell Biol. 2009;184(2):269–280. doi: 10.1083/jcb.200806185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda H, et al. Myosin-dependent endoplasmic reticulum motility and F-actin organization in plant cells. Proc. Natl. Acad. Sci. USA. 2010;107(15):6894–6899. doi: 10.1073/pnas.0911482107. [DOI] [PMC free article] [PubMed] [Google Scholar]