

Abstract
Essential tremor (ET) is a common movement disorder with an estimated prevalence of 5% of the population aged over 65 years. In spite of intensive efforts, the genetic architecture of ET remains unknown. We used a combination of whole-exome sequencing and targeted resequencing in three ET families. In vitro and in vivo experiments in oligodendrocyte precursor cells and zebrafish were performed to test our findings. Whole-exome sequencing revealed a missense mutation in TENM4 segregating in an autosomal-dominant fashion in an ET family. Subsequent targeted resequencing of TENM4 led to the discovery of two novel missense mutations. Not only did these two mutations segregate with ET in two additional families, but we also observed significant over transmission of pathogenic TENM4 alleles across the three families. Consistent with a dominant mode of inheritance, in vitro analysis in oligodendrocyte precursor cells showed that mutant proteins mislocalize. Finally, expression of human mRNA harboring any of three patient mutations in zebrafish embryos induced defects in axon guidance, confirming a dominant-negative mode of action for these mutations. Our genetic and functional data, which is corroborated by the existence of a Tenm4 knockout mouse displaying an ET phenotype, implicates TENM4 in ET. Together with previous studies of TENM4 in model organisms, our studies intimate that processes regulating myelination in the central nervous system and axon guidance might be significant contributors to the genetic burden of this disorder.
Introduction
Essential tremor (ET [MIM 190300]) is a common hyperkinetic movement disorder with an estimated prevalence of 1% in the population. The prevalence increases with age, and 5% of individuals over 65 years of age are affected (1). ET is typically characterized by rhythmic, involuntary shaking of one or more parts of the body, and occurs exclusively during voluntary movements (action tremor) or in positions against gravity (postural tremor) (2). The phenotypic severity of ET is variable, as evidenced by the existence of both highly disabling and milder forms of the disease. ET is considered a complex disorder with a strong genetic component (3,4), since more than half of affected individuals have a positive family history.
The first genetic locus associated with ET was near the leucine-rich repeat and lg domain containing nogo receptor-interacting protein 1 gene (LINGO1 [MIM 609791]), identified by a genome-wide association study (GWAS) in the Icelandic population (5) and replicated in other populations (6). Other studies, including several candidate gene association studies and a recent GWAS (7), revealed potential associations that nonetheless failed to replicate (4). Additionally, three linkage studies conducted in large ET families displaying an autosomal-dominant pattern of inheritance identified only putative candidate loci, but did not find any underlying disease causing gene(s) (8–10). Factors that have confounded the identification of causal genes include non-penetrance of ET; a considerable number of misdiagnoses in the absence of reliable biomarkers; and the existence of phenocopies, as distinct sporadic cases may occur within the same family given the high prevalence in the general population (11). In this regard, numerous ET families have been reported that are characterized by an overrepresentation of affected individuals clearly exceeding the expected 50–50% ratio, which is clearly beyond the classical pattern of autosomal inheritance (11–13).
The first published exome-sequencing study in ET identified a causative nonsense mutation in fused in sarcoma (FUS [MIM 137070]) that was segregating with ET in a large pedigree (14). However, only two additional cases carried other mutations in the same gene, while independent replication studies uncovered few additional mutations, suggesting that FUS is a rare cause of ET (15–18). More recently, a rare variant in HtrA serine peptidase 2 (HTRA2 [MIM 610297]) was shown to cause ET in a large six-generation family. Interestingly, signs of Parkinson's disease (PD) additionally appeared in the middle age in homozygous carriers of this variant (19).
Given the large proportion of familial ET cases without causal mutations, and the low-to-moderate effect size of the identified variant near LINGO1, a large fraction of the heritability of ET remains unexplained. We sought to identify new genes involved in the pathogenesis of ET by studying orphan ET families by exome sequencing. Here we describe the identification of a new gene for ET by the combination of exome and targeted resequencing as well as functional testing using in vitro assays in oligodendrocytes precursor cells and in vivo assays in a zebrafish model.
Results
Exome sequencing
The clinical diagnosis of ET was made initially in the index case (II-5) of a four-generation family (TEF-6) of Spanish origin (Fig. 1A). The phenotypic severity varied among affected family members, ranging from a highly disabling to mild tremor (Supplementary Material, Table S1). Given such common phenotypic variance and transmission of the disease across three generations, we postulated an autosomal-dominant pattern of inheritance in this family. The available data did not allow us to perform linkage analysis, mainly due to limited participation and deceased family members. We therefore performed whole-exome sequencing in four affected family members (II-5, III-2, III-4 and IV-1) assuming that the disease causing variant is located in the protein coding region, cognizant of the fact that a mutation in a regulatory region not captured by this technology cannot be excluded. After alignment and variant calling, we first evaluated variants in genes associated previously with autosomal-dominant ET and PD (in particular FUS [MIM 137070], SNCA [MIM 163890], LRRK2 [MIM 609007]). Despite adequate coverage in these regions, we did not find any candidate pathogenic mutations. We next filtered for single nucleotide variants (SNVs) and indels that (a) were novel and rare [minor allele frequency (MAF) below 0.5%]; (b) were shared by all four individuals sequenced and (c) were predicted computationally to be damaging by Polyphen-2, PROVEAN and CADD (20–22). This yielded a list of rare alleles in four candidate genes, namely RCOR3, CYTL1 [MIM 607930], E2F8 [MIM 612047] and TENM4 [MIM 610084], all of which segregated with the disease phenotype in our pedigree. To avoid bias, we mined expression data for all four genes. We found that RCOR3, CYTL1 and E2F8 are not expressed appreciably in neuronal tissues, while subsequent literature searches intimate an involvement of these transcripts in hepatitis, chondrogenesis and hepatocellular carcinoma (3,23,24). In contrast, TENM4 is expressed primarily in the brain and has been detected in several transcriptomes derived from neuronal tissues (see Materials and Methods). We also noted that Suzuki and colleagues (25) generated a mouse Tenm4 knockout model recently that displays hypomyelination of small-diameter axons in the CNS and a severe action tremor phenotype (as seen in video by Suzuki et al., http://www.youtube.com/watch?v=bcmcjLuB_bs). Taken together, this evidence brought our focus to the missense mutation in TENM4 (c.4100C>A) that leads to a threonine-to-asparagine amino acid substitution (p.T1367N).
Figure 1.

Pedigrees of Spanish essential tremor families TEF-6 (A); FET-3 (B) and TEF-9 (C). (Square) male family members. (Circle) female family members, (filled symbol) individuals with ET. (*) exome-sequenced individuals. (Dashed symbol) deceased individual. TENM4 genotype status: TEF-6—TENM4 c.4100C>A (C/A = heterozygous mutant allele); FET-3: TENM4 c.4324 G>A (G/G = homozygous reference allele, G/A = heterozygous mutant allele; TEF-9: TENM4 c.3412G>A (G/G = homozygous reference allele, G/A = heterozygous mutant allele). If the genetic status is not indicated, the corresponding DNA samples were unavailable.
The c.4100C>A mutation, which has never been detected before (absent in 1000 Genomes and EVS), was present in all affected individuals. We found two healthy offspring in the fourth generation (IV-2 and IV-3) to be mutation carriers as well. However, these subjects are <40 years old and are thus likely presymptomatic carriers that may develop ET, although reduced penetrance remains a possible scenario. copy number variants (CNVs) analysis did not yield any genomic rearrangements that were shared by all four subjects.
Targeted resequencing of TENM4 and segregation analysis in additional ET families
As a first test of the candidacy of TENM4, we asked whether there might be additional ET families with segregating mutations in this gene. To this end, we performed targeted resequencing of TENM4 in 299 index cases of familial ET of Spanish origin (the mean age of onset = 45 ± 22 years). We recorded all novel missense, splice-site or nonsense variants with a MAF below 0.1% according to the EVS and identified 12 missense mutations, of which 11 were predicted to be damaging by Polyphen-2 (20) as well as by PROVEAN and CADD (21,22) (Table 1). DNA samples were available from additional family members of two mutation carriers. One of the two families was FET-3, a four-generation family comprising 11 affected family members (Fig. 1B; Supplementary Material, Table S2). Having identified the predicted damaging variant c.4324 G>A (p.A1442T) in II-1, we evaluated its segregation in all available family members. Strikingly, 9 out of 11 affected carried this variant. Targeted resequencing of TENM4 in III-1 and III-3 excluded other TENM4 mutations, suggesting that these two individuals are potential phenocopies, a phenomenon observed frequently in ET (13). We also detected c.4324 G>A in one unaffected offspring in the third generation (III-7) and another in the fourth generation (IV-2); although both subjects were reported to be healthy, they might still develop the disease at a later age (current age: 42 and 5, respectively). Nevertheless, reduced penetrance cannot be excluded at this stage. The second family was a three-generation pedigree, TEF-9, with five affected family members (Fig. 1C; Supplementary Material, Table S3), for three of which we had DNA available. We found the c.3412G>A variant (p.V1138M) in two of these family members (II-4 and III-1). The third individual, III-2, did not carry this mutation, however, in contrast to her siblings, she was not self-aware of her tremor, which was mild and observed by the clinician only. Taken together, the segregation and phenotyping of individuals from three multigenerational ET pedigrees provided suggestive evidence of involvement of TENM4 mutations. As an alternative means to test the candidacy of TENM4, we interrogated the transmission behavior of the discovered alleles. The null hypothesis (TENM4 alleles transmit randomly) was rejected: among the 15 parent-affected offspring scorable transmissions across the three pedigrees, the TENM4 mutant allele was transmitted 12 times, a significant departure from 50:50 chance (χ2 P < 0.02).
Table 1.
Novel variants (MAF <0.001) found among 300a index cases diagnosed for familial essential tremor
| Chromosomal position | Nucleotide change | Type of mutation | Ref.-allele | Mut.-allele | MAF in EVS (Caucasians) | Number of occurrences in 300 cases | Amino acid change | Polyphen-2 prediction |
|---|---|---|---|---|---|---|---|---|
| 11:78780832 | c.158G>C | M | C | G | NA | 1 | p.R53P | 2 |
| 11:78567058 | c.1421C>A | M | G | T | NA | 1 | p.A474D | 2 |
| 11:78565277 | c.1553G>A | M | C | T | 0.000943 | 1 | p.R518Q | 2 |
| 11:78443446 | c.3053G>A | M | C | T | NA | 1 | p.R1018H | 2 |
| 11:78440445 | c.3382G>A | M | C | T | NA | 1 | p.V1128M | 2 |
| 11:78437262b | c.3412G>A | M | C | T | NA | 2 | p.V1138M | 2 |
| 11:78419515c | c.4100C>A | M | G | T | NA | 1 | p.T1367N | 2 |
| 11:78413334d | c.4324G>A | M | C | T | 0 | 1 | p.A1442T | 2 |
| 11:78413055 | c.4603A>C | M | T | G | NA | 1 | p.K1535Q | 1 |
| 11:78412763 | c.4895G>A | M | C | T | 0.00048 | 1 | p.R1632H | 1 |
| 11:78387406 | c.5287G>A | M | C | T | 0.000942 | 1 | p.G1763R | 2 |
| 11:78380037 | c.7353G>A | M | C | T | 0.0006 | 1 | p.M2451I | 1 |
aIncluding the first mutation identified by exome sequencing in TEF-6.
The variant at chromosomal position marked with ‘c’ was segregating in family TEF-6. The variant at chromosomal position marked with ‘d’ was segregating in family FET-3. The variant at chromosomal position marked with ‘b’ was segregating in family TEF-9. Type of mutations: M = missense. Polyphen-2 prediction: 1 = possibly damaging, 2 = probably damaging. NA = not applicable. All variants are heterozygous.
In vitro testing of TENM4 mutations
Our genetic data suggested a causal link between TENM4 variants and ET. However, because the pathogenic potential of the discovered alleles was predictive, we sought direct evidence. We thus evaluated the impact of the missense mutations segregating with ET in our two largest ET pedigrees on subcellular localization of TENM4 protein. We transfected Oli-neu cells with expression vectors containing full-length human TENM4 (wild-type), negative control c.7933G>A and TENM4 carrying the c.4100C>A, c.4324 G>A and c.3412G>A mutations. In cells transfected with wild-type TENM4 and the negative control c.7933G>A, an allele that has been found in homozygosity in 2/6342 individuals in EVS, the protein localized homogeneously at the cell membrane. In contrast, c.4100C>A (p.T1367N), c.4324 G>A (p.A1442T) and c.3412G>A (p.V1138M) showed clustered TENM4 membrane localization (Fig. 2).
Figure 2.

In vitro characterization of the cellular distribution of human TENM4 mutants. Localization of wild-type (wt) and mutated forms of TENM4 in Oli-neu cells. Wild-type TENM4 as well as the control allele G7933A (c.7933G>A) showed a homogeneous membrane distribution. In contrast, the mutated forms C4100A (c.4100C>A), G4324A (c.4324 G>A) and G3412A (c.3412G>A) showed clustered localization at the membrane. TENM4 is in red; DAPI nuclear dye is in blue.
Modeling of TENM4 in zebrafish
As a second test of allele pathogenicity, as well as a means to determine the direction of effect, we turned to an in vivo zebrafish model. Given previous findings of central nervous system demyelination in a homozygous null Tenm4 mouse model (25), we first assayed myelin levels in the brain by RNA in situ hybridization. Injection of either a translation blocking morpholino (tbMO) or a splice-blocking morpholino (sbMO) (Supplementary Material, Fig. S1) against the sole endogenous copy of zebrafish tenm4 caused modest reduction in intensity of myelin in the brain, likely reflective of the fact that most, but not all, endogenous tenm4 was suppressed by our MO (data not shown). In contrast, we observed a significant increase in the number of aberrant small-diameter neuronal axons along the notochord of morphants (either tb or sbMOs) at 2 days post fertilization (2dpf, Fig. 3A and B). The lesions observed involved extension and pathfinding errors, as well as aberrant branching of the remaining axons, in a pattern reminiscent of the pathfinding defects reported in a Drosophila mutant of the teneurin/tenascin family (26). The number or neuronal somata dorsal to the notochord was also compared across control and morphant embryos, but revealed no differences among the two groups of animals, suggesting that it is not the number or neurons that is affected but the innervation pattern along the peripheral nervous system (PNS). The innervation of the truncal musculature as determined by the neuronal axon aberrations was rescued by co-injection of human TENM4 mRNA, demonstrating that the phenotype was specific (Fig. 3). These data were encouraging but, by themselves, did not constitute proof of TENM4 causality, since they showed only that this transcript/protein is necessary for axonal guiding functions in the PNS. They did, however, offer the opportunity to test the effect of the discovered patient alleles on protein function. We therefore engineered each of the three mutations and injected them into wild-type zebrafish embryos. Injection of TENM4 mRNA carrying the mutations identified in TEF-6, FET-3 and TEF-9 (c.4100C>A, c.4324 G>A, c.3412G>A and c.7933G>A), but not of the wt construct nor the negative control high frequency allele c.7933G>A resulted in a phenotype that phenocopied the one induced by the MO. These data suggested that these alleles adversely impact the function of the protein and exert a likely dominant-negative effect (Fig. 3), supporting our earlier in vitro observations.
Figure 3.
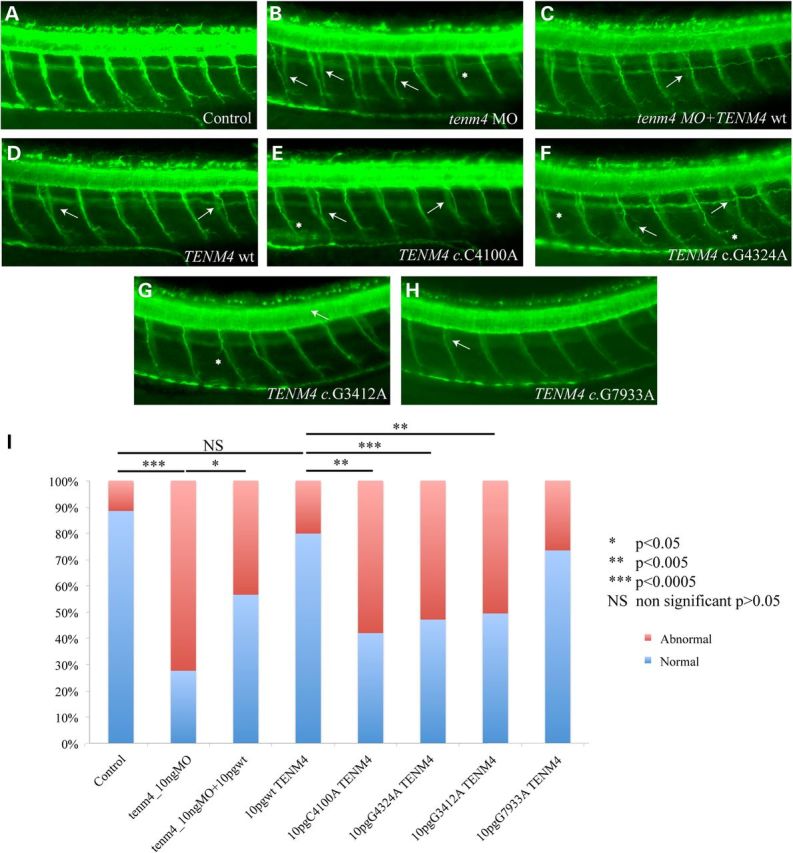
Suppression of tenm4 in zebrafish causes defects in motor axon pathfinding, outgrowth and branching. (A–H) Lateral views of zebrafish embryos at 2dpf stained with acetylated tubulin. In wild-type (wt) embryos (A), motor axons have completed migration from the periphery, along the common path, and extended to the myotome. In tenm4 morphants (B), secondary axons either fail to migrate along their path (white arrows), exit the periphery but fail to extend, or branch abnormally (asterisks). Suppression of tenm4 can be partially rescued by co-injection with human wt RNA (C). Overexpression of human Tenm4 wt causes mild pathfinding errors (D), suggesting dose sensitivity for TENM4, Human Tenm4 mutants C4100A (c.4100C>A), G4324A (c.4324 G>A) and G3412A (c.3412G>A), when overexpressed are significantly more severe than TENM4 wt and have similar effects to suppression of tenm4 by MO knockdown (E–G). Overexpression of control allele G7933A (c.7933G>A) causes mild pathfinding errors like overexpression of wt allele (H). (I) Percentage of normal versus abnormal embryos under the conditions being evaluated above.
Discussion
Here we report pathogenic mutations in TENM4 by exome sequencing in a family with ET, followed by the identification of mutations in two additional families. Although the segregation of the candidate alleles indicates that penetrance is high, in that we did not observe any healthy individuals above the median age of ET with these mutations, we did identify three likely phenocopies across two families. This observation is consistent with recent discussions on the genetic architecture of ET (11–13), but also of other neurological disorders such as restless legs syndrome (27), where the presence of phenocopies has been highlighted as a major confounder in searching for causal alleles in large multiplex families. At the same time, the term ‘phenocopies’ might also be misleading, as the rate of misdiagnosis is known to be high in ET and many other reported families are characterized by an overrepresentation of affected individuals within one pedigree, raising the question whether a rather more complex genetic architecture should be considered in the pathogenesis of ET including di- or multigenetic inheritance as well as epigenetics (11–13). Indeed, the identification of a single causal variant in all six affected siblings in the third generation of family FET-3 seems unlikely. We also draw attention to the fact that in other neurological disorders, such as PD, which has some clinical overlap with ET, the presence of phenocopies is also a known phenomenon, especially in familial PD caused by mutations in LRRK2, a well-established cause of this condition (28). In fact, in ∼14.4% of families with LRRK2 driver mutations, additional affected members carry the reference alleles only (29).
The Teneurin family of genes (TENM1–TENM4) encodes transmembrane proteins that are expressed predominantly in neurons (30). TENM4 is the only member also expressed in the white matter of the cerebellum of adult mice (31) and was identified recently as a regulator of oligodendrocyte maturation and myelination of small-diameter axons in the CNS (25). Interestingly, fractional anisotropy neuroimaging has shown, though not consistently, white matter abnormalities in different parts of the brain, particularly in the cerebellum, in ET patients (32). Additionally, teneurins play a major role in motor neuron guidance and neuronal synapse organization in Drosophila (26) and to affect neurite outgrowth in neuroblastoma cells (33). Our experiments in Oli-neu cells as well as our zebrafish experiments concord with these previous findings. Notably, the two of the three TENM4 point mutations assayed in cells (c.4100C>A, c.4324 G>A), are both located in the NHL-repeat/β propeller domain, which has recently been shown to be crucial for the homophilic interaction between TENM1 and TENM2 (34). Changes in TENM4 localization may contribute to defects in TENM4-mediated regulation of the phosphorylation of the focal adhesion kinase, which activates CDC42 and RAC1, two Rho-GTPases that are required for oligodendroglial process outgrowth (35). Furthermore, our suppression and overexpression experiments in zebrafish show neuronal pathfinding defects, aberrant branching and extension defects of the small-diameter axons that innervate the truncal musculature. These are concordant with the aforementioned studies performed in neuronal cultures and the fruit fly, denoting that TENM4 is essential for the integrity of the axonal outgrowth and neuronal innervation patterns. Furthermore, the disease association of three of the familial TENM4 mutations, but not the benign c.7933G>A allele, is supported by the demonstration of a dominant-negative effect of these alleles. These data also reconcile an apparent discrepancy between homozygous (the presence of tremor) and heterozygous (the absence of tremor) Tenm4 knockout mice: the dominant-negative effect of the discovered mutations is expected to be more severe than haploinsufficiency, but not as severe as the null phenotype. Similar observations have been made recently in other disorders and their corresponding mouse models (36). We are aware that our in vivo functional data provide only a limited insight into an adult onset phenotype. Nonetheless, in addition to our zebrafish studies being concordant with the mouse model, this model organism has been used successfully to model other adult onset disorders, including limb girdle muscular dystrophy (37) and, more relevant to the current phenotype, restless legs syndrome (38).
In conclusion, we provide several lines of evidence for a new gene for ET. First, we have identified three families of Spanish origin, in which three distinct pathogenic mutations in TENM4 both segregated with the phenotype and were over-transmitted in parent–offspring analyses. Second, we showed both in vitro and in vivo a dominant-negative effect of the identified mutations. Third, the existence of a TENM4 knockout mouse displaying a striking and severe ET phenotype with hypomyelination confirms our findings that abolished or strongly reduced activity of TENM4 drives the development of ET by affecting key cellular processes in the nervous system, and substantiates the identification of a major gene in the disease pathogenesis of ET.
We also show that, given the apparent genetic and phenotypic confounders in ET, coupling in vitro and in vivo studies with genomics approaches is critical to improving our ability to interpret the pathogenic effect of rare candidate alleles. The genetic heterogeneity and complex architecture mandate that candidates such as TENM4 should be investigated in additional families and larger cohorts. It is worthwhile to note that LINGO1, the first gene found to be consistently associated with ET, is also known to play an important role in oligodendrocyte maturation and central myelination. Together with this finding, our data suggest that further studies of oligodendrocyte and axonal guidance dysfunction might extend the catalog of genetic drivers of ET and help elucidate the genetic architecture of the disease.
Subjects, Materials and Methods
Study design and subjects
All participants included in the study were Europeans of Spanish origin and provided written informed consent. The study protocol was approved by the local ethics committees of the different hospitals providing the samples. Each patient and unaffected relative of the three families described here were examined by movement disorders specialists (the same neurologist for each family) and diagnoses were made according to the guidelines of the Consensus Statement of the Movement Disorders Society (2). Tremor severity was rated by the Fahn, Tolosa and Marin as well as the Glass scale (39,40). Moreover, the families were pre-screened for known ET and PD mutations (FUS [MIM 137070], SNCA [MIM 163890], LRRK2 [MIM 609007], VPS35 [MIM 601501], PINK1 [MIM 608309] and PARKIN [MIM 602544]).
Exome sequencing
We prepared sequencing libraries using the TruSeq DNA library preparation kit (Illumina) followed by exome capturing with the Nimblegen SeqCap EZ Human Exome Library v3.0 kit according to the manufacturers’ protocols. Five single-indexed libraries were pooled before capturing. We sequenced the multiplexed captured libraries on a HiSeq2000 (Illumina) resulting in a final mean coverage ranging from 39 to 43-fold and aligned the paired-end reads (100 bp) to the Human Genome (UCSC hg19) using bwa (v0.6.2) (41). We then called SNV and indels using three different software packages (GATK (42), SAMtools (43) and SHORE (44)), annotated the variants with ANNOVAR (45) and intersected the output of the three calling programs to obtain a reliable set of variants. After selecting for variants shared by all affected individuals, and establishing population frequency for each variant from the 1000 Genomes database and the Exome Variant Server (46,47), we applied the following filtering steps to obtain a final list of candidate variants: we focused on non-synonymous, splice-site, nonsense and frameshift variants, and performed in silico predictions of the damaging potential of each candidate variant using Polymorphism Phenotyping version 2 (Polyphen-2), Protein Variation Effect Analyzer (PROVEAN) and Combined Annotation Dependent Depletion (CADD) (20–22). We also analyzed the exome data for CNV with Conifer (48).
Targeted resequencing
We applied a recently published method based on the design and usage of molecular inversion probes (MIP) which allows high-throughput resequencing of loci of interest at low cost (49). MIPs were designed to target the coding region of TENM4 [MIM 610084] (probe sequences available on request). After pooling and 5′-phosphorylation, MIPs were hybridized to genomic DNA, followed by gap-filling and circularization. Capture circles were amplified by polymerase chain reaction (PCR) where the Illumina sequencing adaptor and one of 384 sample specific barcodes were appended. After cleanup and pooling of up to 384 single-indexed samples, multiplexed libraries were sequenced on a MiSeq (Illumina) using 150 bp paired-end reads. Paired-end reads were collapsed to one single read with FLASH (50) to reduce potential sequencing errors. Alignment, variant calling and filtering followed the same pipeline as exome sequencing. Identified novel variants (MAF <0.001) were validated by Sanger sequencing (primer sequences available on request).
Production of anti-TENM4 antibodies
A recombinant fusion protein between the Tenascin-C signal peptide and the human teneurin-4 EGF-like repeats was generated by splicing by overlap extension. The resulting PCR fragment was cloned into the expression vector pCEP4 (Invitrogen) and transfected into HEK293 EBNA cells. The recombinant protein was purified from the tissue culture medium using the Probond Purification System (Invitrogen) and sent for injection into rabbits to generate polyclonal antibodies. The antibodies were tested to recognize the recombinant protein as well as the endogenous TENM4 protein in tissue lysates. Using human glioblastoma protein lysates on western blots, the anti-TENM4 antiserum detected a single band above 200 kDa, which was not detectable anymore after preabsorption of the antiserum with the recombinant TENM4 protein used for immunization (not shown).
TENM4 cloning and transfection
We PCR-amplified TENM4 from a human brain cDNA library (Clonetech) and cloned the product into pcDNA 3.1 (Invitrogen). Sanger sequencing validation of the vector uncovered eight synonymous changes and seven point mutations that we retro-mutated using site-directed in vitro mutagenesis (Agilent). We generated TENM4 c.4100C>A, c.4324 G>A, c.3412G>A and c.7933G>A mutants using the same strategy. Murine oligodendroglial precursor cells (Oli-neu) were transfected with wild-type and mutant TENM4 as described (51). Cells were fixed in phosphate-buffered saline containing 4% paraformaldehyde, permeabilized with 0.1% Triton and blocked with 5% goat serum. Cells were then incubated overnight with the primary antiserum (rabbit anti-hTENM4) in 2.5% goat serum followed by goat anti-rabbit-biotin-conjugated and by goat anti-rabbit-streptavidin-Cy3 (Jackson Immunoresearch Laboratories). Nuclei were counterstained with 4',6-diamidino-2-phenylindole (DAPI) and preparations were analyzed with a Leica SP5 AOBS confocal microscope.
In silico analysis of ET candidates
Expression profiles for RCOR3, CYTL1 [MIM 607930], E2F8 [MIM 612047] and TENM4 [MIM 610084] were obtained from the BioGPS database (52). Information on the specific tissues in which each of these genes is detected was retrieved from AceView (53).
Zebrafish functional assay
We used reciprocal BLAST against the Danio rerio genome and identified a sole zebrafish ortholog of TENM4 (ENSDARG00000034264, 87% similarity, 77% identity). To determine the effect of tenm4 suppression in zebrafish embryos, two morpholinos (MO) were designed and obtained from Gene Tools, LLC (Philomath, OR, USA), tenm4 tbMO (AGGGTCTGCGTTCCTTGACTTCCAT), targeting the translation site and tenm4 sbMO (ATGCGGTTTCATACTAACCGATTGC), targeting the splice junction at the 3′ end of exon 10. To determine MO efficiency, total mRNA was extracted from control and MO-injected embryos, reverse-transcribed to produce cDNA and the site targeted by the MO was PCR amplified using the following primers: GGAGGTCGCAGGTCTTTATTG and CTGCAGTCGGGTCCTCTGAAGC, as described (54).
For zebrafish injections, capped human TENM4 mRNA was produced by cloning wild-type TENM4 (NM_001098816.2) cDNA and the four mutant constructs (c.4100C>A, c.4324 G>A, c.3412G>A and c.7933G>A) into the pCS2+ expression vector and reverse transcribing each construct in vitro with the T7 Message Machine kit (Ambion).
All experiments were carried out with the approval of the Institutional Animal Care and Use Committee. Zebrafish were maintained and mated according to standard procedures (55). For knockdown and rescue experiments, we injected 10 ng of sbMO, and/or 10 pg human mRNA into zebrafish embryos at 1–4 cell stage. Injected embryos were fixed at 2 dpf and were stained with anti-α acetylated tubulin primary antibody as described (56). The integrity of the PNS was evaluated by scoring embryos for motor neuron abnormalities involving pathfinding errors, abnormal branching and failure to extend. All the experiments were repeated three times and a Pearson's chi-square test (χ2) was used to determine the significance.
For in situ analysis, a myelin basic protein (mbp) probe was designed using the following primers: AATTAACCCTCACTAAAGGGGCCACTGCAAGCACCTCTGG and TAATACGACTCACTATAGGACGAGGAGAGGACACAAAGC. RNA in situ hybridization was performed as described (57).
Funding
This work was supported by the Spanish Plan Nacional [SAF2008-00357 (NOVADIS) to X.E., SAF2006-10126 (2006–2009) and SAF2010-22329-C02-01 (2011–2013) to P.P. and RD12/0013/0002 (2013–2017) to J.A.G.A.]; the Generalitat de Catalunya (AGAUR 2009 SGR-1502 to X.E.); the European Commission 7th Framework Program [Project No. 261123 (GEUVADIS) and Project No. 262055 (ESGI) to X.E.]; the UTE project Foundation for Applied Medical Research (FIMA) to P.P.; by the Swiss National Science Foundation (PBLAP3-136962 to H.H., 31003A_135735/1 to R.C.); by seed funding from the Center for Human Disease Modeling, Duke University and by P50 MH094268 to N.K. N.K. is a Distinguished Brumley Professor.
Supplementary Material
Acknowledgments
We thank patients and families for their participation in this study. We thank Beth K. Martin and Jay Shendure for technical advice regarding MIP sequencing as well as Elena Lorenzo and Elena Alonso for technical assistance and Jacqueline Trotter for providing us with Oli-neu cells. We also thank our Spanish collaborators Maria A. Pastor, Mario Riverol, Julian Benito-Leon, Ruben Fernandez-Santiago, Mario Ezquerra, Francesc Valldeoriola, Yaroslau Compta and Eduard Tolosa for providing additional DNA samples of ET cases.
Conflict of Interest statement. None declared.
References
- 1.Louis E.D., Ferreira J.J. (2010) How common is the most common adult movement disorder? Update on the worldwide prevalence of essential tremor. Mov. Disord., 25, 534–541. [DOI] [PubMed] [Google Scholar]
- 2.Deuschl G., Bain P., Brin M. (1998) Consensus statement of the Movement Disorder Society on Tremor. Ad Hoc Scientific Committee. Mov. Disord., 13(Suppl. 3), 2–23. [DOI] [PubMed] [Google Scholar]
- 3.Deng Q., Wang Q., Zong W.Y., Zheng D.L., Wen Y.X., Wang K.S., Teng X.M., Zhang X., Huang J., Han Z.G. (2010) E2F8 contributes to human hepatocellular carcinoma via regulating cell proliferation. Cancer Res., 70, 782–791. [DOI] [PubMed] [Google Scholar]
- 4.Jimenez-Jimenez F.J., Alonso-Navarro H., Garcia-Martin E., Lorenzo-Betancor O., Pastor P., Agundez J.A. (2013) Update on genetics of essential tremor. Acta Neurol. Scand., 128, 359–371. [DOI] [PubMed] [Google Scholar]
- 5.Stefansson H., Steinberg S., Petursson H., Gustafsson O., Gudjonsdottir I.H., Jonsdottir G.A., Palsson S.T., Jonsson T., Saemundsdottir J., Bjornsdottir G., et al. (2009) Variant in the sequence of the LINGO1 gene confers risk of essential tremor. Nat. Genet., 41, 277–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jimenez-Jimenez F.J., Garcia-Martin E., Lorenzo-Betancor O., Pastor P., Alonso-Navarro H., Agundez J.A. (2012) LINGO1 and risk for essential tremor: results of a meta-analysis of rs9652490 and rs11856808. J. Neurol. Sci., 317, 52–57. [DOI] [PubMed] [Google Scholar]
- 7.Thier S., Lorenz D., Nothnagel M., Poremba C., Papengut F., Appenzeller S., Paschen S., Hofschulte F., Hussl A.C., Hering S., et al. (2012) Polymorphisms in the glial glutamate transporter SLC1A2 are associated with essential tremor. Neurology, 79, 243–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gulcher J.R., Jonsson P., Kong A., Kristjansson K., Frigge M.L., Karason A., Einarsdottir I.E., Stefansson H., Einarsdottir A.S., Sigurthoardottir S., et al. (1997) Mapping of a familial essential tremor gene, FET1, to chromosome 3q13. Nat. Genet., 17, 84–87. [DOI] [PubMed] [Google Scholar]
- 9.Higgins J.J., Pho L.T., Nee L.E. (1997) A gene (ETM) for essential tremor maps to chromosome 2p22-p25. Mov. Disord., 12, 859–864. [DOI] [PubMed] [Google Scholar]
- 10.Shatunov A., Sambuughin N., Jankovic J., Elble R., Lee H.S., Singleton A.B., Dagvadorj A., Ji J., Zhang Y., Kimonis V.E., et al. (2006) Genomewide scans in North American families reveal genetic linkage of essential tremor to a region on chromosome 6p23. Brain, 129, 2318–2331. [DOI] [PubMed] [Google Scholar]
- 11.Ma S., Davis T.L., Blair M.A., Fang J.Y., Bradford Y., Haines J.L., Hedera P. (2006) Familial essential tremor with apparent autosomal dominant inheritance: should we also consider other inheritance modes? Mov. Disord., 21, 1368–1374. [DOI] [PubMed] [Google Scholar]
- 12.Testa C.M. (2013) Key issues in essential tremor genetics research: Where are we now and how can we move forward? Tremor Other Hyperkinet. Mov. (N. Y.), 3, tre-03-105-1843-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zimprich A. (2012) Phenocopies in families with essential tremor and restless legs syndrome challenge Mendelian laws. Epigenetics might provide answers. Parkinsonism Relat. Disord., 18, 711–716. [DOI] [PubMed] [Google Scholar]
- 14.Merner N.D., Girard S.L., Catoire H., Bourassa C.V., Belzil V.V., Riviere J.B., Hince P., Levert A., Dionne-Laporte A., Spiegelman D., et al. (2012) Exome sequencing identifies FUS mutations as a cause of essential tremor. Am. J. Hum. Genet., 91, 313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bentley D.R., Balasubramanian S., Swerdlow H.P., Smith G.P., Milton J., Brown C.G., Hall K.P., Evers D.J., Barnes C.L., Bignell H.R., et al. (2008) Accurate whole human genome sequencing using reversible terminator chemistry. Nature, 456, 53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ortega-Cubero S., Lorenzo-Betancor O., Lorenzo E., Alonso E., Coria F., Pastor M.A., Fernandez-Santiago R., Marti M.J., Ezquerra M., Valldeoriola F., et al. (2013) Fused in Sarcoma (FUS) gene mutations are not a frequent cause of essential tremor in Europeans. Neurobiol. Aging, 34, e2449–e2441. [DOI] [PubMed] [Google Scholar]
- 17.Rajput A., Rajput A.H., Rajput M.L., Encarnacion M., Bernales C.Q., Ross J.P., Farrer M.J., Vilarino-Guell C. (2013) Identification of FUS p.R377W in essential tremor. Eur. J. Neurol., 21, 361–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zheng W., Deng X., Liang H., Song Z., Gao K., Yang Y., Deng H. (2013) Genetic analysis of the fused in sarcoma gene in Chinese Han patients with essential tremor. Neurobiol. Aging, 34, 2078 e2073–2074. [DOI] [PubMed] [Google Scholar]
- 19.Unal Gulsuner H., Gulsuner S., Mercan F.N., Onat O.E., Walsh T., Shahin H., Lee M.K., Dogu O., Kansu T., Topaloglu H., et al. (2014) Mitochondrial serine protease HTRA2 p.G399S in a kindred with essential tremor and Parkinson disease. Proc. Natl. Acad. Sci. U. S. A., 111, 18285–18290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. (2010) A method and server for predicting damaging missense mutations. Nat. Methods, 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choi Y., Sims G.E., Murphy S., Miller J.R., Chan A.P. (2012) Predicting the functional effect of amino acid substitutions and indels. PLoS One, 7, e46688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kircher M., Witten D.M., Jain P., O'Roak B.J., Cooper G.M., Shendure J. (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet., 46, 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim J.S., Ryoo Z.Y., Chun J.S. (2007) Cytokine-like 1 (Cytl1) regulates the chondrogenesis of mesenchymal cells. J. Biol. Chem., 282, 29359–29367. [DOI] [PubMed] [Google Scholar]
- 24.Xue J.H., Zheng M., Xu X.W., Wu S.S., Chen Z., Chen F. (2011) Involvement of REST corepressor 3 in prognosis of human hepatitis B. Acta Pharmacol. Sin., 32, 1019–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suzuki N., Fukushi M., Kosaki K., Doyle A.D., de Vega S., Yoshizaki K., Akazawa C., Arikawa-Hirasawa E., Yamada Y. (2012) Teneurin-4 is a novel regulator of oligodendrocyte differentiation and myelination of small-diameter axons in the CNS. J. Neurosci., 32, 11586–11599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mosca T.J., Hong W., Dani V.S., Favaloro V., Luo L. (2012) Trans-synaptic Teneurin signalling in neuromuscular synapse organization and target choice. Nature, 484, 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Winkelmann J., Polo O., Provini F., Nevsimalova S., Kemlink D., Sonka K., Hogl B., Poewe W., Stiasny-Kolster K., Oertel W., et al. (2007) Genetics of restless legs syndrome (RLS): state-of-the-art and future directions. Mov. Disord., 22(Suppl 18), S449–S458. [DOI] [PubMed] [Google Scholar]
- 28.Ross O.A., Soto-Ortolaza A.I., Heckman M.G., Aasly J.O., Abahuni N., Annesi G., Bacon J.A., Bardien S., Bozi M., Brice A., et al. (2011) Association of LRRK2 exonic variants with susceptibility to Parkinson's disease: a case-control study. Lancet Neurol., 10, 898–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klein C., Chuang R., Marras C., Lang A.E. (2011) The curious case of phenocopies in families with genetic Parkinson's disease. Mov. Disord., 26, 1793–1802. [DOI] [PubMed] [Google Scholar]
- 30.Tucker R.P., Chiquet-Ehrismann R. (2006) Teneurins: a conserved family of transmembrane proteins involved in intercellular signaling during development. Dev. Biol., 290, 237–245. [DOI] [PubMed] [Google Scholar]
- 31.Zhou X.H., Brandau O., Feng K., Oohashi T., Ninomiya Y., Rauch U., Fassler R. (2003) The murine Ten-m/Odz genes show distinct but overlapping expression patterns during development and in adult brain. Gene Expr. Patterns, 3, 397–405. [DOI] [PubMed] [Google Scholar]
- 32.Passamonti L., Cerasa A., Quattrone A. (2012) Neuroimaging of Essential Tremor: what is the evidence for cerebellar involvement? Tremor Other Hyperkinet. Mov. (N Y), 2, 02-67-421-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suzuki N., Numakawa T., Chou J., de Vega S., Mizuniwa C., Sekimoto K., Adachi N., Kunugi H., Arikawa-Hirasawa E., Yamada Y., et al. (2014) Teneurin-4 promotes cellular protrusion formation and neurite outgrowth through focal adhesion kinase signaling. FASEB J., 28.3, 1386–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beckmann J., Schubert R., Chiquet-Ehrismann R., Muller D.J. (2013) Deciphering teneurin domains that facilitate cellular recognition, cell-cell adhesion, and neurite outgrowth using atomic force microscopy-based single-cell force spectroscopy. Nano Lett., 13, 2937–2946. [DOI] [PubMed] [Google Scholar]
- 35.Hoshina N., Tezuka T., Yokoyama K., Kozuka-Hata H., Oyama M., Yamamoto T. (2007) Focal adhesion kinase regulates laminin-induced oligodendroglial process outgrowth. Genes Cells, 12, 1245–1254. [DOI] [PubMed] [Google Scholar]
- 36.Cross S.H., Macalinao D.G., McKie L., Rose L., Kearney A.L., Rainger J., Thaung C., Keighren M., Jadeja S., West K., et al. (2014) A dominant-negative mutation of mouse Lmx1b causes glaucoma and is semi-lethal via LBD1-mediated dimerisation. PLoS Genet, 10, e1004359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sarparanta J., Jonson P.H., Golzio C., Sandell S., Luque H., Screen M., McDonald K., Stajich J.M., Mahjneh I., Vihola A., et al. (2012) Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat. Genet., 44, 450–455, S451–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schulte E.C., Kousi M., Tan P.L., Tilch E., Knauf F., Lichtner P., Trenkwalder C., Hogl B., Frauscher B., Berger K., et al. (2014) Targeted resequencing and systematic in vivo functional testing identifies rare variants in MEIS1 as significant contributors to restless legs syndrome. Am. J. Hum. Genet., 95, 85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fahn S., Tolosa E., Concepcion M. (1993) Clinical rating scale for tremor. In: Jankovic J., Tolosa E. (eds), Parkinson's Disease and Movement Disorders. 2nd ed. Williams and Wilkins, Baltimore, pp. 271–280. [Google Scholar]
- 40.Gironell A., Martinez-Corral M., Pagonabarraga J., Kulisevsky J. (2010) The Glass scale: a simple tool to determine severity in essential tremor. Parkinsonism Relat. Disord., 16, 412–414. [DOI] [PubMed] [Google Scholar]
- 41.Li H., Durbin R. (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., et al. (2010) The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res., 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ossowski S., Schneeberger K., Clark R.M., Lanz C., Warthmann N., Weigel D. (2008) Sequencing of natural strains of Arabidopsis thaliana with short reads. Genome Res., 18, 2024–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang K., Li M., Hakonarson H. (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res., 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abecasis G.R., Altshuler D., Auton A., Brooks L.D., Durbin R.M., Gibbs R.A., Hurles M.E., McVean G.A. (2010) A map of human genome variation from population-scale sequencing. Nature, 467, 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Callinan P.A., Wang J., Herke S.W., Garber R.K., Liang P., Batzer M.A. (2005) Alu retrotransposition-mediated deletion. J. Mol. Biol., 348, 791–800. [DOI] [PubMed] [Google Scholar]
- 48.Krumm N., Sudmant P.H., Ko A., O'Roak B.J., Malig M., Coe B.P., Quinlan A.R., Nickerson D.A., Eichler E.E. (2012) Copy number variation detection and genotyping from exome sequence data. Genome Res., 22, 1525–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.O'Roak B.J., Vives L., Fu W., Egertson J.D., Stanaway I.B., Phelps I.G., Carvill G., Kumar A., Lee C., Ankenman K., et al. (2012) Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science, 338, 1619–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Magoc T., Salzberg S.L. (2011) FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics, 27, 2957–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hor H., Bartesaghi L., Kutalik Z., Vicario J.L., de Andres C., Pfister C., Lammers G.J., Guex N., Chrast R., Tafti M., et al. (2011) A missense mutation in myelin oligodendrocyte glycoprotein as a cause of familial narcolepsy with cataplexy. Am. J. Hum. Genet., 89, 474–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu C., Orozco C., Boyer J., Leglise M., Goodale J., Batalov S., Hodge C.L., Haase J., Janes J., Huss J.W., 3rd, et al. (2009) BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol., 10, R130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thierry-Mieg D., Thierry-Mieg J. (2006) AceView: a comprehensive cDNA-supported gene and transcripts annotation. Genome Biol., 7(Suppl 1), S12, 11–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Niederriter A.R., Davis E.E., Golzio C., Oh E.C., Tsai I.C., Katsanis N. (2013) In vivo modeling of the morbid human genome using Danio rerio. J. Vis. Exp., e50338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Westerfield M. (2000) The Zebrafish Book: A Guide for the Laboratory use of Zebrafish (Danio Rerio). University of Oregon Press, Eugene, OR. [Google Scholar]
- 56.Margolin D.H., Kousi M., Chan Y.M., Lim E.T., Schmahmann J.D., Hadjivassiliou M., Hall J.E., Adam I., Dwyer A., Plummer L., et al. (2013) Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N. Engl. J. Med., 368, 1992–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zaghloul N.A., Liu Y., Gerdes J.M., Gascue C., Oh E.C., Leitch C.C., Bromberg Y., Binkley J., Leibel R.L., Sidow A., et al. (2010) Functional analyses of variants reveal a significant role for dominant negative and common alleles in oligogenic Bardet–Biedl syndrome. Proc. Natl. Acad. Sci. U. S. A., 107, 10602–10607. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.