

Abstract
Alzheimer's disease (AD) can present with distinct clinical variants. Identifying the earliest neurodegenerative changes associated with each variant has implications for early diagnosis, and for understanding the mechanisms that underlie regional vulnerability and disease progression in AD. We performed voxel‐based morphometry to detect atrophy patterns in early clinical stages of four AD phenotypes: Posterior cortical atrophy (PCA, “visual variant,” n = 93), logopenic variant primary progressive aphasia (lvPPA, “language variant,” n = 74), and memory‐predominant AD categorized as early age‐of‐onset (EOAD, <65 years, n = 114) and late age‐of‐onset (LOAD, >65 years, n = 114). Patients with each syndrome were stratified based on: (1) degree of functional impairment, as measured by the clinical dementia rating (CDR) scale, and (2) overall extent of brain atrophy, as measured by a neuroimaging approach that sums the number of brain voxels showing significantly lower gray matter volume than cognitively normal controls (n = 80). Even at the earliest clinical stage (CDR = 0.5 or bottom quartile of overall atrophy), patients with each syndrome showed both common and variant‐specific atrophy. Common atrophy across variants was found in temporoparietal regions that comprise the posterior default mode network (DMN). Early syndrome‐specific atrophy mirrored functional brain networks underlying functions that are uniquely affected in each variant: Language network in lvPPA, posterior cingulate cortex‐hippocampal circuit in amnestic EOAD and LOAD, and visual networks in PCA. At more advanced stages, atrophy patterns largely converged across AD variants. These findings support a model in which neurodegeneration selectively targets both the DMN and syndrome‐specific vulnerable networks at the earliest clinical stages of AD. Hum Brain Mapp 36:4421–4437, 2015. © 2015 Wiley Periodicals, Inc.
Keywords: Alzheimer's disease, magnetic resonance imaging (MRI), posterior cortical atrophy, logopenic variant primary progressive aphasia, early‐onset dementia, default mode network, language, memory, vision, atrophy, voxel‐based morphometry
Abbreviations
- Aβ
Amyloid‐beta;
- CDR
Clinical dementia rating
- CSF
Cerebrospinal fluid
- DMN
Default mode network
- EOAD
Early‐onset Alzheimer's disease
- LOAD
Late‐onset Alzheimer's disease
- lvPPA
Logopenic variant primary progressive aphasia
- MRI
Magnetic resonance imaging
- PCA
Posterior Cortical Atrophy
- PET
Positron emission tomography
- UCSF
University of California San Francisco
- VUMC
VU University Medical Center
INTRODUCTION
Although amnesia is the most common presenting symptom in Alzheimer's disease (AD), posterior cortical atrophy (PCA, “visual variant”) and logopenic variant primary progressive aphasia (lvPPA, “language variant”) are testament to the great clinical variability of the disease. Both PCA and lvPPA usually manifest at a relatively young age and are strongly associated with underlying AD pathology (i.e. aggregated amyloid‐beta (Aβ) plaques and neurofibrillary tangles) [Grossman 2010; Renner et al., 2004]. Furthermore, while patients with late‐onset AD (LOAD, age‐at‐onset ≥65 years) are more likely to exhibit an amnestic‐predominant presentation, early‐onset AD (EOAD, <65 years) manifests with relatively greater impairment in other cognitive domains at clinical presentation [Koedam et al., 2010; Koss et al., 1996; Mendez et al., 2012; Smits et al., 2012; Stopford et al., 2008]. The remarkable diversity of these phenotypes provides an intriguing model to study clinico‐anatomical heterogeneity in AD.
MRI and [18F]FDG‐PET studies have consistently highlighted distinct neurodegenerative patterns, including those involving visual association areas in PCA [Koedam et al., 2011; Lehmann et al., 2013a; Migliaccio et al., 2009], the language‐dominant left hemisphere in lvPPA [Madhavan et al., 2013; Rabinovici et al., 2008; Rogalski et al., 2014; Teichmann et al., 2013], bilateral temporoparietal cortex in EOAD and medial temporal lobes in LOAD [Frisoni et al., 2007; Moller et al., 2013]. The mechanisms that lead to this anatomic variability are not well understood. The network‐based neurodegeneration theory postulates that neurodegenerative diseases target large‐scale brain networks, with spread of disease following structural and functional connectivity patterns [Seeley et al., 2009; Zhou et al., 2012]. Functional connectivity (resting‐state fMRI) studies, performed primarily in “typical” late‐onset amnestic AD patients, have consistently implicated the default mode network (DMN) as the most vulnerable network in AD [Buckner et al., 2005; Greicius et al., 2004]. The posterior (temporoparietal‐predominant) subdivision may be particularly susceptible in early‐stage AD [Damoiseaux et al., 2012]. Functional connectivity studies in non‐amnestic AD have revealed involvement of both syndrome‐specific networks that correlate with clinical symptoms (visual network in PCA, language network in lvPPA) and shared posterior DMN involvement across AD phenotypes [Lehmann et al., 2013b]. Not yet understood is whether neurodegeneration in non‐amnestic AD originates in the posterior DMN and spreads to more clinically apparent “off‐DMN” networks, or whether the disease begins in peripheral networks in distinct variants, with later convergence in proximal cortical hubs of the posterior DMN. A better understanding of early patterns of spread has important clinical implications, particularly with the push towards early detection and incorporation of topographical markers into diagnostic criteria [Albert et al., 2011; McKhann et al., 2011]. However, measuring brain changes in early clinical stages of non‐amnestic AD has proven challenging due to the low prevalence and the majority of current literature is based on clinically advanced patients.
In this cross‐sectional voxel‐based morphometry study we aimed to investigate the origin and progression of atrophy patterns in four different AD variants at the earliest clinical stages using both a clinical and a neuroimaging proxy of disease severity. Based on the disparate clinical symptoms in each variant, we hypothesized that atrophy would start in syndrome‐specific peripheral regions and would converge across AD variants in posterior DMN regions with advancing disease.
MATERIALS AND METHODS
Participants
A total of 475 subjects were recruited from research cohorts at the University of California San Francisco (UCSF, n = 187) Memory and Aging Center and from the VU University Medical Center (VUMC, n = 288) Amsterdam Dementia Cohort. All patients underwent standard dementia screening that included a medical history and physical examination, a structured caregiver interview, brain MRI and neuropsychological testing. Clinical diagnosis was established by consensus in a multidisciplinary team. All patients fulfilled National Institute on Aging—Alzheimer's Association criteria for probable AD [McKhann et al., 2011] or mild cognitive impairment due to AD [Albert et al., 2011]. Patients with PCA (n = 93) and lvPPA (n = 74) additionally met specific diagnostic criteria for PCA [Mendez et al., 2002] or lvPPA [Gorno‐Tempini et al., 2011]. EOAD patients (n = 114, <65 years at time of diagnosis) were matched for age, sex, disease severity (CDR and MMSE), MRI scanner type and center to PCA and lvPPA patients. Similar matching criteria—except for age—were applied to LOAD patients (n = 114, ≥65 years). EOAD and LOAD did not meet criteria for PCA or lvPPA and had memory‐predominant presentations. Of all patients, 268 (68%) had biomarker (n = 253, positron emission tomography (PET) or cerebrospinal fluid (CSF) analyses) and/or neuropathological confirmation (n = 22) of amyloid pathology. Amyloid status was unknown for 127 patients (32%), and known amyloid‐negative patients were excluded. Assessment of amyloid status was performed according to previously published procedures [Ossenkoppele et al., 2013a; Rabinovici et al., 2007, 2010; Zwan et al., 2014]. For PET, parametric images of [11C]Pittsburgh compound‐B (PIB) were visually assessed by experienced raters and interpreted as positive if there was cortical binding in one or more brain regions and negative in case of predominant binding in the white matter [Ossenkoppele et al., 2013a; Rabinovici et al., 2010]. For CSF Aβ42, we used a threshold of <640 ng/L that previously showed excellent correspondence with global [11C]PIB PET binding [Zwan et al., 2014]. Controls were recruited through advertisements in newspapers and underwent the same diagnostic procedures [Lehmann et al., 2013b; Ossenkoppele et al., 2012]. Because controls were age‐matched to the early‐onset variants (i.e. PCA, lvPPA, and EOAD), we additionally included an older control group (n = 45, mean age: 81 ± 5, available at UCSF only) that was matched with a subset of LOAD patients from UCSF. APOE genotyping was performed in 415 (87%) subjects. Informed consent was obtained from all subjects or their assigned surrogate decision‐makers, and the UCSF and VUMC institutional review boards for human research approved the study.
Neuropsychological Testing
A total of 171 of 187 subjects from UCSF and 215 of 288 subjects from VUMC completed a neuropsychological test battery covering five major cognitive domains [Lehmann et al., 2013a; Ossenkoppele et al., 2014b]: memory (UCSF: delayed recall of the California Verbal Learning Test (nine‐item version) and the modified Rey; VUMC: delayed recall of a Dutch version of the Rey Auditory Verbal Learning Test and the Visual Association Test), language (Category fluency (animals, 1 min) at both sites; UCSF: Boston naming test; VUMC: Visual Association Test Picture Naming), visuospatial function (UCSF: Modified Rey copy and number location from the Visual Object Space and Perception battery; VUMC: Rey Complex Figure copy test), and attention/executive function (Digit span backward and Trailmaking test part B at both sites; UCSF: Letter fluency (D‐words, 1 min); VUMC: Trailmaking Test A and digit span forward).
Image Data Acquisition
At UCSF, T1‐weighted images were acquired on a 1.5 T (Magnetom Avanto System/Magnetom VISION system, Siemens, n = 107) or 3 T (Tim Trio, Siemens n = 80) unit. At VUMC, MRI scans were performed on a 1T (Magnetom Impact, Siemens, n = 52), 1.5 T (Sonata, Siemens, n = 75) or 3 T (SignaHDxt, GE Healthcare, n = 161) unit. The voxel size was 1 mm × 1 mm × 1.5 mm for all scanners. Other acquisition parameters have been published previously [Lehmann et al., 2013a; Moller et al., 2013; Ossenkoppele et al., 2012; Sluimer et al., 2008]. The proportions of subjects studied on each scanner were balanced across the groups, except for UCSF LOAD patients who were predominantly (85%) studied on a 1.5 T scanner. All imaging statistical models included field strength and acquisition site as nuisance variables.
Imaging Data Processing
MRI data were segmented using the New Segment toolbox implemented in the Statistical Parametric Mapping (SPM) 8 software (Wellcome Trust Centre for Neuroimaging, Institute of Neurology at University College London). Diffeomorphic Anatomical Registration Through Exponentiated Lie Algebra (DARTEL) was used to generate a study‐specific template by aligning the gray matter images nonlinearly to a common space. Native gray and white matter images were spatially normalized to the DARTEL template using individual flow fields (modulation was applied to preserve the total amount of signal). Images were smoothed using a 8 mm full width at half maximum (FWHM) isotropic Gaussian kernel. Images were inspected visually after each step in the processing pipeline and the final smoothed‐modulated‐warped gray matter images were checked for sample homogeneity using the VBM8 toolbox to identify potential outliers.
CDR‐Based Approach
Clinical dementia rating
Similar to previous studies [Seeley et al., 2008; Thomas et al., 2014], we used the (global) clinical dementia rating (CDR) [Morris, 1993] as a proxy of disease severity. CDR is a standardized scale that indicates the stage of dementia (ranging from 0 = “none” to 3 = “severe”) based on patient and caregiver information about function of memory, orientation, judgment and problem solving, community affairs, home and hobbies, and personal care. The main objective of this study was to investigate atrophy patterns in the earliest clinical stages, thus we included patients with CDR = 0.5 (“very mild dementia,” n = 172) or CDR = 1 (“mild dementia,” n = 223) in this study.
Analysis
We performed voxelwise contrasts between the four different AD variants (both for CDR 0.5 and 1) and the control group. The model included age, sex, total intracranial volume (TIV), field strength, and acquisition site as nuisance variables. Results are displayed at P < 0.05 (following family wise error correction for multiple comparisons).
W‐Score Neuroimaging Approach
Generation of W‐score maps
The CDR was developed and validated in memory‐predominant AD patients, and due to its emphasis on memory function, it may not be the optimal staging method for nonamnestic AD patients. We therefore additionally applied a neuroimaging method that enables rank ordering of patients based on the extent of whole‐brain atrophy (Fig. 1). First, we performed voxelwise regressions in SPM8 on a set of nuisance factors (age, sex, TIV, field strength and center) to estimate their effect on smoothed‐modulated‐warped gray matter probability images (resulting from VBM processing) in our group of healthy controls. This resulted in beta maps for the nuisance variables as well as a residual map for each healthy control. Next, we used these values to compute a voxelwise map of W‐scores for each patient using the formula: W‐score = (actual – expected)/SD, where actual is the smoothed‐modulated‐warped gray matter probability for a given patient at a given voxel, expected is the predicted gray matter probability for that voxel applying the nuisance factors and beta weights from the regression in healthy controls, and SD is the standard deviation of the residuals for that voxel among the controls. Thus, W‐scores (mean = 0, SD = 1 in the control group, similar to z‐scores) show, at each voxel, where a patient's gray matter probability would fall on the normal distribution of gray matter probabilities in healthy controls, after taking nuisance factors into account [Jack et al., 1997; La Joie et al., 2012]. Our last steps were to binarize the W‐score map for each patient at W <−1.5, and to sum the total number of suprathreshold voxels for every patient. This was used to rank‐order patients within each AD variant, ranging from the lowest 25% (quartile 1) to the highest 25% (quartile 4).
Figure 1.
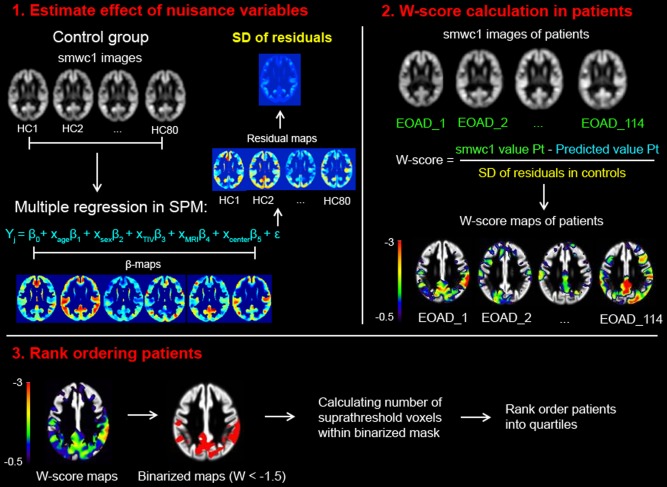
A neuroimaging approach for staging of disease severity. Smoothed, warped, and modulated gray matter images of healthy controls were used to estimate the effect of nuisance variables on voxel intensities (1). Using multiple regressions in SPM, the beta's and residuals were used to determine the predicted value for each patient given their specific covariate (i.e. age, sex, total intracranial volume, MRI field strength, and center). Based on individual raw values in Alzheimer's disease patients, W‐scores can be calculated using the formula shown in (2). Finally, W‐scores maps were binarized at W < −1.5 and the number of voxels below this threshold were calculated for each patient (3). These numbers were rank‐ordered separately for each Alzheimer's disease variant and patients were grouped into quartiles.
Analysis
Within each AD variant, averaged W‐score maps were generated to allow comparison of atrophy patterns for every quartile. In addition, based on [Lehmann et al., 2013b] we selected seven network templates relevant to this study (language, higher‐visual, ventral DMN, posterior dorsal DMN, anterior dorsal DMN, and left and right executive control network) from a set of 15 previously published functional connectivity networks [Damoiseaux et al., 2012; Shirer et al., 2012]. The sensorimotor network, which is typically spared across AD variants, served as control network. Within these functional networks, mean W‐scores were computed and displayed for comparison across quartiles and AD variants.
Statistical Analyses
Differences between groups for demographic, clinical and neuropsychological characteristics were assessed using ANOVA with post hoc Bonferroni tests for continuous variables and χ 2 tests for dichotomous data. Differences in mean W‐scores within each network across AD variants at quartile 1 and 4 were assessed using ANOVA's with post hoc Bonferroni tests. Differences in mean W‐scores between networks in quartile 1 of each AD variant were assessed using independent samples t‐tests. Changes in mean W‐scores within networks with advancing quartiles were assessed using general linear models with W‐score atrophy score within a functional network as the dependent variable and quartile (Q1‐Q4) as fixed factor. Statistical significance was set at P < 0.05.
RESULTS
Participants and Neuropsychological Scores
114 EOAD, 74 lvPPA, 93 PCA, 114 LOAD, and 80 control subjects were included in the present study (Table 1). As expected, cognitive profiles differed across variants with PCA patients showing most prominent visuospatial deficits, lvPPA patients being most impaired in the language domain and LOAD and EOAD patients exhibiting a memory‐predominant neuropsychological profile (Tables 2 and 3). Furthermore, within AD variants most neuropsychological scores were lower for patients with CDR 1 than patients with CDR 0.5.
Table 1.
Patient characteristics
| CDR | Early‐onset Alzheimer's disease | Logopenic variant primary progressive aphasia | Posterior cortical atrophy | Late‐onset Alzheimer's disease | Controls | ||||
|---|---|---|---|---|---|---|---|---|---|
| 0.5 | 1 | 0.5 | 1 | 0.5 | 1 | 0.5 | 1 | 0 | |
| N | 50 | 64 | 41 | 33 | 31 | 62 | 50 | 64 | 80 |
| Age | 62.0 ± 4.5 | 60.7 ± 4.5 | 66.1 ± 7.8 | 65.1 ± 8.3 | 64.1 ± 8.2 | 60.7 ± 5.9 | 79.0 ± 4.2 | 78.2 ± 4.5 | 64.4 ± 7.0a |
| Sex (% male) | 44.0 | 48.4 | 56.1 | 48.5 | 48.4 | 47.5 | 54.0 | 48.4 | 51.3 |
| Education | 16.1 ± 2.4 | 15.6 ± 2.1 | 16.7 ± 2.8 | 16.6 ± 2.5 | 15.4 ± 3.1 | 15.2 ± 2.3 | 15.4 ± 2.6 | 15.1 ± 2.7 | 17.1 ± 2.0b |
| MMSE | 24.2 ± 3.6 | 19.9 ± 3.7f | 23.8 ± 4.0 | 17.0 ± 4.5f | 24.7 ± 3.3 | 20.0 ± 4.2f | 24.6 ± 2.6 | 20.5 ± 4.4f | 29.4 ± 0.8c |
| % APOE ε4 carriers | 64.3 | 77.6 | 35.5 | 56.0 | 70.0 | 54.5 | 62.2 | 69.6 | 20.0d |
| Amyloid status (% POS/unknown/NEG) | 62/38/0 | 72/28/0 | 81/19/0 | 79/21/0 | 94/6/0 | 80/20/0 | 34/66/0** | 56/44/0 | 0/65/35e |
| Total intracranial volume (L) | 1.60 ± 0.15 | 1.58 ± 0.15 | 1.56 ± 0.14 | 1.56 ± 0.16 | 1.58 ± 0.12 | 1.55 ± 0.16 | 1.58 ± 0.12 | 1.59 ± 0.15 | 1.56 ± 0.15 |
Data are presented as mean ± standard deviation unless indicated otherwise. Differences between controls and Alzheimer's disease variants and differences within Alzheimer's disease variants (Clinical dementia rating 0.5 vs. 1) were assessed using ANOVA with post hoc Bonferroni tests (age, education, MMSE, and total intracranial volume) and χ2 (sex and APOE genotype).
Controls < LOAD 0.5/1: P < 0.001; Controls > EOAD 1, PCA 1: P < 0.05.
Controls > EOAD 1, LOAD 0.5/1, PCA 0.5/1: P < 0.05.
Controls < all AD variants: P < 0.001.
Controls < all (except lvPPA ± PA CDR 0.5): P < 0.01.
Controls < all: P < 0.001.
CDR 1 < CDR 0.5: P < 0.05, **CDR 0.5 < CDR 1: P < 0.05.
CDR = Clinical dementia rating; MMSE = Mini‐mental state examination; POS = positive; NEG = Negative.
Table 2.
Neuropsychological test scores (UCSF)
| UCSF CDR | Early‐onset Alzheimer's disease | Logopenic variant primary progressive aphasia | Posterior cortical atrophy | Late‐onset Alzheimer's disease | Controls | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| 0.5 | 1 | 0.5 | 1 | 0.5 | 1 | 0.5 | 1 | 0 | ||
| Episodic memory | ||||||||||
| CVLT‐9 delayed recall (9) | 1.8 ± 1.2 | 1.3 ± 2.0 | 3.5 ± 3.3a | 1.7 ± 1.6 | 4.3 ± 3.3 | 1.4 ± 1.4 | 1.6 ± 2.4 | 0.1 ± 0.3 | 7.4 ± 1.3b | |
| Modified Rey recall (17) | 2.9 ± 2.8 | 2.0 ± 2.3 | 6.4 ± 3.7c | 5.3 ± 3.7 | 5.5 ± 5.2 | 0.8 ± 1.6 | 3.6 ± 3.6 | 1.1 ± 1.4 | 12.4 ± 3.3d | |
| Language | ||||||||||
| Boston Naming Test (15) | 12.2 ± 2.4 | 11.9 ± 3.2 | 9.3 ± 4.8 | 6.1 ± 4.1e | 12.2 ± 3.5 | 9.3 ± 4.3 | 11.6 ± 2.6 | 9.9 ± 4.1 | 14.6 ± 0.9f | |
| Animal fluency (1 min) | 10.7 ± 5.0 | 10.0 ± 5.7 | 10.2 ± 6.5 | 4.5 ± 3.0 | 11.9 ± 6.1 | 6.9 ± 3.6 | 11.7 ± 4.9 | 8.9 ± 5.4 | 24.7 ± 5.1d | |
| Repetitions (3) | 2.7 ± 0.6 | 2.3 ± 0.8 | 1.5 ± 0.9g | 1.2 ± 0.8g | 2.4 ± 0.6 | 2.4 ± 0.7 | 2.5 ± 0.8 | 2.4 ± 0.8 | 2.9 ± 0.3d | |
| Visuospatial | ||||||||||
| Modified Rey copy (17) | 9.7 ± 6.5 | 10.1 ± 5.2 | 15.1 ± 1.5h | 11.5 ± 4.7 | 9.1 ± 6.3 | 2.4 ± 3.2i | 15.1 ± 1.5h | 14.5 ± 1.6j | 15.7 ± 1.2h | |
| VOSP number localization (10) | 5.3 ± 3.2 | 6.8 ± 2.4 | 8.6 ± 1.3k | 6.0 ± 2.4 | 7.1 ± 2.5 | 3.8 ± 2.6l | 8.6 ± 1.4 | 8.1 ± 2.5 | 9.2 ± 1.2m | |
| Calculations (5) | 3.2 ± 1.5 | 3.1 ± 1.5 | 3.0 ± 1.3 | 2.4 ± 1.4 | 3.0 ± 1.7 | 1.2 ± 0.8n | 4.3 ± 0.8o | 3.9 ± 1.3q | 4.9 ± 0.4d | |
| Executive function/attention | ||||||||||
| Digit span backward (8) | 3.3 ± 1.5 | 3.3 ± 1.0 | 2.9 ± 0.8 | 2.5 ± 1.1 | 2.7 ± 0.7 | 2.7 ± 1.1 | 4.6 ± 1.2p | 3.8 ± 0.9 | 5.5 ± 1.3q | |
| Letter fluency (D words, 1 min) | 10.3 ± 4.9 | 10.2 ± 4.8 | 8.1 ± 4.9t | 3.3 ± 2.9r | 11.0 ± 3.7 | 7.6 ± 3.3 | 13.2 ± 5.0 | 9.7 ± 4.1 | 17.1 ± 5.8s | |
| Modified Trails B (120 s) | 89.1 ± 35.8 | 90.7 ± 38.7 | 76.8 ± 34.8 | 117.5 ± 5.7t | 97.8 ± 17.8 | 119.5 ± 1.4u | 78.2 ± 39.7 | 93.4 ± 34.6 | 25.7 ± 10.8v | |
Data are presented as mean ± SD and maximum attainable neuropsychological test scores are presented in parentheses. Differences between groups were assessed using univariate ANOVA with adjustments for age, sex and education and post hoc Bonferroni tests.
lvPPA 0.5 > LOAD 1: P < 0.01
Controls > All (except PCA 0.5): P < 0.001.
lvPPA 0.5 > EOAD 0.5/1, PCA 1 and LOAD 1: P < 0.05.
lvPPA 1 > PCA 1 and LOAD 1: P < 0.05.
Controls > All: P < 0.001.
lvPPA 1 < EOAD 0.5/1, PCA 0.5 and LOAD 0.5: P < 0.01.
Controls > lvPPA 0.5/1, PCA 1 and LOAD 0.5/1: P < 0.05.
lvPPA 0.5/1 < All: P < 0.001.
lvPPA 0.5, LOAD 0.5 and Controls > EOAD 0.5/1 and PCA 0.5/1: P < 0.01.
PCA 1 < All: P < 0.001.
LOAD 1 > EOAD 0.5: P < 0.05.
lvPPA0.5 > EOAD 0.5: P < 0.001.
PCA 1 < lvPPA 0.5 and LOAD 0.5/1: P < 0.05.
Controls > EOAD 0.5, lvPPA1 and PCA 1: P < 0.05.
PCA 1 < All: P < 0.001.
LOAD 0.5 > All (except LOAD 1): P < 0.001; qLOAD 1 > PCA 0.5 and lvPPA 0.5/1: P < 0.05.
LOAD 0.5 > All (except LOAD 1 and Controls): P < 0.05.
Controls > All (except LOAD 0.5): P < 0.001; tlvPPA 0.5 < LOAD 0.5: P < 0.01.
lvPPA 1 < All (except lvPPA 0.5 and PCA 1): P < 0.05.
Controls > All (except LOAD 0.5): P < 0.05.
lvPPA 1 > lvPPA 0.5 and LOAD 0.5: P < 0.05.
PCA 1 > lvPPA 0.5 and LOAD 0.5: P < 0.05.
Controls < All: P < 0.001.
Table 3.
Neuropsychological test scores (VUMC)
| VUMC CDR | Early‐onset Alzheimer's disease | Logopenic variant primary progressive aphasia | Posterior cortical atrophy | Late‐onset Alzheimer's disease | Controls | ||||
|---|---|---|---|---|---|---|---|---|---|
| 0.5 | 1 | 0.5 | 1 | 0.5 | 1 | 0.5 | 1 | 0 | |
| Memory | |||||||||
| RAVLT—delayed recall (15) | 3.2 ± 2.5 | 1.9 ± 2.7 | 4.4 ± 3.7 | 3.2 ± 2.9 | 2.6 ± 2.5 | 2.2 ± 2.7 | 2.1 ± 3.1 | 0.6 ± 0.8a | 8.1 ± 2.5b |
| Visual association test A (12) | 9.1 ± 3.1 | 5.5 ± 2.7c | 10.1 ± 2.4 | 8.2 ± 2.9 | 6.7 ± 3.7 | 7.3 ± 3.1 | 8.9 ± 4.2 | 6.4 ± 2.4d | 11.9 ± 0.4e |
| Language | |||||||||
| VAT naming (12) | 11.9 ± 0.4 | 11.6 ± 1.0 | 10.6 ± 1.9 | 9.4 ± 2.3f | 11.6 ± 0.8 | 10.7 ± 2.2g | 12.0 ± 0 | 11.2 ± 0.6 | 12 ± 0 |
| Animal fluency (1 min) | 16.8 ± 4.5 | 12.5 ± 5.4 | 12.8 ± 5.5 | 9.3 ± 4.2h | 13.9 ± 3.0 | 12.7 ± 5.4 | 16.6 ± 6.8 | 12.9 ± 4.8 | 21.8 ± 6.1 |
| Visuospatial function | |||||||||
| Rey complex figure copy (36) | 30.8 ± 5.8 | 30.8 ± 4.0 | 33.2 ± 4.0 | 27.7 ± 2.5 | 23.7 ± 7.1i | 22.3 ± 6.7i | 33.2 ± 2.6 | 33.2 ± 2.6 | 34.3 ± 2.6 |
| Attention | |||||||||
| TMT‐A (360 s) | 53.4 ± 22.6 | 92.3 ± 68.5j | 62.6 ± 31.3 | 90.5 ± 36.0j | 69.0 ± 25.3 | 129.0 ± 77.5k | 65.4 ± 48.0 | 66.0 ± 37.3 | 40.3 ± 15.6 |
| Digit span forward (21) | 12.1 ± 2.6 | 10.3 ± 2.3 | 9.3 ± 2.1l | 9.1 ± 2.4l | 11.5 ± 3.5 | 11.3 ± 3.0 | 13.2 ± 3.2 | 11.1 ± 3.5 | 12.9 ± 3.1 |
| Executive function | |||||||||
| TMT‐B (500 s) | 130.7 ± 74.7 | 231.2 ± 127.8 | 188.2 ± 116.9 | 265.4 ± 95.6 | 231.6 ± 135.4 | 296.2 ± 171.2m | 157.5 ± 86.4 | 202.8 ± 79.9 | 75.6 ± 23.8n |
| Digit span backward (21) | 7.3 ± 2.9 | 6.2 ± 1.9 | 6.7 ± 2.2 | 5.5 ± 1.4 | 6.3 ± 2.6 | 5.8 ± 2.4 | 8.9 ± 3.1 | 7.0 ± 3.2 | 9.4 ± 2.9o |
Data are presented as mean ± SD and maximum attainable neuropsychological test scores are presented in parentheses. Differences between groups were assessed using univariate ANOVA with adjustments for age, sex and education and post hoc Bonferroni tests.
LOAD 1 < All (except EOAD 1 and PCA 1): P < 0.05.
Controls > All: P < 0.001.
EOAD 1 < EOAD 0.5, lvPPA 0.5 and LOAD 0.5: P < 0.05.
LOAD 1 < EOAD 0.5 and lvPPA 0.5: P < 0.05.
Controls > All (except EOAD 0.5 and lvPPA): P < 0.05.
lvPPA 1 < EOAD 0.5/1, PCA 0.5 and LOAD 0.5: P < 0.05.
PCA 0.5 < Controls: P < 0.05.
lvPPA 1 < EOAD 0.5 and LOAD 0.5: P < 0.01.
PCA 0.5/1 < All (except for lvPPA 1): P < 0.05.
EOAD 1 and lvPPA 1 > Controls: P < 0.05.
PCA 1 > EOAD 0.5, lvPPA 0.5, PCA 0.5 and LOAD 0.5/1: P < 0.05.
lvPPA 0.5/1 < LOAD 0.5 and Controls: P < 0.05.
PCA 1 > EOAD 0.5: P < 0.05.
Controls < All (except for EOAD 0.5 and LOAD 0.5): P < 0.05;
Controls > All (except for EOAD 0.5 and LOAD 0.5/1): P < 0.05.
Voxel Based Morphometry: AD Variants Versus Controls
First, we performed voxelwise contrasts between the different AD variants (separately for CDR 0.5 and 1) and the control group, applying a statistical threshold of P < 0.05 family wise error corrected. At CDR stage 0.5, atrophy patterns in EOAD, PCA, and lvPPA overlapped in several posterior DMN regions in the left hemisphere, including middle temporal gyrus, inferior parietal lobule and posterior cingulate cortex (PCC, Figs. 2 and 3). lvPPA and PCA patients additionally showed syndrome‐specific atrophy in “off‐DMN” regions such as bilateral visual association cortex (PCA), and left superior and inferior temporal cortex implicated in language function (lvPPA). LOAD patients showed relatively focal medial temporal lobe (MTL) atrophy in this early clinical stage, while EOAD patients showed more cortical involvement in the PCC‐hippocampal circuit as well as in lateral temporoparietal cortex. At CDR 1, there was widespread overlap across EOAD, PCA and lvPPA in the lateral temporoparietal cortex, covering large parts of the posterior DMN (Figs. 2 and 3). In addition, syndrome‐specific atrophy further extended into bilateral visual association cortex for PCA and into the PCC‐hippocampal circuit for EOAD, and crossover from language regions in the left hemisphere into the right lateral temporal cortex was appreciated in lvPPA patients. Atrophy in LOAD was restricted to the MTL and medial prefrontal cortex, showing relative sparing of posterior DMN regions. Since the LOAD group was older than the control group, we performed an additional analysis comparing LOAD with an older age‐matched control group, which revealed a comparable atrophy pattern (data not shown). Reversed contrasts showed no areas of increased volume in controls versus patients.
Figure 2.
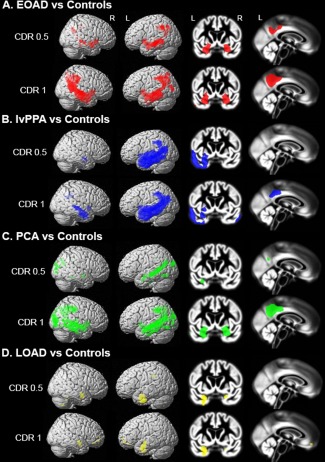
Atrophy maps of Alzheimer's disease variants compared with controls. Regional differences between healthy controls and EOAD (A), lvPPA (B), PCA (C) and LOAD (D) patients at CDR 0.5 and 1. Voxelwise contrasts were adjusted for age, sex, total intracranial volume, MRI field strength and center, and thresholded at P < 0.05 with family‐wise error correction. Results are superimposed on the SPM8 new single‐subject template (left) and the study‐specific DARTEL template (right).
Figure 3.
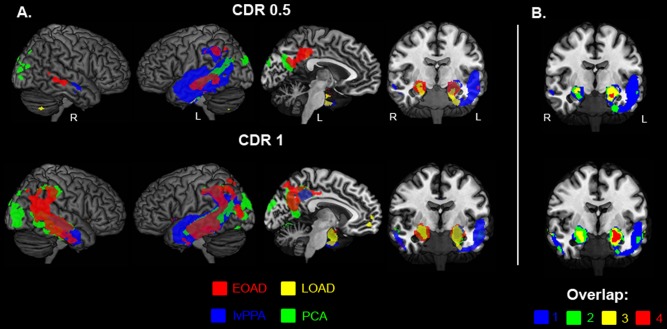
The intersection of distinct Alzheimer's disease phenotypes. Binarized maps of significant voxelwise differences between Alzheimer's disease variants and healthy controls (at P < 0.05 family‐wise error corrected) are overlaid on the MRIcron ch2better template for EOAD (red), lvPPA (blue), PCA (green), and LOAD (yellow) for both CDR 0.5 and 1 (A). (B) Represents a coronal view that displays the overlap between AD variants (blue = only 1 AD variant, green = 2 AD variants, yellow = 3 AD variants and 4 = all AD variants), most prominent in the medial temporal lobe.
W‐Score Approach
An alternative approach to explore the origin and progression of atrophy patterns is to use a neuroimaging—rather than a clinical—staging method. Such a method could help overcome the potential bias inherent in CDR to emphasize memory performance over other cognitive functions. The individual number of suprathreshold voxels (reflecting significant atrophy versus controls) was used to divide patients into quartiles following a rank‐order approach (Fig. 1). This resulted in different groups compared with the CDR‐based approach (Tables 3 and 4). The mean number of suprathreshold voxels did not differ across AD variants: EOAD 38,999 ± 18,063; lvPPA 37,068 ± 15,876; PCA: 42,847 ± 20,213; LOAD: 38,781 ± 23,778 (P = 0.28). Next, mean W‐scores within each quartile were computed and displayed to illustrate changes across quartiles (Fig. 4). In this approach, the first quartiles (Q1) identify those voxels that exceeded the statistical atrophy threshold in the 25% of patients with lowest overall brain atrophy in each variant, thus representing the earliest degenerating regions in each syndrome. Q1 showed overlapping atrophy patterns across AD variants in multiple posterior DMN regions such as PCC, angular gyrus, supramarginal gyrus and middle temporal gyrus (Fig. 4). PCA and lvPPA patients showed additional involvement of syndrome‐specific off‐DMN regions (i.e. lateral occipital cortex in PCA, asymmetric temporoparietal cortex (left > right) in lvPPA), EOAD patients showed marked PCC‐hippocampal atrophy and LOAD patients showed predominantly MTL and medial frontal cortex involvement. There was thus early overlap in posterior DMN regions across AD phenotypes, and even when looking at the 10 patients with the least atrophied brains, both posterior DMN and off‐DMN regions were involved across variants (data not shown). With ascending quartiles (greater extent of whole‐brain atrophy), atrophy progressed along both DMN and off‐DMN regions across phenotypes. An additional analysis comparing LOAD patients with an older age‐matched control group using the W‐score staging method showed comparable MTL involvement but frontal atrophy was less pronounced (data not shown).
Table 4.
Proportions of clinical dementia rating 0.5 in each quartile
| EOAD (n = 50) (%) | LOAD (n = 50) (%) | PCA (n = 31) (%) | lvPPA (n = 41) (%) | |
|---|---|---|---|---|
| Quartile 1 | 53 | 52 | 38 | 61 |
| Quartile 2 | 34 | 34 | 30 | 53 |
| Quartile 3 | 46 | 39 | 26 | 58 |
| Quartile 4 | 39 | 50 | 35 | 50 |
Percentages represent the proportion of patients with clinical dementia rating 0.5 within each quartile for all Alzheimer's disease variants.
EOAD = early‐onset Alzheimer's disease; LOAD = late‐onset Alzheimer's disease; PCA = Posterior cortical atrophy; lvPPA = logopenic variant primary progressive aphasia.
Figure 4.
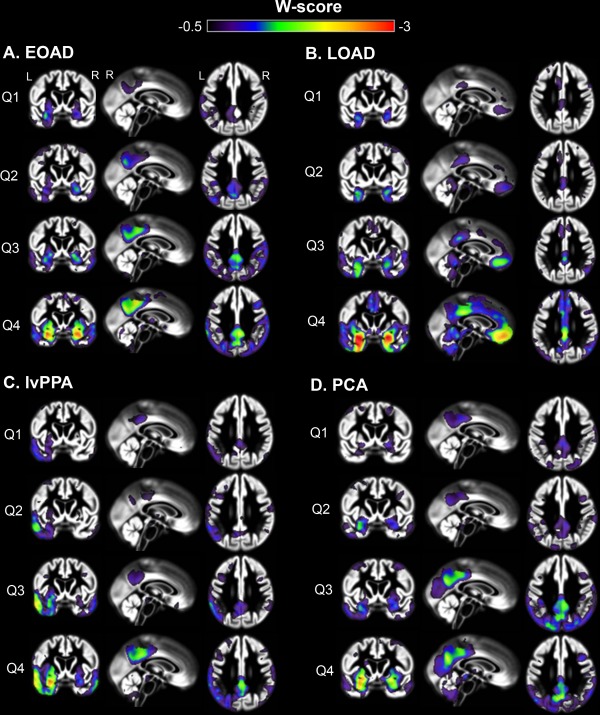
Mean W‐score maps across quartiles for distinct Alzheimer's disease variants. Mean W‐score maps show changes of atrophy patterns with increasing quartiles for each of the Alzheimer's disease variants. W‐score maps are superimposed on the study‐specific template obtained with VBM. Q = quartile (1 = lowest extent of whole‐brain atrophy, 4 = highest).
Structural Changes in Functional Networks
To explore the hypothesis that neurodegeneration spreads through interconnected networks, we calculated mean W‐scores of brain atrophy within eight predefined functional network regions‐of‐interest, based on template connectivity maps [Shirer et al., 2012], across variants and quartiles (Fig. 5). In Q1, most differences in W‐scores found across AD variants involved syndrome‐specific off‐DMN networks: lvPPA patients had lower W‐scores in the left language network than EOAD, LOAD and PCA patients (P < 0.01) and in the left executive control network compared with LOAD (P < 0.01), PCA patients were more affected in the higher visual network and ventral DMN compared with LOAD and lvPPA patients (P < 0.05), and LOAD patients showed lower W‐scores in the anterior DMN compared with PCA patients (P < 0.05). W‐scores in the posterior DMN did not differ across EOAD, lvPPA and PCA patients in Q1, though this network was relatively preserved in the LOAD group (P < 0.05). Compared with Q1, W‐scores were reduced in the majority of networks in Q4 and there were no longer significant group differences in the syndrome‐specific off‐DMN networks or in the posterior DMN, demonstrating convergence across AD variants. The only difference at this stage was that the anterior DMN was more affected in LOAD compared with all other groups (P < 0.001). The sensorimotor network, which is typically spared in AD variants and thus served as a control network, did not differ across groups in any of the quartiles. Table 5 shows the significance of differences in mean W‐scores across functional networks in Q1 for each AD variant. Supporting Information Table 1 indicates changes in W‐scores within networks as a function of advancing quartiles for each AD variant.
Figure 5.
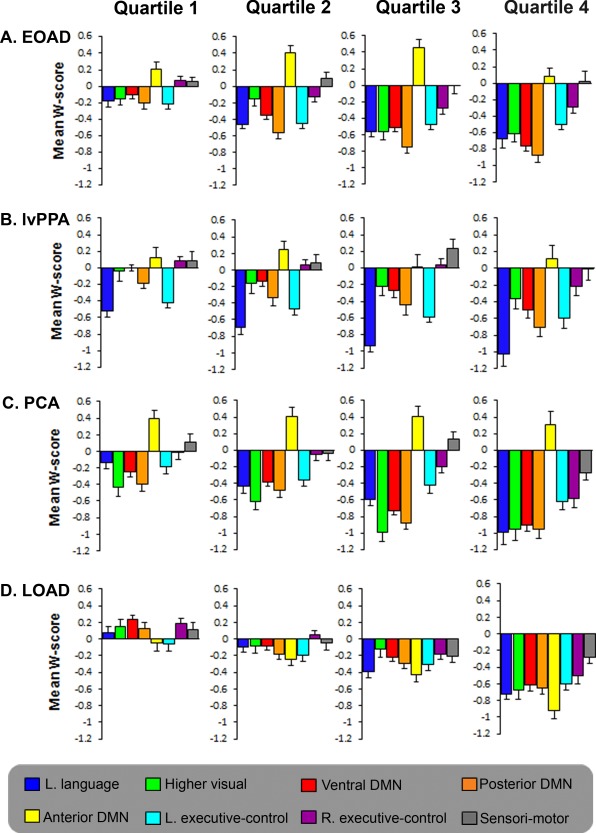
Mean W‐score atrophy values within distinct intrinsic connectivity networks. The y‐axis represents mean W‐scores for brain atrophy extracted within eight functional connectivity network maps for each quartile in EOAD (A), lvPPA (B), PCA (C), and LOAD (D) patients. Statistical comparisons between networks in quartile 1 and within networks across quartiles are shown in Table 3 and Supporting Information Table I.
Table 5.
Differences in atrophy W‐scores between intrinsic connectivity networks in quartile 1
| Language | Visual | vDMN | pdDMN | adDMN | LECN | RECN | SM | |
|---|---|---|---|---|---|---|---|---|
| A. EOAD | ||||||||
| Left language | ||||||||
| Higher visual | ns | |||||||
| Ventral DMN | ns | ns | ||||||
| Posterior dorsal DMN | ns | ns | 0.003 | |||||
| Anterior dorsal DMN | <0.001 | 0.002 | 0.001 | <0.001 | ||||
| Left executive‐control | ns | ns | ns | ns | <0.001 | |||
| Right executive‐control | 0.003 | 0.018 | 0.012 | 0.001 | ns | <0.001 | ||
| Sensori‐motor | 0.015 | 0.028 | 0.025 | 0.001 | ns | 0.001 | ns | |
| B. lvPPA | ||||||||
| Left language | ||||||||
| Higher visual | 0.003 | |||||||
| Ventral DMN | <0.001 | ns | ||||||
| Posterior dorsal DMN | <0.001 | ns | 0.002 | |||||
| Anterior dorsal DMN | 0.001 | ns | ns | 0.049 | ||||
| Left executive‐control | ns | 0.041 | <0.001 | 0.011 | 0.007 | |||
| Right executive‐control | <0.001 | ns | ns | 0.001 | ns | <0.001 | ||
| Sensori‐motor | 0.001 | ns | ns | ns | ns | 0.008 | ns | |
| C. PCA | ||||||||
| Left language | ||||||||
| Higher visual | 0.031 | |||||||
| Ventral DMN | ns | ns | ||||||
| Posterior dorsal DMN | 0.002 | ns | 0.012 | |||||
| Anterior dorsal DMN | <0.001 | <0.001 | <0.001 | <0.001 | ||||
| Left executive‐control | ns | ns | ns | 0.005 | 0.001 | |||
| Right executive‐control | ns | 0.006 | 0.004 | <0.001 | 0.001 | ns | ||
| Sensori‐motor | ns | 0.003 | 0.006 | 0.001 | 0.029 | 0.043 | ns | |
| D. LOAD | ||||||||
| Left language | ||||||||
| Higher visual | ns | |||||||
| Ventral DMN | 0.018 | ns | ||||||
| Posterior dorsal DMN | ns | ns | 0.020 | |||||
| Anterior dorsal DMN | ns | ns | 0.008 | ns | ||||
| Left executive‐control | 0.011 | 0.018 | <0.001 | 0.003 | ns | |||
| Right executive‐control | ns | ns | ns | ns | ns | 0.003 | ||
| Sensori‐motor | ns | ns | ns | ns | ns | ns | ns |
P values for comparisons of mean W‐scores between intrinsic connectivity networks in EOAD (A), lvPPA (B), PCA (C), and LOAD (D) patients in quartile 1 (see Fig. 5). Quartile 1 represents 25% of patients with the lowest number of suprathreshold voxels throughout the brain within each AD variant. P values resulted from independent samples t‐tests.
DMN = default mode network; v = ventral; pd = posterior dorsal; ad = anterior dorsal; LECN = left executive‐control network; RECN = right executive‐control network; ns = nonsignificant.
DISCUSSION
In the present cross‐sectional voxel‐based morphometry study we investigated the origin and spread of brain atrophy in four distinct early‐stage AD variants. In the earliest clinical stages, we found both shared vulnerability in posterior DMN regions across patients and marked syndrome‐specific atrophy patterns in off‐DMN networks that anatomically overlapped with brain networks responsible for critical cognitive functions uniquely affected in each AD variant (language network in lvPPA, PCC‐hippocampal circuit in amnestic‐predominant EOAD and LOAD, and visual network in PCA). Whole‐brain analyses as well as atrophy measurements within distinct functional networks suggested that, with increasing disease severity, atrophy spreads through both posterior DMN and peripheral networks. This supports a network‐based model of neurodegeneration in which vulnerable brain regions are selectively targeted through both syndrome‐specific and common networks across AD variants.
Start and Spread of Brain Atrophy Across AD Variants
Imaging studies have shown that clinical variants of AD are characterized by both common (posterior DMN) and syndrome‐specific (“off‐DMN”) patterns of brain atrophy and functional network disruptions [Adriaanse et al., 2014; Frisoni et al., 2007; Lehmann et al., 2013a,b; Madhavan et al., 2013; Ridgway et al., 2012; Teichmann et al., 2013]. It is unknown, however, whether these clinical phenotypes arise from initial structural alterations in posterior DMN or in off‐DMN regions, and how the disease spreads with advancing severity. Based on the disparate clinical symptoms in each variant, we hypothesized that brain atrophy would (1) start in syndrome‐specific off‐DMN regions in the earliest clinical stage, and (2) converge in posterior DMN regions across AD variants as the disease progresses.
Contrary to our first hypothesis, both posterior DMN and syndrome‐specific off‐DMN regions were affected in the earliest clinically detectable disease stage across AD variants. This may suggest a multifocal onset of disease that targets posterior DMN and peripheral networks simultaneously. Alternatively, the neurodegenerative process may already be too advanced in a clinical population to investigate the sites of earliest atrophy, even at the prodromal stage, and requires subjects in preclinical stages of AD. For example, [Chan et al., 2015] described a cognitively normal research volunteer who developed a PCA syndrome and showed isolated atrophy in the visual association cortex at baseline. This and the present finding of greater extent of atrophy in language and visual areas in PCA and lvPPA compared with posterior DMN regions hints towards earlier involvement of syndrome‐specific off‐DMN regions, but does not rule out initial posterior DMN onset or neurodegenerative processes occurring in parallel.
In line with our second hypothesis, patterns of brain atrophy converged across wide regions of the temporoparietal cortex, including both posterior DMN and off‐DMN regions. Despite significant overlap, there were also uniquely atrophied regions in later stages of the disease that showed strong anatomical correspondence with functional networks closely matching the clinical phenotypes of each AD variant [Lehmann et al., 2012; Rohrer et al., 2013]. This suggests that spread of neurodegeneration follows differential trajectories for each AD variant, and that the functional architecture of the brain, rather than just spatial proximity, is a key determinant for future atrophy [Raj et al., 2012; Seeley et al., 2009; Zhou et al., 2012]. One possibility is that brain atrophy starts in off‐DMN regions and progresses to neighboring networks, rapidly involving the posterior DMN due to its centrality and long‐distance connections [van den Heuvel et al., 2012]. Alternatively, the posterior DMN may serve as a portal for spread of neurodegeneration into highly interconnected regions, thus spreading into posterior functional networks subserving language functions and visual integration.
Posterior DMN as the Common Denominator
The common denominator across AD variants involved reduced gray matter density in wide regions of the posterior DMN, supporting the central role of the DMN in AD [Buckner et al., 2005]. Due to its prominent role in a broad spectrum of brain diseases [Zhang and Raichle, 2010] and relative insusceptibility to lesions in key nodes [Cavanna and Trimble, 2006; Leech and Sharp, 2014], there is no clear consensus about functional consequences of DMN disruptions. Several studies have proposed to fractionate the DMN into distinct subcomponents (i.e. anterior versus posterior or ventral versus dorsal) that potentially better reflect clinical function [Andrews‐Hanna et al., 2010; Damoiseaux et al., 2012; Leech et al., 2011]. It is conceivable, however, that clinico‐anatomical correlates are even more fine‐grained and depend on specific brain structures within subcomponents of the overarching DMN [Nelson et al., 2010]. For example, [Margulies et al., 2009] showed that four small seed regions within the precuneus of both human and monkey brains yielded distinct functional networks (sensorimotor, limbic, cognitive,and visual), suggesting that highly proximate regions within the DMN may engage in different cognitive processes. Remarkably, the precuneus seed that evoked a visual network in Margulies et al. [2009] shows striking overlap with the precuneus cluster that was uniquely atrophied in PCA patients (Fig. 3), while the cognitive and limbic networks show resemblance with the atrophy patterns observed in EOAD and LOAD patients. The diversity of function within small DMN regions may suggest that the role of the DMN is to integrate input from diverse cognitive networks, with different portals subserving each network. Selective vulnerability of DMN subcomponents may thus partially account for clinico‐anatomical heterogeneity in AD.
Neuropsychological Features and Atrophy Patterns
In general, the distinct variants of AD largely showed cerebral atrophy and neuropsychological deficits in the expected brain regions and cognitive domains, respectively. Early syndrome‐specific atrophy patterns mirrored functional brain networks underlying neuropsychological functions that are uniquely affected in each variant, i.e. language for lvPPA, memory for EOAD and LOAD, and visuospatial function for PCA. The convergence in temporoparietal brain regions (involved in a variety of cognitive functions) at CDR 1 corresponded to greater overlap of neuropsychological deficits across AD variants, i.e. relatively greater memory impairment in PCA and lvPPA and decline in non‐memory functions in EOAD and to a lesser extent LOAD. Figure 5 illustrates the increased overlap of atrophy within functional networks across all AD variants, which showed a resemblance with neuropsychological decline with advancing disease. For example, in line with the decline in language scores (Tables 2 and 3) and the commonly observed signs of anomia or aphasia as dementia progresses, the left language network was among the most affected networks in all AD variants in later disease stages (Fig. 5).
Clinical Application
Autopsy and biomarker studies have indicated that the clinical diagnosis, even in the hands of experts, shows only moderate accuracy for predicting the histopathological cause of AD [Beach et al., 2012; Ossenkoppele et al., 2013a]. Possibly due to the young age and atypical presentation, diagnostic accuracy is particularly low in early‐onset and nonamnestic variants of AD [Alladi et al., 2007; Beach et al., 2012], and the diagnosis is often delayed or entirely missed during life [Crutch et al., 2012; van Vliet et al., 2011]. Recent National Institute on Aging—Alzheimer's Association [McKhann et al., 2011] and International Working Group [Dubois et al., 2014] diagnostic criteria for AD included lvPPA and PCA syndromes, and encourage use of biomarkers to facilitate the diagnostic process. The shared selective vulnerability of posterior DMN regions highlights the potential of posterior DMN atrophy as an MRI marker to support the early clinical diagnosis of non‐amnestic AD, as it appeared sensitive to early neurodegeneration in lvPPA, PCA and EOAD. More traditional measures such as hippocampal volumes may lack sensitivity in early stages of non‐amnestic AD variants, but show good performance in LOAD with prominent MTL atrophy.
The W‐Score Method Versus the CDR Approach
The CDR relies heavily on memory function and may not be an ideal marker of disease severity in non‐amnestic AD patients. In the current study, we present an alternative staging method that uses the extent of whole‐brain atrophy as a measure of disease severity. The rationale of the CDR and neuroimaging approaches was to utilize cross‐sectional data to infer where brain atrophy starts and how it may spread as the disease progresses. Tables 2 and 3 indicates that there was only partial agreement between the CDR and W‐score method, as substantial proportions of patients with CDR 1 were found in the lowest quartiles for whole‐brain atrophy and patients with a CDR of 0.5 showed top 25% brain atrophy. This discrepancy between neurodegeneration and disease severity may be mediated by cognitive reserve factors, comorbid conditions like vascular disease or depression, or more focal atrophy in highly strategic brain regions. Alternatively, this discrepancy may represent misclassification of disease severity by the CDR given the bias to more heavily weigh memory‐related symptoms. Contrary to most previous studies which showed a right hemisphere predilection in PCA [Crutch et al., 2012; Migliaccio et al., 2009], visual inspection shows a slight preponderance of left hemisphere brain atrophy in PCA patients (Fig. 2C) using the CDR approach, which was less visible using the W‐score method (Fig. 4D). Conversely, prefrontal cortex atrophy in LOAD was more prominent using the W‐score (Fig. 4B) than the CDR approach (Fig. 2D). It is beyond the scope of this study to perform head‐to‐head statistical comparisons between the two methods but despite the differences, both approaches yielded largely congruent results indicating a certain robustness and reliability.
Strengths and Limitations
A strength of this study is the large sample of AD variants that uniquely cover the earliest clinically detectable stages. A limitation is that, due to low prevalence of some AD variants and scarcity of longitudinal data, we attempted to make inferences about disease progression based on cross‐sectional data. Our findings are in line, however, with small longitudinal series [Lehmann et al., 2012; Rohrer et al., 2013] but need to be confirmed in larger longitudinal studies. Second, data were acquired on different MRI scanners with different field strengths. We have optimized data quality by thorough visual inspection after each step in the pre‐processing pipeline. Additionally, a homogeneity test implemented in the SPM software was used to detect potential outliers, and all analyses were performed using an extensive set of covariates including MRI field strength. Also, a stringent statistical threshold was applied to the data (P < 0.05 family wise error corrected) to minimize false positive findings. Third, while underlying AD pathology was confirmed by autopsy or supported by biomarkers in 68% of patients, the possibility that some of the subjects may harbor non‐AD pathologies that are driving the atrophy patterns cannot be excluded. Finally, the control group was matched with the early‐onset AD variants thus age (though included as a covariate) may have influenced comparisons against significantly older LOAD patients. We therefore additionally included an older control group and voxelwise contrasts indicated that atrophy patterns were independent of the control group, except for brain atrophy in the frontal cortex when applying the W‐score method. This finding should thus to be interpreted with caution.
Future Directions
Future studies should focus on why (in addition to how) heterogeneity occurs in AD, evaluating for potential genetic modifiers, environmental exposures and developmental vulnerabilities that drive neurodegeneration into specific brain regions. For example, individuals with developmental language disabilities such as dyslexia are overrepresented in lvPPA populations [Miller et al., 2013; Rogalski et al., 2008], while Apolipoprotein E ε4‐carriers are more prone to exhibit an amnestic‐predominant phenotype of AD [Lehmann et al., 2014; Ossenkoppele et al., 2013b; van der Flier et al., 2011]. Also, the introduction of novel tau PET tracers [Chien et al., 2013; Maruyama et al., 2013; Okamura et al., 2014] will allow in vivo investigation of the regional distribution of tangle pathology across AD variants, and of its relationships with Aβ, hypometabolism, brain atrophy and cognition in each clinical phenotype. Preliminary findings suggest that neurodegeneration and symptomatology better correspond to the regional distribution of tau than that of Aβ pathology [Ossenkoppele et al., 2015], but further investigation is warranted.
CONCLUSION
In the earliest clinically detectable stages of four distinct AD variants, we found both shared vulnerability in posterior DMN regions across patients and marked syndrome‐specific atrophy patterns in off‐DMN networks that anatomically overlapped with brain networks responsible for critical cognitive functions uniquely affected in each AD variant (language network in lvPPA, PCC‐hippocampal circuit in amnestic‐predominant EOAD and LOAD, and visual network in PCA). This supports a network‐based model of neurodegeneration in which vulnerable brain regions are selectively targeted through both syndrome‐specific and common networks across AD variants.
Supporting information
Supporting Information Table 1.
REFERENCES
- Adriaanse SM, Binnewijzend MA, Ossenkoppele R, Tijms BM, van der Flier WM, Koene T, Smits LL, Wink AM, Scheltens P, van Berckel BN, Barkhof F (2014): Widespread disruption of functional brain organization in early‐onset Alzheimer's disease. PLoS One 9:e102995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, Gamst A, Holtzman DM, Jagust WJ, Petersen RC, Snyder PJ, Carrillo MC, Thies B, Phelps CH (2011): The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 7:270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alladi S, Xuereb J, Bak T, Nestor P, Knibb J, Patterson K, Hodges JR (2007): Focal cortical presentations of Alzheimer's disease. Brain 130:2636–2645. [DOI] [PubMed] [Google Scholar]
- Andrews‐Hanna JR, Reidler JS, Sepulcre J, Poulin R, Buckner RL (2010): Functional‐anatomic fractionation of the brain's default network. Neuron 65:550–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach TG, Monsell SE, Phillips LE, Kukull W (2012): Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer Disease Centers, 2005‐2010. J Neuropathol Exp Neurol 71:266–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner RL, Snyder AZ, Shannon BJ, LaRossa G, Sachs R, Fotenos AF, Sheline YI, Klunk WE, Mathis CA, Morris JC, Mintun MA (2005): Molecular, structural, and functional characterization of Alzheimer's disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci 25:7709–7717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanna AE, Trimble MR (2006): The precuneus: a review of its functional anatomy and behavioural correlates. Brain 129:564–583. [DOI] [PubMed] [Google Scholar]
- Chan LT, Lynch W, De May M, Horton JC, Miller BL, Rabinovici GD. (2015): Prodromal posterior cortical atrophy: clinical, neuropsychological, and radiological correlation. Neurocase 21:44–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien DT, Bahri S, Szardenings AK, Walsh JC, Mu F, Su MY, Shankle WR, Elizarov A, Kolb HC (2013): Early clinical PET imaging results with the novel PHF‐tau radioligand [F‐18]‐T807. J Alzheimers Dis 34:457–468. [DOI] [PubMed] [Google Scholar]
- Crutch SJ, Lehmann M, Schott JM, Rabinovici GD, Rossor MN, Fox NC (2012): Posterior cortical atrophy. Lancet Neurol 11:170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damoiseaux JS, Prater KE, Miller BL, Greicius MD (2012): Functional connectivity tracks clinical deterioration in Alzheimer's disease. Neurobiol Aging 33:828 e19–e830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois B, Feldman HH, Jacova C, Hampel H, Molinuevo JL, Blennow K, DeKosky ST, Gauthier S, Selkoe D, Bateman R, Cappa S, Crutch S, Engelborghs S, Frisoni GB, Fox NC, Galasko D, Habert MO, Jicha GA, Nordberg A, Pasquier F, Rabinovici G, Robert P, Rowe C, Salloway S, Sarazin M, Epelbaum S, de Souza LC, Vellas B, Visser PJ, Schneider L, Stern Y, Scheltens P, Cummings JL (2014): Advancing research diagnostic criteria for Alzheimer's disease: the IWG‐2 criteria. Lancet Neurol 13:614–629. [DOI] [PubMed] [Google Scholar]
- Frisoni GB, Pievani M, Testa C, Sabattoli F, Bresciani L, Bonetti M, Beltramello A, Hayashi KM, Toga AW, Thompson PM (2007): The topography of grey matter involvement in early and late onset Alzheimer's disease. Brain 130:720–730. [DOI] [PubMed] [Google Scholar]
- Gorno‐Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, Ogar JM, Rohrer JD, Black S, Boeve BF, Manes F, Dronkers NF, Vandenberghe R, Rascovsky K, Patterson K, Miller BL, Knopman DS, Hodges JR, Mesulam MM, Grossman M (2011): Classification of primary progressive aphasia and its variants. Neurology 76:1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greicius MD, Srivastava G, Reiss AL, Menon V (2004): Default‐mode network activity distinguishes Alzheimer's disease from healthy aging: evidence from functional MRI. Proc Natl Acad Sci USA 101:4637–4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman M (2010): Primary progressive aphasia: Clinicopathological correlations. Nat Rev Neurol 6:88–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr, Petersen RC, Xu YC, Waring SC, O'Brien PC, Tangalos EG, Smith GE, Ivnik RJ, Kokmen E (1997): Medial temporal atrophy on MRI in normal aging and very mild Alzheimer's disease. Neurology 49:786–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koedam EL, Lauffer V, van der Vlies AE, van der Flier WM, Scheltens P, Pijnenburg YA (2010): Early‐versus late‐onset Alzheimer's disease: more than age alone. J Alzheimers Dis 19:1401–1408. [DOI] [PubMed] [Google Scholar]
- Koedam EL, Lehmann M, van der Flier WM, Scheltens P, Pijnenburg YA, Fox N, Barkhof F, Wattjes MP (2011): Visual assessment of posterior atrophy development of a MRI rating scale. Eur Radiol 21:2618–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koss E, Edland S, Fillenbaum G, Mohs R, Clark C, Galasko D, Morris JC (1996): Clinical and neuropsychological differences between patients with earlier and later onset of Alzheimer's disease: A CERAD analysis, Part XII. Neurology 46:136–141. [DOI] [PubMed] [Google Scholar]
- La Joie R, Perrotin A, Barre L, Hommet C, Mezenge F, Ibazizene M, Camus V, Abbas A, Landeau B, Guilloteau D, de La Sayette V, Eustache F, Desgranges B, Chetelat G (2012): Region‐specific hierarchy between atrophy, hypometabolism, and beta‐amyloid (Abeta) load in Alzheimer's disease dementia. J Neurosci 32:16265–16273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leech R, Kamourieh S, Beckmann CF, Sharp DJ (2011): Fractionating the default mode network: Distinct contributions of the ventral and dorsal posterior cingulate cortex to cognitive control. J Neurosci 31:3217–3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leech R, Sharp DJ (2014): The role of the posterior cingulate cortex in cognition and disease. Brain 137:12–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann M, Barnes J, Ridgway GR, Ryan NS, Warrington EK, Crutch SJ, Fox NC (2012): Global gray matter changes in posterior cortical atrophy: a serial imaging study. Alzheimers Dement 8:502–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann M, Ghosh PM, Madison C, Laforce R Jr, Corbetta‐Rastelli C, Weiner MW, Greicius MD, Seeley WW, Gorno‐Tempini ML, Rosen HJ, Miller BL, Jagust WJ, Rabinovici GD (2013a): Diverging patterns of amyloid deposition and hypometabolism in clinical variants of probable Alzheimer's disease. Brain 136:844–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann M, Madison CM, Ghosh PM, Seeley WW, Mormino E, Greicius MD, Gorno‐Tempini ML, Kramer JH, Miller BL, Jagust WJ, Rabinovici GD (2013b): Intrinsic connectivity networks in healthy subjects explain clinical variability in Alzheimer's disease. Proc Natl Acad Sci USA 110:11606–11611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann M, Ghosh PM, Madison C, Karydas A, Coppola G, O'Neil JP, Huang Y, Miller BL, Jagust WJ, Rabinovici GD (2014): Greater medial temporal hypometabolism and lower cortical amyloid burden in ApoE4‐positive AD patients. J Neurol Neurosurg Psychiatry 85:266–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavan A, Whitwell JL, Weigand SD, Duffy JR, Strand EA, Machulda MM, Tosakulwong N, Senjem ML, Gunter JL, Lowe VJ, et al. (2013): FDG PET and MRI in logopenic primary progressive aphasia versus dementia of the Alzheimer's type. PLoS One 8:e62471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies DS, Vincent JL, Kelly C, Lohmann G, Uddin LQ, Biswal BB, Villringer A, Castellanos FX, Milham MP, Petrides M (2009): Precuneus shares intrinsic functional architecture in humans and monkeys. Proc Natl Acad Sci USA 106:20069–20074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama M, Shimada H, Suhara T, Shinotoh H, Ji B, Maeda J, Zhang MR, Trojanowski JQ, Lee VM, Ono M, Masamoto K, Takano H, Sahara N, Iwata N, Okamura N, Furumoto S, Kudo Y, Chang Q, Saido TC, Takashima A, Lewis J, Jang MK, Aoki I, Ito H, Higuchi M (2013): Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared with normal controls. Neuron 79:1094–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr, Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, Mohs RC, Morris JC, Rossor MN, Scheltens P, Carrillo MC, Thies B, Weintraub S, Phelps CH (2011): The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 7:263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez MF, Ghajarania M, Perryman KM (2002): Posterior cortical atrophy: Clinical characteristics and differences compared with Alzheimer's disease. Dement Geriatr Cogn Disord 14:33–40. [DOI] [PubMed] [Google Scholar]
- Mendez MF, Lee AS, Joshi A, Shapira JS (2012): Nonamnestic presentations of early‐onset Alzheimer's disease. Am J Alzheimers Dis Other Demen 27:413–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliaccio R, Agosta F, Rascovsky K, Karydas A, Bonasera S, Rabinovici GD, Miller BL, Gorno‐Tempini ML (2009): Clinical syndromes associated with posterior atrophy: Early age at onset AD spectrum. Neurology 73:1571–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller ZA, Mandelli ML, Rankin KP, Henry ML, Babiak MC, Frazier DT, Lobach IV, Bettcher BM, Wu TQ, Rabinovici GD, Graff‐Radford NR, Miller BL, Gorno‐Tempini ML (2013): Handedness and language learning disability differentially distribute in progressive aphasia variants. Brain 136:3461–3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller C, Vrenken H, Jiskoot L, Versteeg A, Barkhof F, Scheltens P, van der Flier WM (2013): Different patterns of gray matter atrophy in early‐ and late‐onset Alzheimer's disease. Neurobiol Aging 34:2014–2022. [DOI] [PubMed] [Google Scholar]
- Morris JC (1993): The Clinical Dementia Rating (CDR): Current version and scoring rules. Neurology 43:2412–2414. [DOI] [PubMed] [Google Scholar]
- Nelson SM, Cohen AL, Power JD, Wig GS, Miezin FM, Wheeler ME, Velanova K, Donaldson DI, Phillips JS, Schlaggar BL, et al. (2010): A parcellation scheme for human left lateral parietal cortex. Neuron 67:156–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura N, Furumoto S, Fodero‐Tavoletti MT, Mulligan RS, Harada R, Yates P, Pejoska S, Kudo Y, Masters CL, Yanai K, Rowe CC, Villemagne VL (2014): Non‐invasive assessment of Alzheimer's disease neurofibrillary pathology using 18F‐THK5105 PET. Brain 137:1762–1771. [DOI] [PubMed] [Google Scholar]
- Ossenkoppele R, Tolboom N, Foster‐Dingley JC, Adriaanse SF, Boellaard R, Yaqub M, Windhorst AD, Barkhof F, Lammertsma AA, Scheltens P, van der Flier WM, van Berckel BN (2012): Longitudinal imaging of Alzheimer pathology using [11C]PIB, [18F]FDDNP and [18F]FDG PET. Eur J Nucl Med Mol Imaging 39:990–1000. [DOI] [PubMed] [Google Scholar]
- Ossenkoppele R, Prins ND, Pijnenburg YA, Lemstra AW, van der Flier WM, Adriaanse SF, Windhorst AD, Handels RL, Wolfs CA, Aalten P, Verhey FR, Verbeek MM, van Buchem MA, Hoekstra OS, Lammertsma AA, Scheltens P, van Berckel BN (2013a): Impact of molecular imaging on the diagnostic process in a memory clinic. Alzheimers Dement 9:414–421. [DOI] [PubMed] [Google Scholar]
- Ossenkoppele R, van der Flier WM, Zwan MD, Adriaanse SF, Boellaard R, Windhorst AD, Barkhof F, Lammertsma AA, Scheltens P, van Berckel BN (2013b): Differential effect of APOE genotype on amyloid load and glucose metabolism in AD dementia. Neurology 80:359–365. [DOI] [PubMed] [Google Scholar]
- Ossenkoppele R, van der Flier WM, Verfaillie SC, Vrenken H, Versteeg A, van Schijndel RA, Sikkes SA, Twisk J, Adriaanse SM, Zwan MD, Boellaard R, Windhorst AD, Barkhof F, Scheltens P, Lammertsma AA, van Berckel BN (2014): Long‐term effects of amyloid, hypometabolism, and atrophy on neuropsychological functions. Neurology 82: 1768–1775. [DOI] [PubMed] [Google Scholar]
- Ossenkoppele R, Schonhaut DR, Baker SL, O'Neil JP, Janabi M, Ghosh PM, Santos M, Miller ZA, Bettcher BM, Gorno‐Tempini ML, Miller BL, Jagust WJ, Rabinovici GD (2015): Tau, amyloid and hypometabolism in a patient with posterior cortical atrophy. Ann Neurol 77:338–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovici GD, Seeley WW, Kim EJ, Gorno‐Tempini ML, Rascovsky K, Pagliaro TA, Allison SC, Halabi C, Kramer JH, Johnson JK, Weiner MW, Forman MS, Trojanowski JQ, Dearmond SJ, Miller BL, Rosen HJ (2007): Distinct MRI atrophy patterns in autopsy‐proven Alzheimer's disease and frontotemporal lobar degeneration. Am J Alzheimers Dis Other Demen 22:474–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovici GD, Jagust WJ, Furst AJ, Ogar JM, Racine CA, Mormino EC, O'Neil JP, Lal RA, Dronkers NF, Miller BL, Gorno‐Tempini ML (2008): Abeta amyloid and glucose metabolism in three variants of primary progressive aphasia. Ann Neurol 64:388–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovici GD, Furst AJ, Alkalay A, Racine CA, O'Neil JP, Janabi M, Baker SL, Agarwal N, Bonasera SJ, Mormino EC, Weiner MW, Gorno‐Tempini ML, Rosen HJ, Miller BL, Jagust WJ (2010): Increased metabolic vulnerability in early‐onset Alzheimer's disease is not related to amyloid burden. Brain 133:512–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, Kuceyeski A, Weiner M (2012): A network diffusion model of disease progression in dementia. Neuron 73:1204–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner JA, Burns JM, Hou CE, McKeel DW Jr, Storandt M, Morris JC (2004): Progressive posterior cortical dysfunction: A clinicopathologic series. Neurology 63:1175–1180. [DOI] [PubMed] [Google Scholar]
- Ridgway GR, Lehmann M, Barnes J, Rohrer JD, Warren JD, Crutch SJ, Fox NC (2012): Early‐onset Alzheimer disease clinical variants: Multivariate analyses of cortical thickness. Neurology 79:80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogalski E, Johnson N, Weintraub S, Mesulam M (2008): Increased frequency of learning disability in patients with primary progressive aphasia and their first‐degree relatives. Arch Neurol 65:244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogalski E, Cobia D, Martersteck A, Rademaker A, Wieneke C, Weintraub S, Mesulam MM (2014): Asymmetry of cortical decline in subtypes of primary progressive aphasia. Neurology 83:1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrer JD, Caso F, Mahoney C, Henry M, Rosen HJ, Rabinovici G, Rossor MN, Miller B, Warren JD, Fox NC, Ridgway GR, Gorno‐Tempini ML (2013): Patterns of longitudinal brain atrophy in the logopenic variant of primary progressive aphasia. Brain Lang 127:121–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeley WW, Crawford R, Rascovsky K, Kramer JH, Weiner M, Miller BL, Gorno‐Tempini ML (2008): Frontal paralimbic network atrophy in very mild behavioral variant frontotemporal dementia. Arch Neurol 65:249–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD (2009): Neurodegenerative diseases target large‐scale human brain networks. Neuron 62:42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirer WR, Ryali S, Rykhlevskaia E, Menon V, Greicius MD (2012): Decoding subject‐driven cognitive states with whole‐brain connectivity patterns. Cereb Cortex 22:158–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluimer JD, van der Flier WM, Karas GB, Fox NC, Scheltens P, Barkhof F, Vrenken H (2008): Whole‐brain atrophy rate and cognitive decline: longitudinal MR study of memory clinic patients. Radiology 248:590–598. [DOI] [PubMed] [Google Scholar]
- Smits LL, Pijnenburg YA, Koedam EL, van der Vlies AE, Reuling IE, Koene T, Teunissen CE, Scheltens P, van der Flier WM (2012): Early onset Alzheimer's disease is associated with a distinct neuropsychological profile. J Alzheimers Dis 30:101–108. [DOI] [PubMed] [Google Scholar]
- Stopford CL, Snowden JS, Thompson JC, Neary D (2008): Variability in cognitive presentation of Alzheimer's disease. Cortex 44:185–195. [DOI] [PubMed] [Google Scholar]
- Teichmann M, Kas A, Boutet C, Ferrieux S, Nogues M, Samri D, Rogan C, Dormont D, Dubois B, Migliaccio R (2013): Deciphering logopenic primary progressive aphasia: A clinical, imaging and biomarker investigation. Brain 136:3474–3488. [DOI] [PubMed] [Google Scholar]
- Thomas JB, Brier MR, Bateman RJ, Snyder AZ, Benzinger TL, Xiong C, Raichle M, Holtzman DM, Sperling RA, Mayeux R, et al. (2014): Functional connectivity in autosomal dominant and late‐onset Alzheimer disease. JAMA Neurol 71:1111–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Heuvel MP, Kahn RS, Goni J, Sporns O (2012): High‐cost, high‐capacity backbone for global brain communication. Proc Natl Acad Sci USA 109:11372–11377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Flier WM, Pijnenburg YA, Fox NC, Scheltens P (2011): Early‐onset versus late‐onset Alzheimer's disease: the case of the missing APOE epsilon4 allele. Lancet Neurol 10:280–288. [DOI] [PubMed] [Google Scholar]
- van Vliet D, de Vugt ME, Bakker C, Koopmans RT, Pijnenburg YA, Vernooij‐Dassen MJ, Verhey FR (2011): Caregivers' perspectives on the pre‐diagnostic period in early onset dementia: A long and winding road. Int Psychogeriatr 23:1393–1404. [DOI] [PubMed] [Google Scholar]
- Zhang D, Raichle ME (2010): Disease and the brain's dark energy. Nat Rev Neurol 6:15–28. [DOI] [PubMed] [Google Scholar]
- Zhou J, Gennatas ED, Kramer JH, Miller BL, Seeley WW (2012): Predicting regional neurodegeneration from the healthy brain functional connectome. Neuron 73:1216–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwan M, van Harten A, Ossenkoppele R, Bouwman F, Teunissen C, Adriaanse S, Lammertsma A, Scheltens P, van Berckel B, van der Flier W (2014): Concordance between cerebrospinal fluid biomarkers and [11C]PIB PET in a memory clinic cohort. J Alzheimers Dis 41:801–807. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Table 1.