

Abstract
We present an extremely rare case of malignant peripheral nerve sheath tumour (MPNST) in a 30-year-old woman without associated neurofibromatosis 1. The patient presented with an 8 cm×4 cm lesion extending from 46 to the retro molar region involving the ramus of the right mandible associated with regional paraesthesia. Incisional biopsy revealed spindle cells with vesicular nuclei arranged in fascicles leading to a diagnosis of spindle cell lesion. Posterior segmental mandibulectomy was performed under general anaesthesia. On excisional biopsy, a definitive diagnosis of low-grade MPNST was established on the basis of immunohistochemistry. The patient was then lost to follow-up.
Background
To the best of our knowledge, only two cases of malignant peripheral nerve sheath tumour (MPNST) of the mandible without association of neurofibromatosis have been reported in the literature to date. The aim of this paper is to highlight, in depth, its various clinicopathological characteristics and the role of immunohistochemistry findings that differentiate MPNST from other commonly encountered spindle cell malignancies.
Case presentation
A 30-year-old woman was referred to our department of oral and maxillofacial pathology with a tender swelling on the right side of the mandible of about 2 months’ duration. The swelling had gradually increased in size since she first noticed it, and had abruptly increased postextraction of 47 1 month prior (figure 1). She presented no relevant family, medical or personal history.
Figure 1.
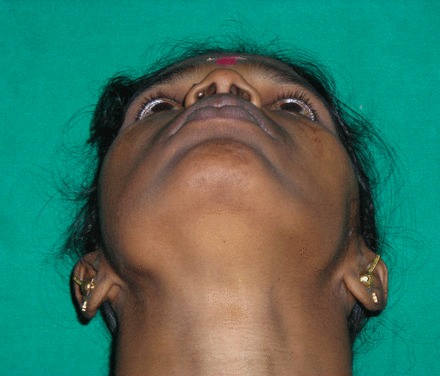
Extraoral view.
The swelling on the right side of the mandible extended horizontally from first molar up to the sigmoid notch, and vertically to the inferior border of the mandible, pushing 48 inferiorly. Intraoral examination revealed a swelling from 46 to retro molar region. Mild cortical expansion was noticed (figure 2). The lesion was covered with normal-appearing mucosa. On palpation, the lesion was slightly tender, non-fluctuant and soft-to-firm in consistency. Paraesthesia of the same side was noted. The cervical lymph nodes were apparently normal. Incisional biopsy revealed spindle cells with vesicular nuclei arranged in fascicles leading to a diagnosis of spindle cell lesion.
Figure 2.
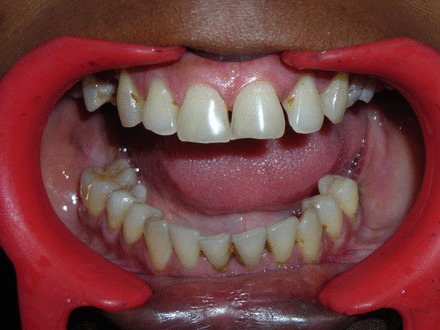
Intraoral view.
Posterior segmental mandibulectomy was performed under general anaesthesia. Histopathological examination aided with immunohistochemistry of the resected specimen confirmed the diagnosis of low-grade MPNST. The patient was advised chemotherapy and radiotherapy; however, the patient was subsequently lost to follow-up.
Investigations
Radiographic analysis showed a well-defined radiolucency involving body and ramus of the right side of the mandible extending from 46 up to the sigmoid notch (figures 3 and 4).
Figure 3.
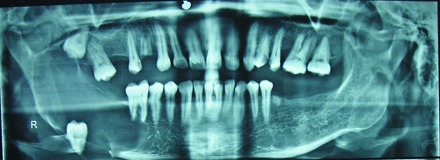
Orthopantomograph view showing well-defined radiolucency with displaced 48.
Figure 4.
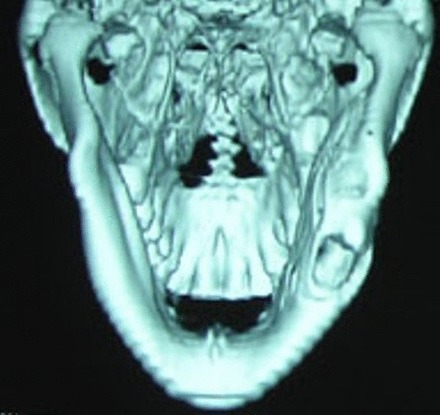
Three-dimensional reformatted CT image showing mild cortical expansion with inferiorly displaced 48.
Histopathological findings showed a partially encapsulated lesion having a fasciculated growth pattern with alternate hypocellular and hypercellular areas. Under higher magnification, fusiform or spindle-shaped cells were arranged in short fascicles in a haphazard fashion. These cells had scanty cytoplasm with hyperchromatic nuclei demonstrating mild-to-moderate pleomorphism and minimal mitotic activity. The tumour cells were also seen invading into the capsule and surrounding soft tissues (figures 5–7).
Figure 5.
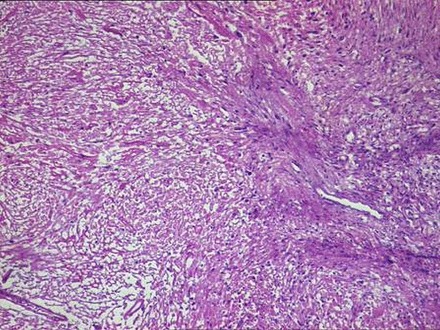
Lesion showing fasciculated growth pattern with alternate hypocellular and hypercellular areas (H&E ×10).
Figure 6.
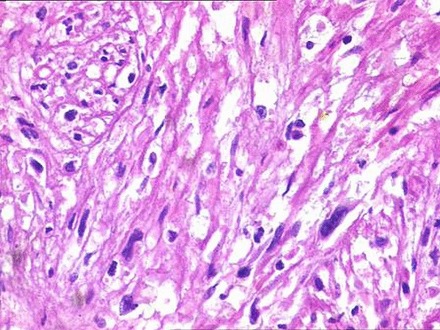
Fusiform or spindle-shaped cells with scanty cytoplasm with hyperchromatic nuclei.
Figure 7.
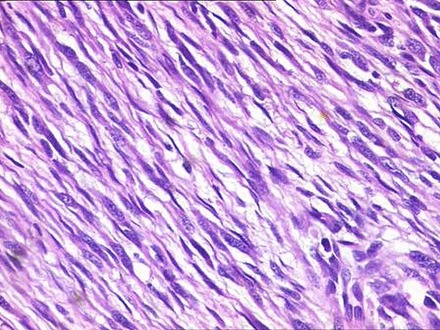
Tumour cells demonstrating mild-to-moderate pleomorphism and minimal mitotic activity.
Immunohistochemical analysis showed intense and diffuse positivity for vimentin, S-100 and Bcl-2 (figures 8–10). The tumour cells were non-reactive for desmin, cytokeratin (CK-7), epithelial membrane antigen (EMA), smooth muscle antigen (SMA), CD99 and melanocytic markers (melan-A, HMB-45).
Figure 8.
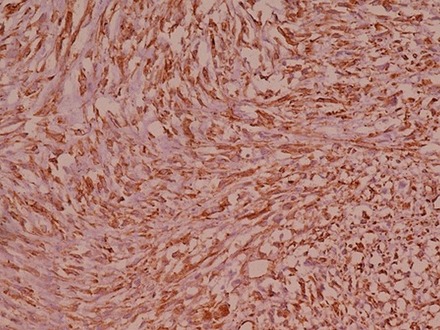
Diffuse and intense immune reactivity for vimentin.
Figure 9.
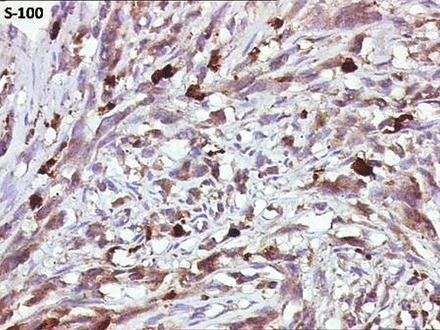
Diffuse immune reactivity for S-100.
Figure 10.
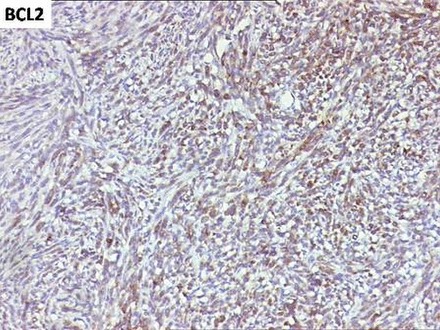
Diffuse and intense immune reactivity for Bcl-2.
Differential diagnosis
Most MPNSTs are histologically easily recognised as malignant tumours, but they may often resemble non-neural soft tissue spindle cell malignancies such as monophasic spindle cell variants of synovial sarcoma, fibrosarcoma, leiomyosarcoma, metastatic melanoma, and certain benign and reactive spindle cell lesions such as solitary fibrous tumour and nodular fascitis.1
Synovial sarcoma and fibrosarcoma show a uniform fasciculated growth pattern and contain fibroblast-like spindle cells. They may also contain hyalinised collagen bands and, occasionally, calcified areas. Most synovial sarcomas (about 90%) display immunoreactivity for CKs and EMA, at least in a few isolated areas, coexpressed along with S-100, which will be focally positive in about 30% of synovial sarcomas. CD99, a product of MIC-2, can be used as a differentiation marker for synovial sarcoma and is positive in 60–70% of cases. Fibrosarcomas lack evidence of nerve sheath differentiation and characteristically show cells arranged in long, sweeping fascicles. Fibrosarcomas usually lack immunoreactivity for CK, EMA and S-100; scattered cells in some fibrosarcomas may also stain for smooth muscle or muscle-specific actin. Leiomyosarcoma can usually be distinguished from MPNSTs based on classical histopathological features, cells with centrally placed blunt-ended/cigar-shaped nuclei, having deeply eosinophilic cytoplasm, with juxtranuclear vacuoles. In addition, leiomyosarcoma shows immunoreactivity to most of the smooth muscle markers, unlike MPNST. Metastatic melanomas clinically present as a soft tissue mass and share similar histopathological features with MPNST. Features including history of primary cutaneous melanomas, lymph node involvement and immunoreactivity to melanocytic markers such as HMB-45, melan-A, tyrosinase and microphthalmia transcription factor easily excludes melanoma from MPNST. Other spindle cell lesions such as solitary fibrous tumour and nodular fasciitis have limited growth potential with specific histopathological features indicating lesions of a benign nature. Clinical and histopathological features may suffice in differentiating these lesions from MPNST.1 2
Treatment
Posterior segmental mandibulectomy was performed under general anaesthesia (figure 11).
Figure 11.
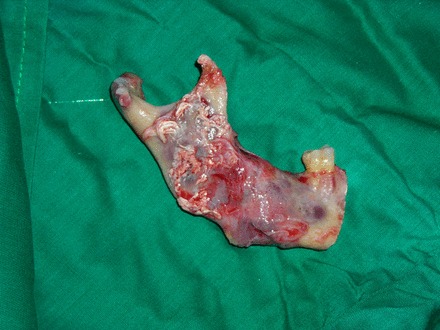
Excised gross specimen.
Outcome and follow-up
The patient was lost to follow-up.
Discussion
MPNST, otherwise known as malignant schwannoma or neurogenic sarcoma is an aggressive entity, with an origin from any cell of the nerve sheath: Schwann cell, perineural fibroblast or endoneurial fibroblast. It accounts for about 5–10% of soft tissue sarcomas, of which, in general, only about 8–16% occur in the head and neck region with very rare presentation in the jaws (maxilla>mandible).1 3–5 MPNST can arise from pre-existing neurofibroma or de novo. Aetiopathogenesis of MPNST with neurofibromatosis is believed to be associated with loss of chromosomal arm 17q sequence including complete inactivation of neurofibromatosis-1(NF-1) gene. A solitary variant of MPNST also occurs in 50–60% of cases. In the present case, association with NF-1 is excluded on the basis of major criteria laid down by the National Institutes of Health consensus development conference of 1988. The diagnostic major criteria for NF-1 are six or more café au lait macules (>0.5 cm in children or >1.5 cm in adults), two or more cutaneous/subcutaneous neurofibromas or one plexiform neurofibroma, axillary or groin freckling, optic pathway glioma, two or more Lisch nodules (iris hamartomas seen on slit lamp examination), bony dysplasia (sphenoid wing dysplasia, bowing of long bone±pseudarthrosis) and a first degree relative with NF1.6 In the majority of cases, close association of the nerve sheath is thought to be a diagnostic criterion for MPNST. If not associated with a peripheral nerve, aetiology of de novo intraosseous solitary MPNST is still obscure. One author suggested the possibility of odontogenic or neuroectodermal origin based on its prevalent periapical and gnathic involvement.3–5 Until now, only two cases of intraosseous (mandible) MPNST without association of neurofibromatosis have been reported (table 1).
Table 1.
Summary of clinical data of patients with intraosseous (mandible) malignant peripheral nerve sheath tumour without neurofibromatosis
| Author | Year | Age/sex | Site/radiological features | Treatment | Clinical outcome/follow-up period |
|---|---|---|---|---|---|
| Che et al5 Sham et al4 |
2006 2009 |
15/F 18/F |
Anterior mandible/ill-defined radiolucent lesion Body and angle multilocular/irregular radiolucent of mandible lesion |
Wide excision Segmental mandibulectomy and tumour resection with wide margin Bilateral comprehensive functional neck dissection (type-III) |
Not mentioned No recurrences/17 months |
MPNSTs generally present as a painless, enlarging mass with associated numbness along the distribution of the affected nerve. They mainly affect the 20–50-year-old age group and have an equal gender distribution. Radiographic presentation of intraosseous MPNST includes a complete destructive pattern with bony expansion, erosion and widening of mandibular canal/mental foramen with or without irregular destruction of surrounding bone.4 5 7 In the present case, the 30-year-old patient presented a solitary intraosseous MPNST as a well-defined radiolucent lesion in the body and ramus of the mandible.
A diagnosis of MPNST should be made on the basis of following widely accepted criteria, such as origin from a peripheral nerve, arising from a pre-existing benign nerve sheath tumour (neurofibroma) or the tumour displaying a constellation of histological features, including the dense and hypodense fascicles alternating in a marble-like pattern consisting of asymmetrically tapered spindled cells with irregular buckled nuclei. Other subtle and less-specific features such as nuclear palisading, increased perivascular cellularity and storiform or herringbone, curlicue growth patterns, may also be seen. Areas of geographic necrosis may be present. The most striking feature of MPNST is its degree of morphological heterogeneity, not only in terms of cellular pleomorphism and nuclear atypia but also in its abrupt transition of patterns, cellularity and grade. Certain histological variants such as epithelioid, glandular, rhabdomyosarcomatous (malignant triton tumour), pigmented and perineural have also been described in the literature but have little effect on prognosis.1 Diagnosis of MPNSTs is usually based on histopathology aided by immunohistochemistry, which reflects the Schwann cell differentiation in this neoplasm. Approximately 50–90% of MPNSTs are positive for S-100 protein. Occasionally, they may also express glial fibrillary acidic protein, EMA and CK expression.1 2 4 5 In the present case, histopathological features of the lesion, which included fasciculated growth pattern with alternate hypodense and hyperdense areas, and spindle cells with minimal cytological atypia, were seen infiltrating into the surrounding hard and soft tissue structures. These features were suggestive of spindle cell malignancy. The major challenge was to differentiate this neoplasm from other mimicking spindle cell malignancy, necessitating the aid of immunohistochemistry for a definitive diagnosis. The tumour cells showed immune reactivity for S-100, vimentin and Bcl-2, suggestive of neural origin. Immunonegativity of desmin and SMA ruled out the possibility of muscle-specific spindle cell malignancies. Non-reactivity to CD99, EMA, CK-7 and melanocytic markers eliminated other plausible neoplasms including synovial sarcoma and melanoma.
The treatment of MPNSTs of the jaws is wide surgical excision; however, local recurrence is common. The most common metastatic site for MPNST is the lung followed by bone and pleura. Regional lymph node metastasis is also not uncommon. Haematogenous metastasis occurs in at least half of cases.1 7–11
Prognosis of MPNST is generally poor. Prognostic variables for MPNST are the size of lesion, location, stage and grade, status of margins, necrosis and use of adjuvant radiation. Among these, the status of surgical margin and history of irradiation are independent negative prognostic factors.1 5 Overall survival rate is 40–70%.4
Learning points.
Diagnosis of malignant peripheral nerve sheath tumours (MPNSTs) in the head and neck region is difficult as there are no standardised radiological and histological criteria demarcating them from other spindle cell neoplasms and this often poses a great challenge for pathologists.
Definitive diagnosis of MPNSTs is usually based on histopathology aided by immunohistochemistry.
Surgical margin status and history of irradiation are strong negative predictors for prognosis of MPNST.
Footnotes
Contributors: SP contributed to the review of literature and design. SP, JP and KD participated in the preparation of the manuscript. NM and KD performed editing of the manuscript.
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Weiss SW, Goldblum JR. Enzinger and Weiss's soft tissue tumors. 5th edn St. Louis: Mosby, 2008:903–41. [Google Scholar]
- 2.Hirose T, Hasegawa T, Kudo E et al. MPNST an immuno-histochemical study in relation to ultrastructural features. Hum Pathol 1992;23(8):865–870. 10.1016/0046-8177(92)90396-K [DOI] [PubMed] [Google Scholar]
- 3.Ellis GL, Abrams AM, Melrose RJ. Intraosseous benign neural sheath neoplasms of the jaws. Report of seven new cases and review of the literature. Oral Surg Oral Med Oral Pathol 1977;44:731–43. 10.1016/0030-4220(77)90383-8 [DOI] [PubMed] [Google Scholar]
- 4.Sham ME, Ghorpande, Shetty A et al. Malignant peripheral nerve cell tumor. J Maxillofac Oral Surg 2009;9:68–71. 10.1007/s12663-010-0019-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Che Z, Nam W, Park W-S et al. Intraosseous nerve sheath tumors in the jaws. Yonsei Med J 2006;47:264–70. 10.3349/ymj.2006.47.2.264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferner RE, Huson SM, Thomas N et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet 2007;44:81–8. 10.1136/jmg.2006.045906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bullock MJ, Bedard YC, Bell RS et al. Intraosseous malignant peripheral nerve sheath tumor. Report of a case and review of the literature. Arch Pathol Lab Med 1995;119:367–70. [PubMed] [Google Scholar]
- 8.Polak M, Polak G, Brocheriou C et al. Solitary neurofibroma of the mandible: case report and review of the literature. J Oral Maxillofac Surg 1989;47:65–8. 10.1016/0278-2391(89)90127-4 [DOI] [PubMed] [Google Scholar]
- 9.Fernandes AM, Johann ACBR, Da Silveira-Juniour JB et al. Malignant peripheral nerve cell tumour of tongue. Oral Oncology Extra 2006;42:210–12. 10.1016/j.ooe.2005.12.003 [DOI] [Google Scholar]
- 10.Conley J, Janecka IP. Neurilemmoma of the head and neck. Trans Sect Otolaryngol Am Acad Ophthalmol Otolaryngol 1975;80:459–64. [PubMed] [Google Scholar]
- 11.Dorfman HD, Czerniak B. Bone tumors. St Louis: Mosby, 1998:839–41. [Google Scholar]