

Abstract
For aggregation tests of genes or regions, the set of included variants often have small total minor allele counts (MACs), and this is particularly true when the most deleterious sets of variants are considered. When MAC is low, commonly used asymptotic tests are not well calibrated for binary phenotypes and can have conservative or anti-conservative results and potential power loss. Empirical 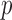 -values obtained via resampling methods are computationally costly for highly significant
-values obtained via resampling methods are computationally costly for highly significant 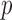 -values and the results can be conservative due to the discrete nature of resampling tests. Based on the observation that only the individuals containing minor alleles contribute to the score statistics, we develop an efficient resampling method for single and multiple variant score-based tests that can adjust for covariates. Our method can improve computational efficiency
-values and the results can be conservative due to the discrete nature of resampling tests. Based on the observation that only the individuals containing minor alleles contribute to the score statistics, we develop an efficient resampling method for single and multiple variant score-based tests that can adjust for covariates. Our method can improve computational efficiency 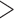 1000-fold over conventional resampling for low MAC variant sets. We ameliorate the conservativeness of results through the use of mid-
1000-fold over conventional resampling for low MAC variant sets. We ameliorate the conservativeness of results through the use of mid- -values. Using the estimated minimum achievable
-values. Using the estimated minimum achievable 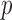 -value for each test, we calibrate QQ plots and provide an effective number of tests. In analysis of a case–control study with deep exome sequence, we demonstrate that our methods are both well calibrated and also reduce computation time significantly compared with resampling methods.
-value for each test, we calibrate QQ plots and provide an effective number of tests. In analysis of a case–control study with deep exome sequence, we demonstrate that our methods are both well calibrated and also reduce computation time significantly compared with resampling methods.
Keywords: Rare variants, Next generation sequencing, Resampling methods
1. Introduction
Recent advances in sequencing technologies have made it possible to investigate the role of rare variants in complex diseases, and numerous statistical methods have been developed to identify rare variant associations. Many of the currently popular gene- or region-based multiple variants tests are based on individual variant score statistics, which provide rapid computation and natural adjustment for covariates (Lee and others, 2014). For example, variance component tests use the weighted sum of squared individual variant score statistics as in C-alpha (Neale and others, 2011), SSU (Pan, 2009), and SKAT (Wu and others, 2011). Many versions of burden tests (Li and Leal, 2008; Lin and Tang, 2011; Madsen and Browning, 2009) are essentially equivalent to collapsing the individual variant score statistics. Other examples include SKAT-O (Lee, Emond, and others, 2012; Lee, Wu, and others, 2012) and Fisher method (Derkach and others, 2012; Sun and others, 2013).
For a given gene or region, the number of variants tested together and the total of their minor allele counts (MACs) can vary due to the sequence or genotyping coverage of the gene, the class of variants tested, and the sample size. In the context of gene-based tests, we use MAC to refer to the total MAC of all variants in a tested set (i.e. the sum of the MAC of the rare, low, and common frequency variants in the set) and in the context of single variant tests, MAC refers to the single variant MAC.
In exome-sequencing studies, one approach (among many) is to test disruptive or predictively damaging variants (Zuk and others, 2014). These tend to be very rare and tests based on these variants often have sets of variants with very small total MACs 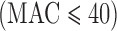 . Asymptotic-based score tests for a single variant with small MAC, however, yield conservative results under a balanced case–control design, and anti-conservative results under an unbalanced case–control design (Ma and others, 2013). This lack of calibration can lead to lack of calibration in gene- or region-based asymptotic score tests. A moment-based adjustment (MA) was developed to improve the Type I error control when testing variant sets with low MAC; however, this approach is also based on the asymptotic properties of the tests (Lee, Emond, and others, 2012; Lee, Wu, and others, 2012) and may be less well calibrated, when testing for very low MAC variant sets. An alternative approach would be to perform experiment-wise permutation to control family wise error rate by obtaining the empirical distribution of asymptotic
. Asymptotic-based score tests for a single variant with small MAC, however, yield conservative results under a balanced case–control design, and anti-conservative results under an unbalanced case–control design (Ma and others, 2013). This lack of calibration can lead to lack of calibration in gene- or region-based asymptotic score tests. A moment-based adjustment (MA) was developed to improve the Type I error control when testing variant sets with low MAC; however, this approach is also based on the asymptotic properties of the tests (Lee, Emond, and others, 2012; Lee, Wu, and others, 2012) and may be less well calibrated, when testing for very low MAC variant sets. An alternative approach would be to perform experiment-wise permutation to control family wise error rate by obtaining the empirical distribution of asymptotic 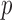 -values across variant sets (Kiezun and others, 2012). However, because the degree of miscalibration for asymptotic
-values across variant sets (Kiezun and others, 2012). However, because the degree of miscalibration for asymptotic  -values can vary by MAC, this approach may have reduced power to detect specific classes of causal multiple variant sets.
-values can vary by MAC, this approach may have reduced power to detect specific classes of causal multiple variant sets.
Resampling methods, such as permutation tests, do not rely on the asymptotic properties of the test (Efron and Tibshirani, 1994). Permutation tests for genetic data often permute case and control status without regard to differential odds of individual being a case based on covariates. This approach can result in the inflated Type I error rates in the presence of confounding covariates, such as population stratification (Epstein and others, 2012). In a more nuanced approach, permutations can be performed within strata of one or more covariates, such as geographical region, so the underlying null distribution provides a better match to the observed test statistic (Purcell and others, 2007). In the presence of continuous covariates, such as principal components which are used to adjust for population stratification, Fisher's noncentral hypergeometric distribution-based permutation can be performed, allowing for individuals to have different odds of being selected as a case (Efron and Tibshirani, 1994; Fog, 2008). The major limitation of the permutation approach is that disease status is permuted across all study participants, requiring significant computational cost, which increases as sample sizes become larger. Adaptive permutation procedures can reduce computational time for the estimation of large or moderate 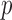 -values (Efron and Tibshirani, 1994), but substantial time is still required to estimate highly significant
-values (Efron and Tibshirani, 1994), but substantial time is still required to estimate highly significant  -values. In addition, permutation
-values. In addition, permutation 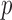 -values tend to be conservative for binary traits with small MAC, since test statistics are discrete (Lancaster, 1961).
-values tend to be conservative for binary traits with small MAC, since test statistics are discrete (Lancaster, 1961).
In this paper, we develop an efficient resampling (ER) method for score statistic-based single and multiple variant tests that improves computational efficiency. Our method is based on the insight that only individuals with minor alleles (assuming the minor allele is coded as one) contribute to the score test. Instead of permuting case–control status across all individuals, resampling can be performed by resampling the case–control status of individuals with a minor allele at a given variant (for a single variant test), and similarly, individuals with minor alleles at any included variants (for a multiple variant test). Within the group of individuals with minor alleles, we allow for covariate adjustment through the use of Fisher's noncentral hyper-geometric distribution (Epstein and others, 2012; Fog, 2008). The computational time for the ER method increases as the MAC increases, so we developed a method for moderate to high variant set MAC  in which quantiles of the test statistics are estimated through ER (based on a more limited number of permutations) and then used to better-calibrate our moment-matching approximation quantile adjustment (QA).
in which quantiles of the test statistics are estimated through ER (based on a more limited number of permutations) and then used to better-calibrate our moment-matching approximation quantile adjustment (QA).
Furthermore, we develop statistical approaches to calibrate the discrete nature of test statistics. Using the ER method, we obtain mid- -values (Lancaster, 1961). We estimate the lower limit of
-values (Lancaster, 1961). We estimate the lower limit of 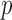 -values for each variant set (minimum achievable
-values for each variant set (minimum achievable 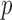 -values (MAP)) (Kiezun and others, 2012), using the exact resampling distribution. We use the MAP to estimate the effective number of tests and to calibrate quantile–quantile (QQ) plots. Through simulation-based work and analysis of deep exome sequencing data, we demonstrate that the ER-based methods and calibration approaches are computationally efficient, control the false-positive rate (FPR) and can improve power.
-values (MAP)) (Kiezun and others, 2012), using the exact resampling distribution. We use the MAP to estimate the effective number of tests and to calibrate quantile–quantile (QQ) plots. Through simulation-based work and analysis of deep exome sequencing data, we demonstrate that the ER-based methods and calibration approaches are computationally efficient, control the false-positive rate (FPR) and can improve power.
2. Methods
2.1. Statistical model and rare variant tests
To understand currently used rare variant tests, suppose that 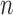 subjects are sequenced with
subjects are sequenced with 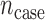 diseased individuals. The region being tested has
diseased individuals. The region being tested has 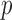 variant loci. For the
variant loci. For the 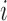 th subject, let
th subject, let 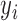 denote a binary phenotype,
denote a binary phenotype,  the number of copies of the minor allele (
the number of copies of the minor allele ( ), and
), and 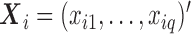 the covariates. MAC is defined as the sum of all genotype values,
the covariates. MAC is defined as the sum of all genotype values, 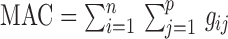 . To relate genotypes to binary phenotypes, we posit the logistic regression model,
. To relate genotypes to binary phenotypes, we posit the logistic regression model, 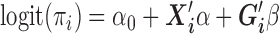 , where
, where 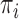 is a disease probability,
is a disease probability, 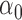 is the intercept, and
is the intercept, and 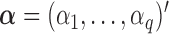 and
and 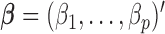 are regression coefficients of covariates and genetic variants, respectively. A score statistic from a marginal model of variant
are regression coefficients of covariates and genetic variants, respectively. A score statistic from a marginal model of variant  is
is
 |
(2.1) |
where  is an estimate of
is an estimate of 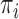 under the null model
under the null model  . For single variant tests,
. For single variant tests, 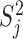 is the score test statistic of variant
is the score test statistic of variant 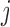 and follows a (scaled)
and follows a (scaled) 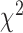 distribution with
distribution with 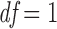 . Many popular gene- or region-based tests are also based on
. Many popular gene- or region-based tests are also based on 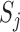 . For example, Burden and SKAT test statistics can be written as a weighted linear and quadratic sum of
. For example, Burden and SKAT test statistics can be written as a weighted linear and quadratic sum of 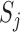
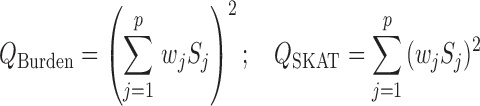 |
where 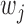 is a weight for variant
is a weight for variant  . SKAT-O combines Burden test and SKAT using the following framework as
. SKAT-O combines Burden test and SKAT using the following framework as  . Since the optimal
. Since the optimal 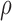 is not known in prior, SKAT-O uses the minimum
is not known in prior, SKAT-O uses the minimum 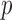 -values over a grid of
-values over a grid of 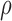 as a test statistic.
as a test statistic.
2.2. ER method
In this section, we present the ER method for rare variant score tests with binary traits. We describe the generation of 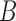 resamples to estimate the following four probabilities of the gene or region-based association test statistic
resamples to estimate the following four probabilities of the gene or region-based association test statistic 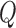 , which is a function of
, which is a function of 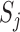 (
(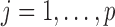 ), given genotypes (
), given genotypes (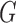 ), phenotypes (
), phenotypes (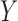 ) and covariates (
) and covariates (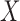 ):
):
ER
 -value:
-value: 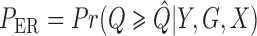
ER mid-
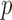 -value:
-value: 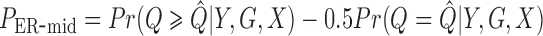
ER minimum achievable
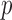 -value:
-value: 
ER minimum achievable mid-
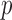 -value:
-value: 
where  is a test statistic from the original phenotype, and
is a test statistic from the original phenotype, and 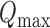 is the maximum of all possible permutation test statistics. Let
is the maximum of all possible permutation test statistics. Let 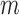 (
(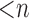 ) be the number of individuals with minor alleles in the gene or region,
) be the number of individuals with minor alleles in the gene or region, 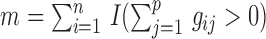 , where
, where 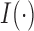 is an indicator function. It is apparent that
is an indicator function. It is apparent that 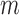 is smaller than or equal to MAC. From Equation (2.1), only individuals with minor alleles contribute to
is smaller than or equal to MAC. From Equation (2.1), only individuals with minor alleles contribute to  , since the remaining individuals have zero genotype values for all of their loci. This observation allows us to reduce the computation time by restricting resampling to the case–control status of those
, since the remaining individuals have zero genotype values for all of their loci. This observation allows us to reduce the computation time by restricting resampling to the case–control status of those 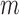 individuals only, rather than using all
individuals only, rather than using all 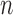 individuals. To estimate ER
individuals. To estimate ER 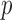 -values, we use a two-step approach that is based on the fact that
-values, we use a two-step approach that is based on the fact that 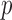 -value can be factorized as
-value can be factorized as
 |
where  is the number of cases among
is the number of cases among 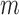 individuals carrying a minor allele in the tested region.
individuals carrying a minor allele in the tested region.
Step 1 is to estimate  . If there are no covariates to adjust for,
. If there are no covariates to adjust for,  follows the central-hypergeometric distribution. When there are covariates to adjust for, we use Fisher's noncentral hypergeometric distribution, which allows each individual to have different odds of being a case (Fog, 2008). Since estimating
follows the central-hypergeometric distribution. When there are covariates to adjust for, we use Fisher's noncentral hypergeometric distribution, which allows each individual to have different odds of being a case (Fog, 2008). Since estimating 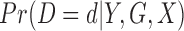 while allowing all individuals to have different odds is computationally challenging, we propose to stratify the
while allowing all individuals to have different odds is computationally challenging, we propose to stratify the 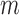 individuals into groups based on
individuals into groups based on 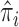 and to assume an average common odds for all individuals within the same stratum. The
and to assume an average common odds for all individuals within the same stratum. The 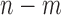 individuals without variants are treated as a single group (Supplementary Appendix A). The only use of this stratification is to estimate
individuals without variants are treated as a single group (Supplementary Appendix A). The only use of this stratification is to estimate 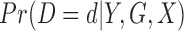 for the
for the 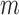 individuals in Step 1. We used 10 strata for the
individuals in Step 1. We used 10 strata for the 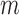 individuals, for a total of 11 strata.
individuals, for a total of 11 strata.
In Step 2, we estimate 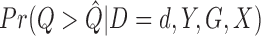 by generating
by generating  permutations of the case–control status of
permutations of the case–control status of 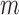 individuals. Suppose
individuals. Suppose 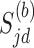 is the
is the 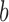 th resample of
th resample of 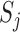 given
given 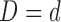 , and
, and  is the resulting test statistic
is the resulting test statistic 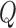 . Examples of
. Examples of  include the resampled Burden and SKAT test statistics,
include the resampled Burden and SKAT test statistics,  and
and  . The probability for the
. The probability for the 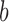 th resample given
th resample given  , say
, say 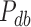 , is also calculated using Fisher's noncentral hypergeometric distribution at the level of each individual in
, is also calculated using Fisher's noncentral hypergeometric distribution at the level of each individual in 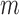 (rather than the level of strata as in Step 1). Then the estimator of
(rather than the level of strata as in Step 1). Then the estimator of 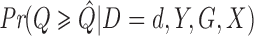 is
is  and the ER
and the ER 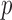 -value is
-value is
 |
The estimator of ER-mid 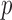 -value is
-value is
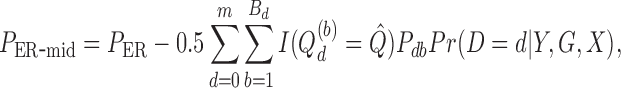 |
where the second term is an estimator of the tie probability. Suppose 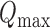 is the maximum of over all
is the maximum of over all  and
and  (i.e.
(i.e. 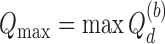 ). Then, estimators of
). Then, estimators of 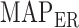 and
and 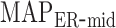 are
are
 |
The detailed derivations of Steps 1 and 2 are given in Supplementary Appendix A.
The computational complexity of the proposed method is  (Bmp) for SKAT and SKAT-O, and
(Bmp) for SKAT and SKAT-O, and 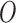 (Bm) for single variant and Burden tests, respectively. The computation complexity can be further reduced if the total number of configurations of case–control status (
(Bm) for single variant and Burden tests, respectively. The computation complexity can be further reduced if the total number of configurations of case–control status (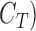 is small. For example, the total number of configurations of case–control status is 1024 when
is small. For example, the total number of configurations of case–control status is 1024 when  , indicating that we only need to evaluate 1024 possible configurations to obtain the exact resampling distribution. We note that we estimate MAPs when the exact resampling distribution is obtained (i.e.
, indicating that we only need to evaluate 1024 possible configurations to obtain the exact resampling distribution. We note that we estimate MAPs when the exact resampling distribution is obtained (i.e.  ); otherwise, the MAP estimates are not accurate. Since the computational cost of ER increases as
); otherwise, the MAP estimates are not accurate. Since the computational cost of ER increases as  increases, it may not be practical to use ER for variant sets with moderate or large MAC. We develop ER-based QA moment matching (Supplementary Appendix B) for these variant sets, which produces more accurate
increases, it may not be practical to use ER for variant sets with moderate or large MAC. We develop ER-based QA moment matching (Supplementary Appendix B) for these variant sets, which produces more accurate 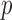 -values than the moment matching adjustment and yet provides fast computation for moderate or large MAC variant sets.
-values than the moment matching adjustment and yet provides fast computation for moderate or large MAC variant sets.
Because Bonferroni correction and QQ plots assume that  -values have a uniform distribution, they cannot correctly account for the fact that resampling
-values have a uniform distribution, they cannot correctly account for the fact that resampling 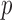 -values have lower limits, i.e., the MAPs. Kiezun and others (2012) proposed a heuristic approach in which to first identify variant sets with
-values have lower limits, i.e., the MAPs. Kiezun and others (2012) proposed a heuristic approach in which to first identify variant sets with 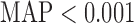 , and to count only these variant sets as the effective number of tests. We developed an alternative statistical approach to estimate the effective number of test and calibrating QQ plots using MAP (Supplementary Appendix C).
, and to count only these variant sets as the effective number of tests. We developed an alternative statistical approach to estimate the effective number of test and calibrating QQ plots using MAP (Supplementary Appendix C).
2.3. Numerical simulations
We generated 10 000 sequence haplotypes for an 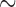 250 kbps region using a coalescent simulator FTEC (Reppell and others, 2012) with a faster-than-exponential growth model. In order to make variant sets having wide-ranges of MAC, we randomly selected a regions ranging from 125 to 12 500 bps, and then generated genotypes of variant sets using the simulated haplotypes. Three different case–control ratios were considered (1000:1000, 500:1500, and 500:1500). The binary phenotypes were generated from the logistic regression model:
250 kbps region using a coalescent simulator FTEC (Reppell and others, 2012) with a faster-than-exponential growth model. In order to make variant sets having wide-ranges of MAC, we randomly selected a regions ranging from 125 to 12 500 bps, and then generated genotypes of variant sets using the simulated haplotypes. Three different case–control ratios were considered (1000:1000, 500:1500, and 500:1500). The binary phenotypes were generated from the logistic regression model:
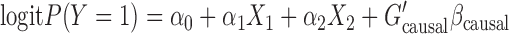 |
(2.2) |
where  is a genotype vector containing causal variants,
is a genotype vector containing causal variants, 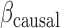 is a vector of genetic effect coefficients,
is a vector of genetic effect coefficients,  was a binary covariate of Bernoulli (0.5), and
was a binary covariate of Bernoulli (0.5), and 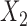 was a continuous covariate of
was a continuous covariate of 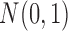 . The intercept
. The intercept  was chosen for the disease prevalence of 0.05. The non-genetic covariate coefficients
was chosen for the disease prevalence of 0.05. The non-genetic covariate coefficients 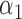 and
and 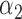 were 0 without covariates and 0.5 with covariates.
were 0 without covariates and 0.5 with covariates.
We applied five different methods to compute 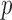 -values for each of the Burden, SKAT and SKAT-O tests: (i) ER with a
-values for each of the Burden, SKAT and SKAT-O tests: (i) ER with a 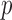 -value (ER); (ii) ER with a mid-
-value (ER); (ii) ER with a mid-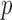 -value (ER-mid); (iii) QA moment matching; (iv) moment matching adjustment (MA); and (v) unadjusted (UA) asymptotic tests. To verify that ER and the whole-sample permutation methods produce essentially identical
-value (ER-mid); (iii) QA moment matching; (iv) moment matching adjustment (MA); and (v) unadjusted (UA) asymptotic tests. To verify that ER and the whole-sample permutation methods produce essentially identical  -values, we generated 20 000 variants sets and compared the
-values, we generated 20 000 variants sets and compared the 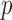 -values from ER and the permutation methods with and without covariates by generating 10
-values from ER and the permutation methods with and without covariates by generating 10 resamples (Supplementary Appendix E). We also compared computation times of SKAT-ER with whole-sample permutation for
resamples (Supplementary Appendix E). We also compared computation times of SKAT-ER with whole-sample permutation for 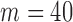 and total sample sizes ranging from 100 to 50 00 0 (Supplementary Appendix E).
and total sample sizes ranging from 100 to 50 00 0 (Supplementary Appendix E).
To compare the FPR for different ranges of total MAC, we considered six total MAC bins: 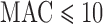 ;
; 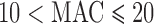 ;
;  ;
; 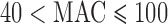 ;
;  ; and
; and  . For each bin, we used ranges of the number of variant sets
. For each bin, we used ranges of the number of variant sets  to 20 000, corresponding to candidate gene studies to genome-wide studies. In addition to FPR simulations, we carried out simulations to evaluate the power of ER and other tests. Details of FPR and power simulations can be found in Supplementary Appendix E.
to 20 000, corresponding to candidate gene studies to genome-wide studies. In addition to FPR simulations, we carried out simulations to evaluate the power of ER and other tests. Details of FPR and power simulations can be found in Supplementary Appendix E.
3. Results
3.1. Numerical simulations
We examine the FPR control, power, and computational time of two existing approaches, the MA and UA 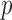 -value, and three newly developed ER-based methods, ER with
-value, and three newly developed ER-based methods, ER with 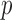 -value (ER), ER with mid-
-value (ER), ER with mid-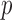 -value (ER-mid), and the ER-based quantile adjustment (QA) for single variant and multiple variant tests across a range of MAC and case–control imbalance. For simulation-based data, we generated sequence haplotypes with a European demographic model that mimics the MAF spectrum and linkage-disequilibrium (LD) structure of the current European population (Reppell and others, 2012). The MAF spectrum of simulated haplotypes was similar to that observed for the GoT2D exome sequencing data (Supplementary Figure S1).
-value (ER-mid), and the ER-based quantile adjustment (QA) for single variant and multiple variant tests across a range of MAC and case–control imbalance. For simulation-based data, we generated sequence haplotypes with a European demographic model that mimics the MAF spectrum and linkage-disequilibrium (LD) structure of the current European population (Reppell and others, 2012). The MAF spectrum of simulated haplotypes was similar to that observed for the GoT2D exome sequencing data (Supplementary Figure S1).
3.1.1. Comparison of p-values obtained using ER or whole-sample permutations
We compared SKAT  -values for 20 000 variant sets with total
-values for 20 000 variant sets with total 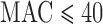 using the ER method to those obtained from whole-sample-based permutation, either in the absence of covariates (permutation of case–control status) or in the presence of covariates (using Fishers noncentral hypergeometric distribution). The
using the ER method to those obtained from whole-sample-based permutation, either in the absence of covariates (permutation of case–control status) or in the presence of covariates (using Fishers noncentral hypergeometric distribution). The  log 10
log 10  -values were very highly correlated (
-values were very highly correlated (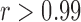 ) for tests with and without covariates, indicating that the ER-based results mirror those obtained from whole-sample-based permutation methods (Figure 1). We observed equally concordant
) for tests with and without covariates, indicating that the ER-based results mirror those obtained from whole-sample-based permutation methods (Figure 1). We observed equally concordant  -values for Burden and SKAT-O tests (data not shown).
-values for Burden and SKAT-O tests (data not shown).
Fig. 1.

Comparison of SKAT  -values obtained using ER or whole-sample
permutations. In the absence of covariates, SKAT
-values obtained using ER or whole-sample
permutations. In the absence of covariates, SKAT 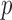 -values were obtained
through ER or whole-sample permutation (Perm) of disease status (top panel).
In the presence of covariates, SKAT
-values were obtained
through ER or whole-sample permutation (Perm) of disease status (top panel).
In the presence of covariates, SKAT 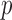 -value were obtained through ER or
Fisher's noncentral hypergeometric distribution based whole-sample permutation
(FNHPerm) implemented in the BiasedUrn R-package (bottom panel). From left
to the right, the plots consider case:
-value were obtained through ER or
Fisher's noncentral hypergeometric distribution based whole-sample permutation
(FNHPerm) implemented in the BiasedUrn R-package (bottom panel). From left
to the right, the plots consider case: :1000, 500:1500, and 200:1800, respectively.
The
:1000, 500:1500, and 200:1800, respectively.
The  -axis represents
-axis represents 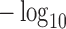 SKAT-ER
SKAT-ER  -values and
-values and 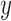 -axis represents
-axis represents 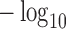 SKAT-Perm or
SKAT-FNHPerm
SKAT-Perm or
SKAT-FNHPerm 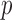 -values. Variant sets were randomly simulated, 20 000
sets with
-values. Variant sets were randomly simulated, 20 000
sets with  selected, and 10
selected, and 10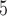 resamples were generated to compute
resamples were generated to compute 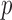 -values for each
method.
-values for each
method.
3.1.2. Comparison of computational times for the estimation of a significant gene-based p-value
To compare the computation times for a significant gene-based 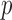 -value (0.05/20 000 genes), we generated 10
-value (0.05/20 000 genes), we generated 10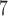 resamples for each method for a single variant set. This allows us to estimate a
resamples for each method for a single variant set. This allows us to estimate a 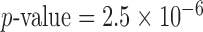 with a standard error
with a standard error 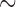 0.2 of
0.2 of  . When 40 individuals have minor alleles (MAC equal or slightly higher than 40), SKAT-ER with no covariates ran in
. When 40 individuals have minor alleles (MAC equal or slightly higher than 40), SKAT-ER with no covariates ran in 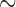 10 s and the computation times were invariant to sample size (100–50 000 samples). In contrast, for SKAT whole-sample permutations (SKAT-Perm), the computation time increased linearly with total sample size, from 0.35 to 10 h for 2000 and 50 000 samples, respectively (Figure 2(a)). With covariates, SKAT-ER also ran in
10 s and the computation times were invariant to sample size (100–50 000 samples). In contrast, for SKAT whole-sample permutations (SKAT-Perm), the computation time increased linearly with total sample size, from 0.35 to 10 h for 2000 and 50 000 samples, respectively (Figure 2(a)). With covariates, SKAT-ER also ran in 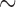 10 s and was invariant to sample size, whereas SKAT Fisher's noncentral hypergeometric distribution-based whole-sample permutations (SKAT-FNHPerm) using the BiasedUrn R-package took
10 s and was invariant to sample size, whereas SKAT Fisher's noncentral hypergeometric distribution-based whole-sample permutations (SKAT-FNHPerm) using the BiasedUrn R-package took  10 h for 2000 samples (Figure 2(b)). The running times for SKAT-ER-mid were nearly identical to those for SKAT-ER (data not shown). In existing programs, 10
10 h for 2000 samples (Figure 2(b)). The running times for SKAT-ER-mid were nearly identical to those for SKAT-ER (data not shown). In existing programs, 10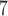 resamples of 2000 (50 000) samples with no covariates took 6 min (3.6 h) for C-alpha in PLINK/SEQ (and substantially longer for SKAT), and with covariates, took 6.4 h (
resamples of 2000 (50 000) samples with no covariates took 6 min (3.6 h) for C-alpha in PLINK/SEQ (and substantially longer for SKAT), and with covariates, took 6.4 h ( 240 h) in SCORE-Seq using the offered set of 5 gene-based tests (Supplementary Table S1).
240 h) in SCORE-Seq using the offered set of 5 gene-based tests (Supplementary Table S1).
Fig. 2.

Comparison of computation times for the estimation of a significant
gene-based 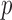 -value using ER and existing methods. Estimated computation
time for 10
-value using ER and existing methods. Estimated computation
time for 10 resamples of a single variant set for 40 individuals with minor
alleles (
resamples of a single variant set for 40 individuals with minor
alleles (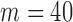 ) and varying numbers of total samples (balanced case:control) using
SKAT-ER or SKAT-Perm in the absence of covariates (a) or using SKAT-ER or
SKAT-FNHPerm in the presence of covariates (b). The BiasedUrn R-package was
used for SKAT-FNHPerm. Estimated computation time for 10
) and varying numbers of total samples (balanced case:control) using
SKAT-ER or SKAT-Perm in the absence of covariates (a) or using SKAT-ER or
SKAT-FNHPerm in the presence of covariates (b). The BiasedUrn R-package was
used for SKAT-FNHPerm. Estimated computation time for 10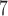 resamples of a single
variant set for 2000 samples (balanced case:control) in the presence of covariates
for SKAT-O, SKAT, or Burden test for
resamples of a single
variant set for 2000 samples (balanced case:control) in the presence of covariates
for SKAT-O, SKAT, or Burden test for 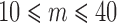 individuals with minor alleles using ER (c)
or for
individuals with minor alleles using ER (c)
or for 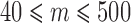 individuals with minor alleles using ER and QA (d). Each point represents
a median of 10 experiments. When
individuals with minor alleles using ER and QA (d). Each point represents
a median of 10 experiments. When 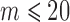 , the number of all possible configurations of
the case–control status of individuals with minor alleles was smaller than 10
, the number of all possible configurations of
the case–control status of individuals with minor alleles was smaller than 10 ; ER,
therefore, obtained the exact resampling
; ER,
therefore, obtained the exact resampling 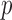 -values. The number of variant loci was
30 when
-values. The number of variant loci was
30 when 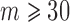 , otherwise, it was the same as
, otherwise, it was the same as 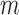 .
.
In contrast to the invariance by sample size, the computation time for ER increased with increasing number of individuals with minor alleles. For a single test with covariates, when the number of individuals with minor alleles 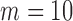 , 40, 100, and 500, SKAT-ER took 0.01, 10, 58, and 310 s; the burden test was faster and SKAT-O slower (Figures 2(c) and (d); Supplementary Table S2). When
, 40, 100, and 500, SKAT-ER took 0.01, 10, 58, and 310 s; the burden test was faster and SKAT-O slower (Figures 2(c) and (d); Supplementary Table S2). When 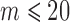 , computation took substantially less time because the total number of configurations of cases and controls among those
, computation took substantially less time because the total number of configurations of cases and controls among those 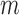 individuals was
individuals was  . The increase in computation time with increasing
. The increase in computation time with increasing 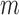 led us to develop a substantially faster (
led us to develop a substantially faster ( 6- to 18-fold) QA asymptotic method based on ER (QA) (Figure 2(d) and Supplementary Table S2). QA was essentially linear in
6- to 18-fold) QA asymptotic method based on ER (QA) (Figure 2(d) and Supplementary Table S2). QA was essentially linear in 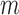 and invariant to sample size (data not shown). For comparison, with covariates for
and invariant to sample size (data not shown). For comparison, with covariates for 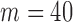 and sample size of 2000, the existing MA method for Burden, SKAT and SKAT-O took
and sample size of 2000, the existing MA method for Burden, SKAT and SKAT-O took  0.2 s (and was invariant to
0.2 s (and was invariant to  ), and UA for Burden, SKAT and SKAT-O took
), and UA for Burden, SKAT and SKAT-O took 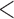 0.02 s (and was invariant to
0.02 s (and was invariant to 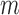 ) (data not shown).
) (data not shown).
3.1.3. FPRs for existing and ER-based methods
We compared empirical FPRs for variant sets for these five methods. We define the best-calibrated test as the one that had the FPR closest to but, at most, slightly exceeding the expected FPR at the Bonferroni corrected level  . Figure 3 shows the FPRs for SKAT in the presence of covariates using Bonferroni corrected
. Figure 3 shows the FPRs for SKAT in the presence of covariates using Bonferroni corrected 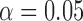 for 5–20 000 sets of variants and
for 5–20 000 sets of variants and 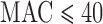 . Over the MAC and case–control imbalance scenarios, ER-mid had the best-calibrated FPRs, though it was conservative when
. Over the MAC and case–control imbalance scenarios, ER-mid had the best-calibrated FPRs, though it was conservative when 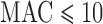 for balanced case–control studies. ER was slightly more conservative than ER-mid when
for balanced case–control studies. ER was slightly more conservative than ER-mid when 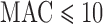 , but otherwise behaved similarly. QA was designed to speed the computation for moderate or large MAC. For MAC between 10 and 40 QA was conservative for balanced studies, and slightly anti-conservative for imbalanced studies. MA had conservative or anti-conservative FPRs depending on the scenario, and UA was both the most conservative for balanced studies at
, but otherwise behaved similarly. QA was designed to speed the computation for moderate or large MAC. For MAC between 10 and 40 QA was conservative for balanced studies, and slightly anti-conservative for imbalanced studies. MA had conservative or anti-conservative FPRs depending on the scenario, and UA was both the most conservative for balanced studies at 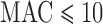 , and the most anticonservative for imbalanced studies. We observed similar trends for the Burden test (Supplementary Figure S2) and SKAT-O (Supplementary Figure S3).
, and the most anticonservative for imbalanced studies. We observed similar trends for the Burden test (Supplementary Figure S2) and SKAT-O (Supplementary Figure S3).
Fig. 3.

False positive rates (FPRs) for SKAT using ER-based and existing methods
to compute  -values for variant sets with
-values for variant sets with 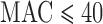 . From top to bottom the plots show
variant sets with
. From top to bottom the plots show
variant sets with  ;
;  and
and 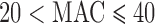 . From left to the right, the plots consider case:
. From left to the right, the plots consider case: :1000,
500:1500, and 200:1800. In each plot, the
:1000,
500:1500, and 200:1800. In each plot, the 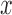 -axis is the number of variant sets
(
-axis is the number of variant sets
(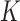 ) and their corresponding Bonferroni corrected level
) and their corresponding Bonferroni corrected level  , and the
, and the  -axis is the
empirical FPRs divided by the expected FPR. A well-calibrated test should have
empirical/expected
-axis is the
empirical FPRs divided by the expected FPR. A well-calibrated test should have
empirical/expected 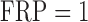 (gray dashed line).
(gray dashed line).
ER-mid based 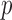 -values are conservative for variant sets with
-values are conservative for variant sets with 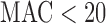 because many of the variant sets cannot reach Bonferroni-corrected thresholds. To improve the calibration of ER-mid, we used a mixture model (Supplementary Appendix C) to estimate the effective number of tests (
because many of the variant sets cannot reach Bonferroni-corrected thresholds. To improve the calibration of ER-mid, we used a mixture model (Supplementary Appendix C) to estimate the effective number of tests ( ) defined as the number of independent tests that yields the expected Bonferroni corrected FPR (Figure 4). For SKAT-ER-mid, when
) defined as the number of independent tests that yields the expected Bonferroni corrected FPR (Figure 4). For SKAT-ER-mid, when 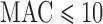 ,
, 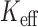 was substantially smaller than the number of variant sets, especially for balanced studies. The
was substantially smaller than the number of variant sets, especially for balanced studies. The 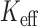 -based Bonferroni correction had a slightly anti-conservative FPR for balanced case–control samples but well-calibrated FPRs for imbalanced case–control samples. The computation time for the
-based Bonferroni correction had a slightly anti-conservative FPR for balanced case–control samples but well-calibrated FPRs for imbalanced case–control samples. The computation time for the  -based multiple test adjustment are essentially the sum of the computation time to test each variant set, as fitting the mixture model requires little additional computation. We observed similar patterns of results for Burden test (Supplementary Figure S4) and SKAT-O (Supplementary Figure S5).
-based multiple test adjustment are essentially the sum of the computation time to test each variant set, as fitting the mixture model requires little additional computation. We observed similar patterns of results for Burden test (Supplementary Figure S4) and SKAT-O (Supplementary Figure S5).
Fig. 4.

Estimated effective number of tests (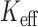 ) and FPRs for SKAT-ER-mid for
variant sets with
) and FPRs for SKAT-ER-mid for
variant sets with  . Variant sets with
. Variant sets with 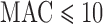 (top row) and
(top row) and 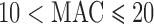 (bottom row) are shown. From
left to the right, the plots consider case:
(bottom row) are shown. From
left to the right, the plots consider case: :1000, 500:1500, and 200:1800. In each
plot, the top panel shows a bar plot of the estimated effective number of tests (
:1000, 500:1500, and 200:1800. In each
plot, the top panel shows a bar plot of the estimated effective number of tests (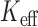 )
divided by the number of variant sets (
)
divided by the number of variant sets ( ), and the bottom panel shows the empirical
false positive rate (FPR) divided by the expected FPR of SKAT-ER-mid based on
), and the bottom panel shows the empirical
false positive rate (FPR) divided by the expected FPR of SKAT-ER-mid based on
 (square) or
(square) or 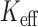 (circle). A well-calibrated test should have empirical/expected
(circle). A well-calibrated test should have empirical/expected 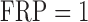 (black
dashed line). The
(black
dashed line). The  -axis shows the number of variant sets (
-axis shows the number of variant sets (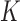 ).
).
Next, we examined the FPRs for sets of variants with 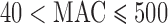 in the presence of covariates. SKAT-ER-mid was generally well calibrated, although it was slightly conservative or anti-conservative at
in the presence of covariates. SKAT-ER-mid was generally well calibrated, although it was slightly conservative or anti-conservative at  (Supplementary Figure S6). SKAT-QA was slightly conservative for balanced studies and slightly anti-conservative for studies with case–control imbalance. SKAT-MA was well calibrated or slightly anti-conservative for balanced studies, and was anti-conservative for imbalanced studies. SKAT-UA was not well calibrated in any of these scenarios. For Burden tests, all methods had close to the expected FPRs for balanced studies and Burden-QA was best calibrated for unbalanced studies (Supplementary Figure S7). We observed similar patterns of results for SKAT-O (Supplementary Figure S8).
(Supplementary Figure S6). SKAT-QA was slightly conservative for balanced studies and slightly anti-conservative for studies with case–control imbalance. SKAT-MA was well calibrated or slightly anti-conservative for balanced studies, and was anti-conservative for imbalanced studies. SKAT-UA was not well calibrated in any of these scenarios. For Burden tests, all methods had close to the expected FPRs for balanced studies and Burden-QA was best calibrated for unbalanced studies (Supplementary Figure S7). We observed similar patterns of results for SKAT-O (Supplementary Figure S8).
Overall, the results were quantitatively the same in the absence of covariates or when, instead of testing a set of variants, we tested single variants (a test which very similar to a Burden test with equal weights for all variants) (data not shown). To test for the robustness of our methods in the presence of population stratification, we simulated African American and European ancestry samples with a differential disease risk and adjusted for stratification in the analysis. The Type 1 error rates (Supplementary Appendix F and Supplementary Figures S9–S11) were quantitatively similar to those in Figure 3 and Supplementary Figures S2, S3 for European ancestry only.
Over a range of MAC and case–control ratios, no approach yielded an optimal mix of control of FPR and efficient computation. Based on our findings, we propose an ER-based hybrid approach (ER-mid when variant set 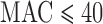 ; MA when variant set
; MA when variant set 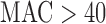 and balanced case–control; and QA when variant set
and balanced case–control; and QA when variant set 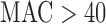 and imbalanced case–control) to provide a balance of well-calibrated FPRs and computation time.
and imbalanced case–control) to provide a balance of well-calibrated FPRs and computation time.
3.1.4. Comparison of power to identify associations between low MAC variant sets and binary phenotypes
We next compared power for the ER-based hybrid approach using either experiment-wide permutations of the total sample or the effective number of tests (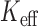 ) based Bonferroni correction, and power for the MA or UA tests using experiment-wide permutations. We estimated the power to detect one causal variant set
) based Bonferroni correction, and power for the MA or UA tests using experiment-wide permutations. We estimated the power to detect one causal variant set 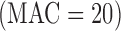 out of a background of 19 999 non-causal variant sets with the MAC distribution of
out of a background of 19 999 non-causal variant sets with the MAC distribution of 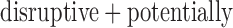 damaging variants observed in NHLBI ESP data (Supplementary Appendix D and Table 1). Our causal variant set had 50% causal variants, either all increasing risk or with half the variants increasing and half decreasing risk. Over the different gene-based tests approaches and varying case control ratios, we observed similar power for ER-based hybrid approach using experiment wide permutations or
damaging variants observed in NHLBI ESP data (Supplementary Appendix D and Table 1). Our causal variant set had 50% causal variants, either all increasing risk or with half the variants increasing and half decreasing risk. Over the different gene-based tests approaches and varying case control ratios, we observed similar power for ER-based hybrid approach using experiment wide permutations or 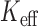 -based Bonferroni correction (Supplementary Figure S12). For SKAT and SKAT-O, the ER-based hybrid approach had higher power than MA or UA. For the burden test, MA or UA had similar or slightly higher power to the ER-hybrid approach, but neither test was consistently higher power. We observed similar trends for causal
-based Bonferroni correction (Supplementary Figure S12). For SKAT and SKAT-O, the ER-based hybrid approach had higher power than MA or UA. For the burden test, MA or UA had similar or slightly higher power to the ER-hybrid approach, but neither test was consistently higher power. We observed similar trends for causal 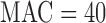 (Supplementary Figure S13).
(Supplementary Figure S13).
Table 1.
Number of genes by MAC of selected variants in NHLBI-ESP whole-exome data and in chromosome 2 GoT2D-exome data
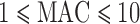 |
 |
 |
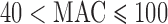 |
 |
Total | |
|---|---|---|---|---|---|---|
| NHLBI ESP | ||||||
| Disruptive | 7261 (62%) | 1425 (12%) | 1313 (11%) | 1306 (11%) | 485 (4%) | 11 790 |
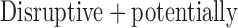 damaging damaging |
4250 (25%) | 2636 (15%) | 3135 (18%) | 4034 (23%) | 3185 (18%) | 17 240 |
| All nonsynonymous | 1699 (9%) | 1579 (9%) | 2568 (14%) | 4791 (27%) | 7371 (41%) | 18 008 |
| GoT2D Chr2 | ||||||
| Disruptive | 312 (92%) | 17 (5%) | 5 (1%) | 6 (2%) | 0 (0%) | 340 |
 damaging damaging |
481 (46%) | 174 (17%) | 186 (18%) | 161 (15%) | 37 (4%) | 1039 |
| All nonsynonymous | 284 (26%) | 165 (15%) | 208 (19%) | 330 (30%) | 123 (11%) | 1110 |
Each cell has the number (percent) of genes in each MAC bin for genes with 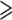 1
variant. “Total” indicates the total number of genes with
1
variant. “Total” indicates the total number of genes with  1 variant. Nonsense,
splicing, and frame-shift variants are classified as “disruptive” variants, and
possibly and probably damaging variants by Polyphen2 and disruptive variants
together are classified as “
1 variant. Nonsense,
splicing, and frame-shift variants are classified as “disruptive” variants, and
possibly and probably damaging variants by Polyphen2 and disruptive variants
together are classified as “ damaging” variants.
damaging” variants.
3.2. GoT2D data analysis
We performed single and multiple variant tests using GoT2D chromosome 2 deep exome sequence data (1326 cases and 1331 controls) (Supplementary Appendix G). 35 576 (84%) of 42 045 chromosome 2 variants had 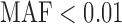 (corresponding
(corresponding  ). For single variant tests of
). For single variant tests of  variants, the estimated effective number of tests (
variants, the estimated effective number of tests (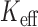 ) was 2762, giving an order of magnitude less stringent threshold than the family-wise error rate 0.05. No variants were significant at
) was 2762, giving an order of magnitude less stringent threshold than the family-wise error rate 0.05. No variants were significant at 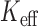 -based Bonferroni-corrected
-based Bonferroni-corrected  . The unadjusted QQ plot for single variant results showed a substantial
. The unadjusted QQ plot for single variant results showed a substantial 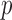 -value deflation compared with the expected
-value deflation compared with the expected 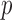 -value (Figure 5(a)); though the deflation was less pronounced when testing was restricted to variants with
-value (Figure 5(a)); though the deflation was less pronounced when testing was restricted to variants with  (Figure 5(b)). In contrast, in QQ plots based on a mixture model of the minimum achievable
(Figure 5(b)). In contrast, in QQ plots based on a mixture model of the minimum achievable  -values, no
-values, no 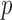 -value deflation was observed (Figures 5(a) and (b)).
-value deflation was observed (Figures 5(a) and (b)).
Fig. 5.

MAP-adjusted and un-adjusted QQ plots of single variant and
SKAT-ER-hybrid 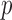 -values from analysis of GoT2D chromosome 2 exome data. QQ
plots of single variant tests with all rare variants
-values from analysis of GoT2D chromosome 2 exome data. QQ
plots of single variant tests with all rare variants  (a) and rare variants with
(a) and rare variants with
 (b). QQ plots of ER-hybrid SKAT
(b). QQ plots of ER-hybrid SKAT 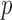 -values with disruptive variants (c) and
-values with disruptive variants (c) and 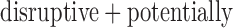 damaging variants (d). In each plot, the
damaging variants (d). In each plot, the 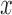 -axis is the MAP-adjusted or un-adjusted
expected quantile of
-axis is the MAP-adjusted or un-adjusted
expected quantile of  log
log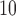
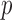 -values, and the
-values, and the 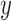 -axis is observed quantiles of
-axis is observed quantiles of  log
log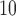
 -values.
Observed
-values.
Observed 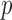 -values are plotted against the MAP-adjusted expected quantiles (black
dots) and un-adjusted expected quantiles (gray dots). The dashed line represents
a 95% confidence band based on 500 random draws from the MAP-based mixture
distribution.
-values are plotted against the MAP-adjusted expected quantiles (black
dots) and un-adjusted expected quantiles (gray dots). The dashed line represents
a 95% confidence band based on 500 random draws from the MAP-based mixture
distribution.
In the chromosome 2 GoT2D data, 334 of 340 (98%) genes with at least one disruptive variant had 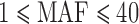 , and 841 of 1039 (81%) genes with at least one
, and 841 of 1039 (81%) genes with at least one 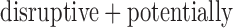 damaging variant had
damaging variant had 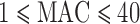 (Table 1). Even in the whole-exome data from the larger NHLBI-ESP sample, 85% and 58% of genes with at least one disruptive or
(Table 1). Even in the whole-exome data from the larger NHLBI-ESP sample, 85% and 58% of genes with at least one disruptive or 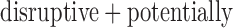 disruptive variant, respectively, had
disruptive variant, respectively, had 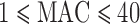 (Supplementary Appendix D and Table 1). We used SKAT-ER-hybrid to perform gene-based tests for disruptive and
(Supplementary Appendix D and Table 1). We used SKAT-ER-hybrid to perform gene-based tests for disruptive and 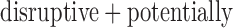 damaging variants (
damaging variants ( and 540, respectively) in the chromosome 2 GoT2D exome data. No gene was significant at the
and 540, respectively) in the chromosome 2 GoT2D exome data. No gene was significant at the 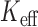 -based Bonferroni corrected
-based Bonferroni corrected 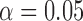 . In unadjusted QQ plots, we observed deflation of the gene-based
. In unadjusted QQ plots, we observed deflation of the gene-based  -values, whereas in MAP adjusted QQ plots the
-values, whereas in MAP adjusted QQ plots the  -values were not deflated and results for disruptive variants were near the upper 95% confidence bound (Figures 5(c) and (d)). We observed similar results for ER-hybrid Burden and SKAT-O tests (Supplementary Figures S14 and S15).
-values were not deflated and results for disruptive variants were near the upper 95% confidence bound (Figures 5(c) and (d)). We observed similar results for ER-hybrid Burden and SKAT-O tests (Supplementary Figures S14 and S15).
Within the  damaging variant tests, YSK4 Sps1/Ste20-related kinase homolog (YSK4) was the most significant gene for the Burden-ER-mid test (
damaging variant tests, YSK4 Sps1/Ste20-related kinase homolog (YSK4) was the most significant gene for the Burden-ER-mid test (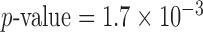 ,
, 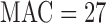 ) and the second most significant gene for SKAT-O-ER-mid (
) and the second most significant gene for SKAT-O-ER-mid ( ). Recent large-scale meta-analysis has shown that a common variant in YSK4 is associated with fasting insulin (Scott and others, 2012).
). Recent large-scale meta-analysis has shown that a common variant in YSK4 is associated with fasting insulin (Scott and others, 2012).
To assess the ER method using dosage data, we compared the results of ER and whole-sample permutations for variant set-based testing using dosage data from non-exomal GOT2D low-pass sequencing and found very similar  -values (Supplementary Appendix H and Supplementary Figure 16).
-values (Supplementary Appendix H and Supplementary Figure 16).
4. Discussion
In this paper, we develop an ER method for binary traits for score statistic-based tests of variant sets with low MAC that allows inclusion of covariates in analysis. The ER methods are necessary because the existing asymptotic (UA) or asymptotic-based adjustment methods (MA) have poor calibration of FPRs at lower MAC and imbalanced case control ratios. As in whole-sample permutations, the ER method preserves the correlation structure or LD among variants in the tested set. Across almost all tested MAC bins and case–control ratios, we found that one or more of the ER-based methods were well calibrated. Based on these observations and the computational time considerations, we recommend a hybrid approach using ER-mid for small variant set MAC  ; MA for moderate or large variant set MAC with balanced case–control and QA for moderate or large variant set MAC with unbalanced case–control. Use of a threshold of
; MA for moderate or large variant set MAC with balanced case–control and QA for moderate or large variant set MAC with unbalanced case–control. Use of a threshold of  is a practical compromise between computational time and Type 1 error rate; a slightly lower threshold would result in faster computation time but at the risk of slightly higher Type 1 error rate, particularly for the SKAT and SKAT-O. If a permutation approach is desired, then ER-mid is (substantially) faster than whole-sample permutations even for large MAC.
is a practical compromise between computational time and Type 1 error rate; a slightly lower threshold would result in faster computation time but at the risk of slightly higher Type 1 error rate, particularly for the SKAT and SKAT-O. If a permutation approach is desired, then ER-mid is (substantially) faster than whole-sample permutations even for large MAC.
Estimation of the effective number of tests, 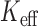 , using MAP is a simple and fast alternative to performing experiment-wise permutation of the total sample to control the family-wise error rate. One limitation of the MAP approach is that it cannot account for correlations among tests, and may result in conservative FPRs in the presence of the strong correlations of variants between genes. However, we expect that gene-based tests will be less correlated than single variant tests, since they involve multiple variants and genes located further away from each other than individual variants.
, using MAP is a simple and fast alternative to performing experiment-wise permutation of the total sample to control the family-wise error rate. One limitation of the MAP approach is that it cannot account for correlations among tests, and may result in conservative FPRs in the presence of the strong correlations of variants between genes. However, we expect that gene-based tests will be less correlated than single variant tests, since they involve multiple variants and genes located further away from each other than individual variants.
When MAC is extremely small, MAP is unlikely to reach genome-wide significance. One approach to increase power would be to construct larger sets by combining adjacent regions or including more classes of potentially functional variants.
The ER method can be used for imputed dosage, as well as genotype data; permutations are performed within the individuals with non-zero genotype or dosage values. If many individuals have very small dosage values (e.g. 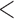 0.1), the number of individuals with minor alleles can be larger than MAC (i.e.
0.1), the number of individuals with minor alleles can be larger than MAC (i.e. 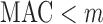 ). Thus, for the same MAC, computational time can be higher with dosage data than with genotype data; however, the ER method still takes substantially less time than whole-sample permutation method.
). Thus, for the same MAC, computational time can be higher with dosage data than with genotype data; however, the ER method still takes substantially less time than whole-sample permutation method.
QQ plots comparing observed vs. expected 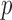 -value distributions are used in genetic association studies to assess both the presence of confounding (or misimplimented/misspecified test) and the presence of significant association signals. However, when MAC is small, the expected
-value distributions are used in genetic association studies to assess both the presence of confounding (or misimplimented/misspecified test) and the presence of significant association signals. However, when MAC is small, the expected 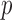 -value distribution of the resampling-based test is not uniform (0,1), and hence the (unadjusted) QQ plot cannot be used to accurately assess the concordance (or departure) of the observed
-value distribution of the resampling-based test is not uniform (0,1), and hence the (unadjusted) QQ plot cannot be used to accurately assess the concordance (or departure) of the observed 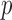 -value distribution from the expected. In the spirit of experiment wide permutations (Kiezun and others, 2012), we use the MAP-adjusted
-value distribution from the expected. In the spirit of experiment wide permutations (Kiezun and others, 2012), we use the MAP-adjusted  -value distribution to model the expected distribution of ER-hybrid
-value distribution to model the expected distribution of ER-hybrid  -values. In the MAP-adjusted QQ plot, the GoT2D gene-based
-values. In the MAP-adjusted QQ plot, the GoT2D gene-based 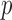 -value distribution for disruptive variants lies near the top of the 95% confidence band. This view allows better assessment of potentially interesting results than the unadjusted QQ plot in which the
-value distribution for disruptive variants lies near the top of the 95% confidence band. This view allows better assessment of potentially interesting results than the unadjusted QQ plot in which the 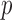 -value distribution is deflated.
-value distribution is deflated.
Most of variant sets in whole-exome or whole-genome data will not require 10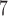 resampling since their
resampling since their 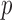 -values will be substantially higher than exome-wide (or genome-wide) significant levels. Hence, an adaptive resampling procedure, which reduces the number of resamples when a test has a moderate or large
-values will be substantially higher than exome-wide (or genome-wide) significant levels. Hence, an adaptive resampling procedure, which reduces the number of resamples when a test has a moderate or large 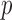 -value, can substantially reduce computation time and has been implemented for the ER method. However, the use of adaptive resampling precludes the calculation of the effective number of test and the use of MAP-adjusted QQ plots, and thus we recommend the adaptive resampling procedure only for the case where case–control combinations among individuals with minor alleles are substantially larger than the number of resamples performed (for example,
-value, can substantially reduce computation time and has been implemented for the ER method. However, the use of adaptive resampling precludes the calculation of the effective number of test and the use of MAP-adjusted QQ plots, and thus we recommend the adaptive resampling procedure only for the case where case–control combinations among individuals with minor alleles are substantially larger than the number of resamples performed (for example,  for 10
for 10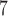 resamples).
resamples).
Our work has focused on providing well-calibrated gene-based tests for single studies across a range of MAC and case–control imbalance. Meta-analysis of gene-based tests can increase the power to detect genes of interest, but meta-analysis is sensitive to the calibration of the underlying tests (Ma and others, 2013), and may be particularly sensitive to the inclusion of studies with highly imbalanced case–control ratios. Further work will be needed to determine how best to combine results or data from across studies with a variety of case–control ratios.
5. Software
ER-mid, ER, QA, and MA methods are implemented in the SKAT R-package.
Supplementary material
Supplementary Material is available at http://biostatistics.oxfordjournals.org.
Funding
This work was supported by grants R00 HL113264 (S.L.), the Austrian Science Fund (F.W.F.) grant J-3401 (C.F.), R01 HG000376 and RC2 DK088389 (L.S.).
Supplementary Material
Acknowledgments
We thank investigators of GoT2D project for access to the chromosome 2 exome sequence data. We also thank M. Boehnke for discussion and insightful comments and Phoenix Kwan for her initial insights into the behavior of gene-based tests in the GOT2D data. Conflict of Interest: None declared.
References
- Derkach A., Lawless J. F., Sun L. (2012). Robust and powerful tests for rare variants using Fisher's method to combine evidence of association from two or more complementary tests. Genetic Epidemiology 37, 110–121. [DOI] [PubMed] [Google Scholar]
- Efron B., Tibshirani R. J. (1994) An Introduction to the Bootstrap. CRC press. [Google Scholar]
- Epstein M. P., Duncan R., Jiang Y., Conneely K. N., Allen A. S., Satten G. A. (2012). A permutation procedure to correct for confounders in case–control studies, including tests of rare variation. American journal of human genetics 91, 215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fog A. (2008). Calculation methods for Wallenius' noncentral hypergeometric distribution. Communications in Statistics—Simulation and Computation 37, 258–273. [Google Scholar]
- Kiezun A., Garimella K., Do R., Stitziel N. O., Neale B. M., McLaren P. J., Gupta N., Sklar P., Sullivan P. F., Moran J. L. (2012). Exome sequencing and the genetic basis of complex traits. Nature Genetics 44, 623–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster H. (1961). Significance tests in discrete distributions. Journal of the American Statistical Association 56, 223–234. [Google Scholar]
- Lee S., Abecasis G. R., Boehnke M., Lin X. (2014). Rare-variant association analysis: study designs and statistical tests. American Journal of Human Genetics 95, 5–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S., Emond M. J., Bamshad M. J., Barnes K. C., Rieder M. J., Nickerson D. A., Christiani D. C., Wurfel M. M., Lin X. (2012). Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. American Journal of Human Genetics 91, 224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S., Wu M. C., Lin X. (2012). Optimal tests for rare variant effects in sequencing association studies. Biostatistics 13, 762–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Leal S. M. (2008). Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. American Journal of Human Genetics 83, 311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D. Y., Tang Z. Z. (2011). A general framework for detecting disease associations with rare variants in sequencing studies. American Journal of Human Genetics 89, 354–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C., Blackwell T., Boehnke M., Scott L. J. (2013). Recommended joint and meta-analysis strategies for case–control association testing of single low-count variants. Genetic Epidemiology 37, 539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen B. E., Browning S. R. (2009). A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genetics 5, e1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale B. M., Rivas M. A., Voight B. F., Altshuler D., Devlin B., Orho-Melander M., Kathiresan S., Purcell S. M., Roeder K., Daly M. J. (2011). Testing for an unusual distribution of rare variants. PLoS Genetics 7, e1001322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan W. (2009). Asymptotic tests of association with multiple SNPs in linkage disequilibrium. Genetic Epidemiology 33, 497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M. A., Bender D., Maller J., Sklar P., De Bakker P. I., Daly M. J. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. American Journal of Human Genetics 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reppell M., Boehnke M., Zöllner S. (2012). FTEC: a coalescent simulator for modeling faster than exponential growth. Bioinformatics 28, 1282–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott R. A., Lagou V., Welch R. P., Wheeler E., Montasser M. E., Luan J. A., Gustafsson S. (2012). Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nature Genetics 44, 991–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J., Zheng Y., Hsu L. (2013). A Unified Mixed-Effects Model for Rare-Variant Association in Sequencing Studies. Genetic Epidemiology 37, 334–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M. C., Lee S., Cai T., Li Y., Boehnke M. C., Lin X. (2011). Rare variant association testing for sequencing data wsing the sequence kernel association test (SKAT). American Journal of Human Genetics 89, 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuk O., Schaffner S. F., Samocha K., Do R., Hechter E., Kathiresan S., Daly M. J., Neale B. M., Sunyaev S. R., Lander E. S. (2014). Searching for missing heritability: designing rare variant association studies. Proceedings of the National Academy of Sciences 111, E455–E464. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.