

Abstract
The posttranscriptional control of gene expression by microRNAs (miRNAs) is highly redundant, and compensatory effects limit the consequences of the inactivation of individual miRNAs. This implies that only a few miRNAs can function as effective tumor suppressors. It is also the basis of our strategy to define functionally relevant miRNA target genes that are not under redundant control by other miRNAs. We identified a functionally interconnected group of miRNAs that exhibited a reduced abundance in leukemia cells from patients with T cell acute lymphoblastic leukemia (T-ALL). To pinpoint relevant target genes, we applied a machine learning approach to eliminate genes that were subject to redundant miRNA-mediated control and to identify those genes that were exclusively targeted by tumor-suppressive miRNAs. This strategy revealed the convergence of a small group of tumor suppressor miRNAs on the Myb oncogene, as well as their effects on HBP1, which encodes a transcription factor. The expression of both genes was increased in T-ALL patient samples, and each gene promoted the progression of T-ALL in mice. Hence, our systematic analysis of tumor suppressor miRNA action identified a widespread mechanism of oncogene activation in T-ALL.
INTRODUCTION
Genomic studies have catalogued chromosomal lesions and somatic mutations in T cell acute lymphoblastic leukemia (T-ALL) (1–3). Increased expression of microRNAs (miRNAs) represents an additional mechanism of oncogenesis, and several highly abundant miRNAs promote leukemogenesis through the dominant repression of critical tumor suppressor genes. Of these, the miR-17~92 cluster may be the best-characterized example. Expression of this cluster is occasionally enhanced in Burkitt’s lymphoma, and it promotes lymphoma and T-ALL development in experimental models (4–6). The oncogenic action of miRNAs is further increased by overlapping and cooperative effects on target genes. In T-ALL, for example, five oncogenic miRNAs (miR-19b, miR-20a, miR-26a, miR-92, and miR-223) converge on a small set of tumor suppressor genes (for example, Pten and Fbxw7) and markedly reduce their expression (7). Hence, increases in the abundances of individual miRNAs have substantial effects on gene expression and cause various phenotypes, including malignant transformation.
On the other hand, the effects of miRNAs on target genes are highly redundant, an effect that attenuates the consequences of losing individual miRNAs. For example, the average human transcript harbors 70 to 100 3′ untranslated region (UTR)–binding sites, typically representing 16 different miRNAs (8, 9). Concordantly, comprehensive miRNA knockout studies in Caenorhabditis elegans or in mice often show minor or no apparent phenotypes (10–12). Similarly, only a few miRNAs act as tumor suppressors in loss-of-function studies. The best-characterized tumor-suppressive miRNAs are miR-15 and miR-16, which are lost in chronic lymphocytic leukemia (CLL) (13, 14), as well as miR-146, whose inactivation in mice stimulates a similar proliferation of myeloid cells (15). Here, we performed a comprehensive analysis of tumor suppressor miRNAs in cancer, and through a computational strategy, we identified a convergence of five miRNAs on MYB and HBP1 oncogenes in T-ALL.
RESULTS
Identification of tumor suppressor miRNAs in T-ALL
We took a systematic and stepwise approach to identify candidate tumor suppressor miRNAs and their unique targets (Fig. 1A). First, we catalogued all miRNAs that were differentially decreased in abundance in T-ALL patient specimens compared to normal T cells and their precursors. We then tested these miRNAs in gain- and loss-of-function studies and developed a machine learning strategy to identify nonredundant miRNA targets.
Fig. 1. Identification of miRNAs that are decreased in abundance in T-ALL.
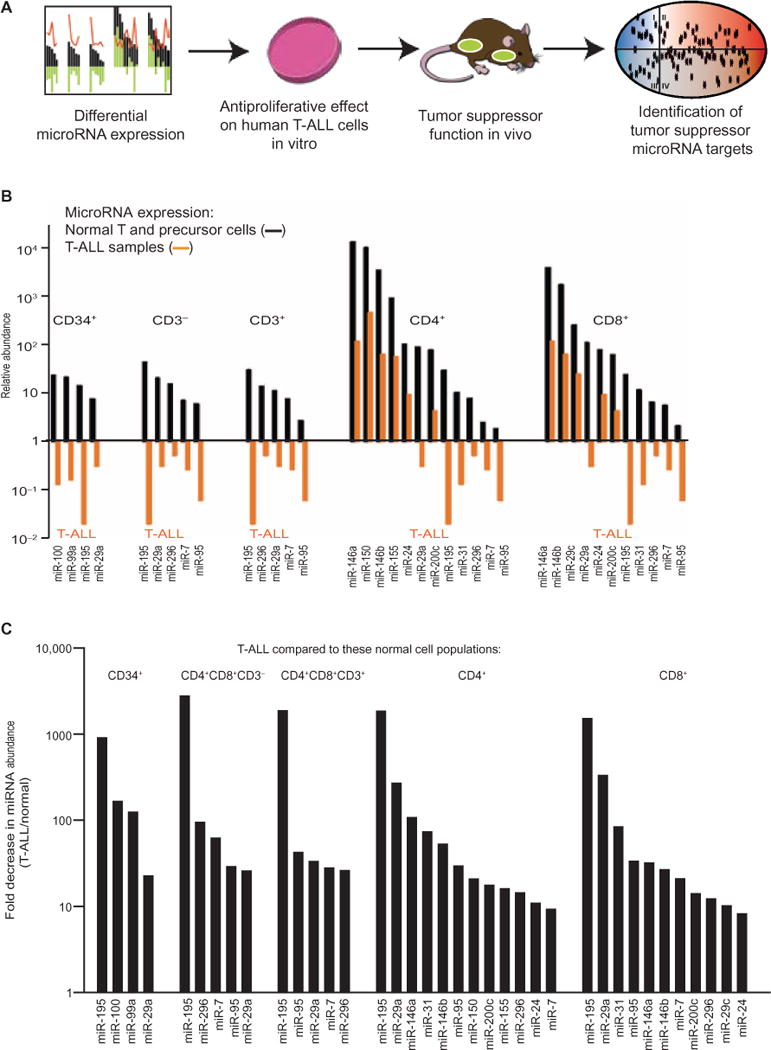
(A) Schematic of the overall experimental strategy to define biologically active tumor suppressor miRNAs and their targets in T-ALL. (B) The relative abundances of the indicated miRNAs were determined by qRT-PCR analysis of 50 T-ALL samples and the indicated populations of normal T cells and precursor cells. The relative abundances of miRNAs in T-ALL samples are indicated by orange bars. The relative abundances of miRNAs in normal cell populations sorted by fluorescence-activated cell sorting (FACS) are indicated by black bars. All values are normalized to the mean abundance of all detected miRNAs in a given sample. All miRNAs with a relative expression value >1.0 and >2-fold change in abundance compared to normal subsets and with an FDR < 0.05 are shown. The miRNAs are presented in order from highest to lowest abundance in each normal cell population. Data are from triplicate measurements, and all FDR values are <0.05. (C) Analysis of the fold decrease in the abundances of the indicated miRNAs in 50 primary T-ALL samples compared to their abundances in the indicated populations of FACS-purified normal T cell and precursor cell populations. Data in (B) and (C) are from the same experiments shown in (A). (See also tables S1 and S2 for all measurements.)
First, we compared the amounts of miRNAs in 50 T-ALL samples to those in different normal T cell and precursor populations. We used quantitative reverse transcription polymerase chain reaction (qRT-PCR) assays to measure the relative abundances of 430 miRNAs in normal progenitor cells (CD34+ cells and CD4+CD8+CD3− cells) and differentiated T cell populations (CD4+CD8+CD3+ and CD4+ or CD8+ cells) and compared them to those in 50 T-ALL specimens, which included all major cytogenetic subgroups, including MLL (n = 4 patients), CALM-AF10 (n = 3), inversion (7) (n = 5), LMO2 (n = 7), SIL-TAL (n = 8), TLX3 (n = 10), TLX1 (n = 5), and unknown (n = 8), as well as in 18 T-ALL cell lines (table S1) (16). Overall, the major cytogenetic groups showed broadly similar miRNA abundances (7). To identify miRNAs that were decreased in abundance in T-ALL cells compared to in normal cells, we used the following thresholds and requirements: (i) the miRNA had to be abundant in any one of the normal cell populations (that is, its relative abundance was >1.0); (ii) the miRNA had to be decreased in abundance by at least 10-fold in T-ALL samples compared to that in normal cells; and (iii) the change in abundance had to be statistically significant [that is, there should be a false discovery rate (FDR) < 0.05]. These thresholds were designed to be inclusive pending subsequent functional filtering; however, we identified only 12 miRNAs that met these criteria: miR-7, miR-24, miR-29, miR-31, miR-95, miR-100, miR-146, miR-150, miR-155, miR-195, miR-200c, and miR-296 (Fig. 1, B and C, and table S2). Some miRNAs that have been described as tumor suppressors in other cancers were either unchanged in abundance in T-ALL cells (for example, miR-15, miR-16, and Let7) or even increased in abundance in T-ALL cells compared to those in normal T cells (miR-34 and miR-451) (fig. S1, A to D, and table S3). Hence, we identified a set of miRNAs that were differentially decreased in abundance in T-ALL.
Functional assessment of candidate tumor suppressor miRNAs
Next, we tested the effect of enforced expression of the 12 miRNAs that we identified in human T-ALL cell lines. Briefly, we transduced KoptK1, RPMI-8402, DND41, and T-ALL cells (with 20 to 50% transduction efficiency) with retroviruses expressing the individual miRNAs transcriptionally tethered to complementary DNA (cDNA) encoding green fluorescent protein (GFP), which acted as a reporter, and monitored changes in the proportion of cells in each population that contained GFP (GFP+) over time (Fig. 2A). An increase in GFP+ cells indicates that the coexpressed miRNAs provided a proliferative advantage to the transduced cells and vice versa. As expected, all 12 miRNAs were detected, albeit at low abundance, in all of the cell lines (table S1). Of the 12 miRNAs tested, the enforced expression of only miR-29, miR-31, miR-150, miR-155, and miR-200 produced the most consistent antiproliferative effects. Our criteria for defining tumor-suppressive miRNAs were reproducibility and statistical significance (P < 0.05) in triplicate experiments, as well as an antiproliferative effect in three of the four cell lines tested (Fig. 2B). These criteria were designed to be exclusive in preparation for subsequent loss-of-function studies in vivo; however, miRNAs that did not meet these criteria might still have relevant functions under other specific circumstances (fig. S2).
Fig. 2. Analysis of miRNAs with antileukemic potential.
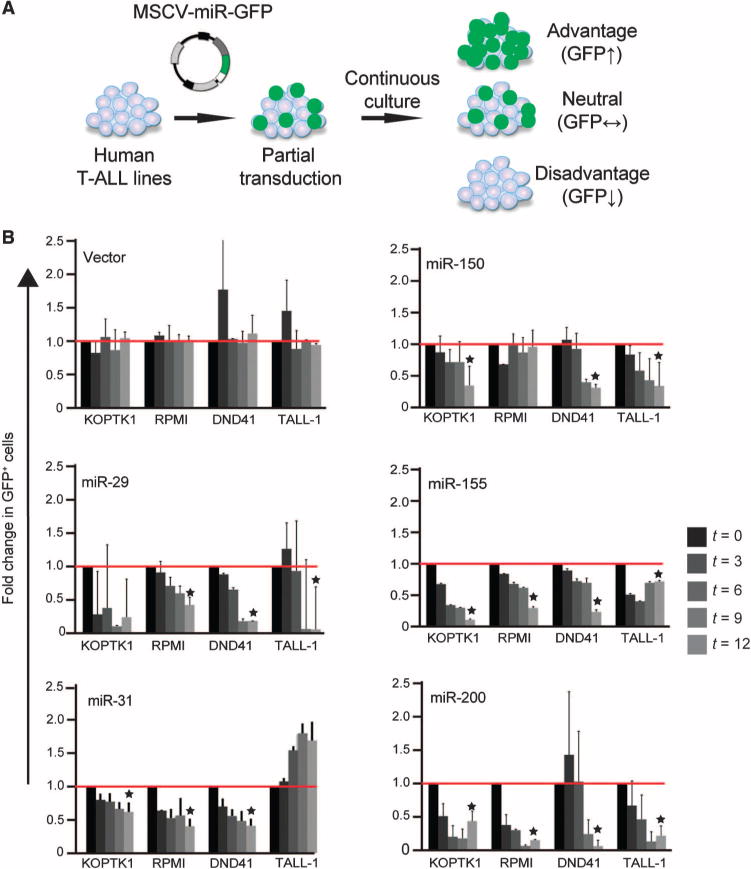
(A) Schematic of the competition assay in which human T-ALL cell lines were transduced with retroviruses expressing individual miRNAs and GFP, and the proportion of miRNA-expressing cells (as determined by GFP fluorescence) within each population was monitored by flow cytometric analysis. Those miRNAs that provided a proliferative advantage to the transduced cells resulted in an increased proportion of GFP+ cells. (B) A panel of the indicated human T-ALL cell lines were transduced with retrovirus expressing the indicated miRNAs and GFP, and the proportions of each cell population that expressed each miRNA (defined as GFP+ cells) at the indicated times (t, in days) were determined by flow cytometric analysis. The proportions of GFP+ cells at each time point are expressed relative to those on day 3 after infection. Data are means ± SD of three independent experiments. Only those miRNAs that showed a statistically significant effect on proliferation (*P < 0.05 by Student’s t test) in at least three of the four cell lines tested were selected for subsequent experiments.
We then performed miRNA loss-of-function studies in a murine T-ALL model. We used a mouse model based on the adoptive transfer of hematopoietic progenitor cells (HPCs) expressing the intracellular domain of Notch1 (ICN) into irradiated syngeneic mice (Fig. 3A) (17, 18). ICN is a dominant oncogene product, and it induced T-ALL in 100% of mice with a median latency of 2 months (median survival = 59 days; n = 6 mice); however, we still detected a statistically significant increase in the rate of T-ALL development upon miRNA inactivation. For example, expression of ICN in miR-150−/− or miR-155−/− HPCs statistically significantly increased the rate of onset of leukemia in recipient mice. The median survival time for mice receiving ICN-expressing miR-150−/− HPCs was 39 days (n = 7 mice), whereas the median survival time for mice that received ICN-expressing miR-155−/− HPCs was 42 days (n = 10 mice) (Fig. 3B, P < 0.02 compared to ICN-expressing, wild-type HPCs). On the other hand, loss of miR-146 (in miR-146−/− HPCs) had no effect on tumor onset. The median survival time for mice receiving ICN-expressing miR-146−/− HPCs was 72 days (n = 5) (Fig. 3B, P = 0.8 compared to ICN-expressing, wild-type HPCs).
Fig. 3. Loss-of-function studies define tumor suppressor miRNAs in T-ALL.
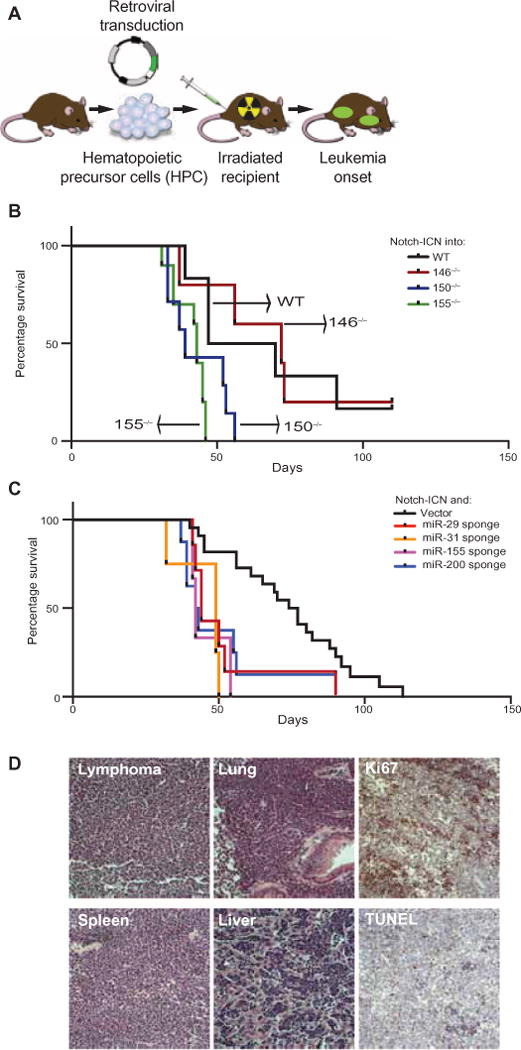
(A) Schematic of the mouse T-ALL model based on the adoptive transfer of mouse HPCs transduced with retrovirus expressing Notch1-ICN. (B) Irradiated C57BL/6 mice were subjected to transplantation with Notch1-ICN–expressing HPCs from wild-type (WT), miR-146−/−, miR-150−/−, or miR-155−/− mice. The percentages of the recipient mice that survived were measured over time and subjected to Kaplan-Meier analysis. (C) Irradiated C57BL/6 mice were subjected to transplantation with WT HPCs transduced with retrovirus expressing Notch1-ICN together with the indicated miRNA-specific sponges or with empty vector as a control. See fig. S3 for the functional characterization of the miRNA sponges. The percentages of the recipient mice that survived were measured over time and subjected to Kaplan-Meier analysis. For the miR-29 sponge, n = 7 mice and P = 0.04 compared to vector; for the miR-31 sponge, n = 4 mice and P = 0.001; for the miR-155 sponge, n = 3 mice and P = 0.0003; and for the miR-200 sponge, n = 8 mice and P = 0.01. (D) Representative microphotographs of ICN-driven murine T-ALL. All images are at ×100 magnification and are from a representative mouse. The pathologic appearances of T-ALLs harboring different miRNA lesions were identical. See fig. S3 for surface marker analysis.
We used miRNA sponges to confirm these effects and to test additional candidate miRNAs for which knockout animals were not available (19). Briefly, the sponges encode four miRNA seed–specific binding elements, and they efficiently repressed the on-target activities of their cognate miRNAs in reporter assays (fig. S3, A and B). Cotransduction of wild-type HPCs with ICN and the sponges against miR-29 (median survival, 44 days), miR-31 (median survival, 49 days), miR-155 (median survival, 42 days), and miR-200 (median survival, 43 days) accelerated the onset of T-ALL in recipient mice compared to that in mice that received control vectors (median survival, 76 days) (Fig. 3C). Similarly, sponge-mediated inhibition of these miRNAs in MOHITO cells, a murine T cell line, increased their proliferation compared to that of cells that received a nontargeting sponge (fig. S3C).
A third approach to target miRNAs is based on lentiviral miRZips, which act as RNA interference constructs that target specific miRNAs (20). We found that coexpression of ICN and miRZips specific for miR-29a (median survival, 48 days), miR-150 (survival, 60 days), miR-155 (median survival, 62 days), or miR-200 (median survival, 47 days) accelerated leukemia development and confirmed the observations from the experiments with knockout cells and sponges (fig. S3D). To exclude any nonspecific effect by the miR-150 miRZip, we introduced the construct into cells from miR-150−/− mice, but it had no additional effect on leukemia development in recipient mice compared to that in mice that received HPCs from miR-150−/− mice without miRZip (fig. S3E).
Pathologically, all ICN-driven leukemias were indistinguishable from each other, because they were THY-1+ and CD4 and CD8 double-positive and they showed minimal infiltration of B cells; aggressive invasion into solid organs; a high proliferative index, as determined by Ki-67 staining; and the absence of apoptosis, as determined by terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick end labeling (TUNEL) staining (Fig. 3D and fig. S3, F and G). Hence, we identified miR-29, miR-31, miR-150, miR-155, and miR-200 as tumor suppressor miRNAs whose abundance was decreased in T-ALL and whose loss promoted leukemogenesis in vivo.
A strategy to identify nonredundant miRNA targets in loss-of-function studies
The large number of genes targeted by miRNAs sometimes obscures the biologically relevant miRNA targets. However, we reasoned that in a loss-of-function setting, this pleiotropy should facilitate target identification. Briefly, among all of the predicted target genes of a given miRNA, only a small number will be affected by loss of that miRNA, whereas the expression of most predicted gene targets will not change and they will be controlled by other miRNAs (Fig. 4A). In practice, we computationally identified all of the predicted targets of our set of tumor suppressor miRNAs (Fig. 4B). We then quantified the interaction of all targets with these miRNAs by adding their mirSVR scores, which is a computational prediction of miRNA-mRNA binding (21), and then we did the same for the 30 most highly abundant miRNAs in T-ALL. The difference in mirSVR score is expressed on the x axis and identifies preferential binding by tumor suppressor miRNAs (toward the right) or by abundant miRNAs (to the left) (Fig. 4B). Next, we excluded genes whose mRNA abundance was not increased in T-ALL samples compared to that in normal T cells (expressed on the y axis) on the basis of published gene expression data (22). This approach left genes represented in quadrant I as the likely relevant targets of the tumor suppressor miRNAs (Fig. 4B and table S4).
Fig. 4. Tumor suppressor miRNAs converge on the Myb oncogene in T-ALL.
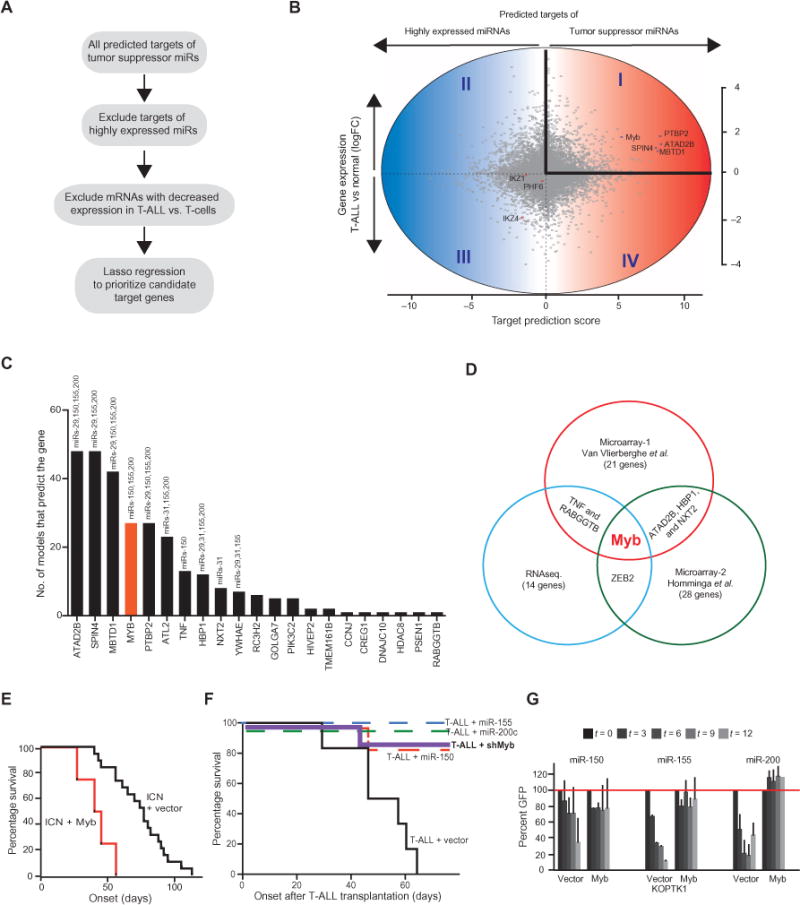
(A) Schematic of the strategy used to identify relevant miRNA targets. (B) Cloud diagram showing all predicted targets of the tumor suppressor miRNAs. The y axis reflects differential gene expression in T-ALL. Quadrants I and II include genes whose expression is increased in T-ALL compared to that in normal subsets. The x axis reflects the difference in cumulative binding efficiencies (mirSVR score) for tumor suppressor miRNAs and the 30 most highly abundant miRNAs. Genes in quadrants I and IV are more efficiently targeted by tumor suppressor miRNAs than by highly abundant miRNAs. (C) Discriminative machine learning prioritizes genes in quadrant I. The graph indicates how many models out of 100 runs identified the indicated gene as a discriminator between tumor suppressor miRNAs and random sets of 30 highly abundant miRNAs. (D) Myb was identified as a converging target of tumor-suppressive miRNAs in three independent analyses described in (B) and (C). (E) Analysis of leukemia-free survival after transplantation of mice with HPCs expressing Notch1-ICN and either vector (black, n = 19 mice) or Myb (red, n = 4 mice, P = 0.0005). The percentages of the recipient mice that survived were measured over time and subjected to Kaplan-Meier analysis. (F) Analysis of the time of leukemia initiation in mice injected intravenously with 25,000 murine T-ALL cells (ICN/Ikaros-6) transduced with empty vector (n = 6 mice; median survival of 50.5 days) or with retrovirus expressing a Myb-specific short hairpin RNA (shMyb-2, n = 7 mice; P < 0.005), miR-150 (miR-150, n = 7 mice; P < 0.005), miR-155, (miR-155, n = 5 mice; P < 0.005), or miR-200 (miR-200, n = 7 mice; P < 0.005), as indicated. The percentages of the recipient mice that survived were measured over time and subjected to Kaplan-Meier analysis. (G) Competition experiment comparing the antiproliferative effects of the indicated miRNAs in parental KoptK1 cells transfected with empty vector or with plasmid expressing Myb. Data are means of three independent infections.
Next, we applied a discriminative machine learning approach that used sparse logistic regression to prioritize genes in quadrant I. The machine learning algorithm learned to discriminate between tumor-suppressive miRNAs and random sets of 30 expressed miRNAs on the basis of their mirSVR-predicted target genes (21). The use of a lasso constraint resulted in only a few target genes being included in the model with nonzero regression coefficients. To account for the small number of positive training examples, we performed a stability analysis by repeating the learning procedure 100 times with different random miRNA sets (Fig. 4C and table S5). We also applied this same model to another publically available gene expression data set (23) and to our RNAseq-based gene expression analysis of eight T-ALL and four normal T cell samples. The Myb oncogene emerged from this analysis as among the top candidate genes and was the only gene identified by all three analyses (Fig. 4D). In detail, the machine learning approach identified Myb in 38% of iterative calculations in our RNAseq-based data set, in 27% in the expression data from Van Vlierberghe et al. (22), and in 45% in the expression data from Homminga et al. (23) (fig. S4). Hence, our computational strategy to discriminate unique from common miRNA targets identified Myb as a key effector of miRNA-mediated tumor suppression in T-ALL.
Characterization of Myb as a biologically relevant target of tumor suppressor miRNAs
The 3′UTR of Myb has three miR-150–binding sites (24), as well as two binding sites each for miR-155 and miR-200c (fig. S5A). Accordingly, murine T-ALLs expressing miRZips or sponges specific for miR-150, miR-155, or miR-200 exhibited increased amounts of Myb mRNA and protein compared to control tumors (fig. S5, B to D). Furthermore, luciferase reporter assays performed with constructs expressing the 3′UTR of wild-type Myb or binding site mutant forms of the Myb 3′UTR showed direct and site-specific repression by miR-150, miR-155, and miR-200 (fig. S5, E to G). In vivo, overexpression of Myb in HPCs substantially accelerated the rate of development of ICN-dependent T-ALL in transplanted mice (median survival of 42.5 days) compared to mice that received HPCs transduced with empty vector (median survival of 77 days) (Fig. 4E). Conversely, knockdown of Myb expression (fig. S5H) or overexpression of miR-150, miR-155, or miR-200 in established murine leukemic cells completely abrogated their leukemia-initiating capacity upon transplantation into syngeneic animals (25,000 cells via tail vein injection). Finally, expression of a cDNA encoding a mutant Myb deficient in all miRNA-binding sites prevented the antiproliferative effects of miR-150, miR-155, and miR-200 on human T-ALL cells (Fig. 4G and fig. S5I). Finally, sponge-mediated inhibition of miRNA155 and, to a lesser extent, miR-200 caused an increase in Myb abundance (fig. S5J).
Characterization of HBP1 as an oncogenic target of tumor suppressor miRNAs in T-ALL
Inhibition of miR-29 and miR-31 promoted leukemogenesis in vivo (Fig. 3C and fig. S3D). With the same machine learning strategy, we found that in two of the three data sets, the gene encoding high mobility group boxtranscription factor (HBP1) was a target of both miR-29 and miR-31 (together with miR-155 and miR-200) (Fig. 4, C and D). The binding sites for these tumor-suppressive miRNAs are conserved in the 3′UTR of murine Hbp1 (fig. S6A), and consequently, mouse tumors arising as a result of sponge-mediated repression of these miRNAs showed a statistically significant increase in Hbp1 expression (fig. S6B). We examined the potential oncogenic effect of Hbp1 expression and found a statistically significant increase in the rate of disease development in an ICN-dependent T-ALL model (median survival: 40 days) (fig. S6, C and D). HBP1 has been implicated in the suppression of the cell cycle regulator p21 (25) as well as in the induction of CD2 expression in T cells (26, 27). Hence, our analysis of the target genes of tumor suppressor miRNAs has uncovered a previously uncharacterized leukemogenic function for HBP1.
Coordinate control of tumor suppressor miRNAs by c-Myc in T-ALL
Chang et al. (28) reported the control of miRNA expression by MYC, and they identified several miRNAs whose expression was repressed by MYC, including miR-29 and miR-150. Myc is a direct effector of NOTCH1 (29, 30). We took advantage of a tamoxifen-inducible dominant-negative MYC protein (omomycER) to test whether MYC was required for the expression of tumor suppressor miRNAs in T-ALL (31, 32). Briefly, omomyc forms transcriptionally inactive heterodimers with all endogenous MYC proteins (c-MYC, N-MYC, and L-MYC) by blocking the interaction of MYC with its binding partner MAX (32). We introduced omomycER into KoptK1 T-ALL cells and, after tamoxifen treatment, measured the abundances of miRNAs of interest by qRT-PCR analysis. We found that the abundances of miR-31, miR-150, and miR-155 were statistically significantly reduced in cells with omomycER compared to those in control cells (fig. S6E). MYC is under the transcriptional control of NOTCH1, and accordingly, treating KoptK1 cells with Compound E, a γ-secretase inhibitor (which prevents cleavage of NOTCH1 and generation of the ICN), led to a statistically significant increase in the abundances of miR-150 and miR-155, as well as a trend toward increased amounts of miR-31 (fig. S6F). Hence, repression of the expression of a subset of tumor suppressor miRNAs in T-ALL cells was a consequence of NOTCH and MYC activation.
DISCUSSION
Our study provides insights into the pathogenesis of T cell leukemia. We report a large-scale analysis of miRNA expression profiles, and we identified and functionally validated five tumor suppressor miRNAs and their biological targets in T-ALL. Among a larger set of differentially expressed miRNAs, only miR-29, miR-31, miR-150, miR-155, and miR-200 behaved as tumor suppressors whose inactivation promoted leukemogenesis. The activities of these miRNAs are tissue-specific, and miR-29 and miR-155 have tumor-promoting activities in B lymphocytes and myeloid cells (33–37). This tissue specificity is consistent with the notion that lineage-specific gene expression programs determine the target repertoire, and therefore the biological consequences, of miRNA inactivation. Conversely, we found that miRNAs also controlled the abundance of lineage-restricted transcription factors, such as Myb. Specifically, the convergence of three tumor suppressor miRNAs that were lost (or decreased in abundance) across the T-ALL samples that we analyzed reveals a widespread mechanism of Myb activation and suggests an alternative oncogenic mechanism to that of rare chromosomal gains affecting the Myb locus (38–40). An oncogenic function of Myb has been described in certain leukemias, including acute myeloid leukemia (41) and T-ALL (38). In T-ALL, Myb overexpression blocks the differentiation of immature T cells, whereas its knockdown reverses this effect (38). Multiple mechanisms lead to Myb overexpression in T-ALL, including mutation or miR-223–mediated repression of the E3 ubiquitin ligase Fbxw7 (7, 42–44), increased transcription (45), and increased eIF4A (eukaryotic initiation factor 4A)–dependent translation of Myb mRNA (46). These events are not mutually exclusive, which is suggestive of additive effects in T-ALL pathogenesis. MYB has been an elusive drug target; however, histone deacetylase inhibitors such as SAHA (suberoylanilide hydroxamic acid) (47), which can impede Myb expression, and the natural compound Silvestrol, which potently blocks the translation of Myb mRNA (46), might be effective in the treatment of T-ALL.
Our study revealed a Myc-dependent network of tumor suppressor miRNAs in T-ALL. Studies of Dicer-deficient mice have suggested a widespread role for miRNAs in cancer (48, 49); however, only a small number of miRNAs are targets of genomic deletions or behave as tumor suppressors in loss-of-function studies (13–15). Conceivably, the overlapping and pleiotropic on-target effects of miRNAs have evolved to minimize the effects of losing individual miRNAs. This notion is borne out in systematic knockout studies in which the loss of miRNAs is most associated with minimal or absent phenotypes (10, 11). Our results identify a small number of functionally validated tumor suppressor miRNAs. The expression of these miRNAs is repressed upon MYC activation, and this links their expression directly to a key oncogenic driver in T-ALL. Notably, these miRNAs show substantial convergence on Myb and HBP1, and their loss largely alleviates miRNA-mediated control of the expression of these genes. To identify such distinct miRNA targets, we developed a discriminatory machine learning tool. This method is specifically useful in deciphering the effects of miRNA inactivation and takes advantage of the overlapping target gene repression to identify a small number of unique targets whose expression is expected to change upon miRNA inactivation. In summary, we report a functionally interconnected set of tumor suppressor miRNAs that converge on key T-ALL–associated oncogenes.
MATERIALS AND METHODS
T-ALL patient samples and cell lines
Diagnostic bone marrow samples of 50 T-ALL patients were obtained from different European centers (UZ Ghent, Ghent; UZ Leuven, Leuven; Hôpital Purpan, Toulouse; CHU de Nancy-Brabois, Vandoeuvre-Les-Nancy). The resulting T-ALL patient cohort consisted of 15 TAL/LMO (7 LMO2 and 8 SIL-TAL), 12 HOXA [4 MLL, 6 inv(7)(p15q35), and 2 CALM-AF10], 10 TLX3, 5 TLX1, and 8 cases that could not be categorized. Total RNA was isolated from the bone marrow samples with the miRNAeasy Mini Kit (Qiagen). This study (2008/531) was approved by the Medical Ethical Commission of Ghent University Hospital (Belgium, registration B67020084745). Eighteen T-ALL cell lines were cultured in RPMI 1640 (Invitrogen), 15% fetal calf serum, 1% penicillin, 1% streptomycin, 1% kanamycin, and 1% glutamine.
Subsets of normal T cell populations and miRNA profiling
Normal T cell populations from pediatric thymuses (CD34+, CD4+CD8+CD3−, CD4+CD8+CD3+, CD3+CD4+CD8−, and CD3+CD4−CD8+) were obtained by FACS and MACS (magnetic cell sorting). CD34+ cells were isolated with CD34 microbeads (Miltenyi Biotec) (50). Double-positive thymocytes were isolated by staining with fluorescein isothiocyanate (FITC)–conjugated anti-CD3, phycoerythrin (PE)–conjugated anti-CD8, and allophycocyanin (APC)–conjugated anti-CD4 antibodies. Single-positive thymocytes were depleted of CD1+ thymocytes by Dynabeads (Dynal Biotech) and subsequently labeled with anti-CD3–FITC, anti-CD8–PE, and anti-CD4–APC. The purity of the different subsets of normal T cell populations was always at least 98%. Profiling of miRNAs was performed with stem-loop reverse transcription primers for the synthesis of cDNA from miRNA, followed by a preamplification step and real-time PCR analysis (Applied Biosystems) as described previously (51). Briefly, 20 ng of total RNA was reverse-transcribed with the Megaplex reverse transcription stem-loop primer pool for the cDNA synthesis of 448 small RNAs, including 430 miRNAs and 18 small RNA controls. Preamplification of cDNA was performed in a 14-cycle PCR with TaqMan PreAmp Master Mix (2×) and the PreAmp Primer Mix (5×) (Applied Biosystems) consisting of a miRNA-specific forward primer and a universal reverse primer (52). Finally, the 448 small RNAs were profiled for each sample with a 40-cycle PCR protocol. SDS software version 2.1 was used to calculate the raw quantification cycle (Cq) values with automatic baseline settings and a threshold of 0.05. For normalization of the qRT-PCR data of the miRNA expression profiles, we used the mean expression value of all expressed miRNAs in a given sample as the normalization factor; miRNAs with a Cq value >32 were considered to be below the limit of detection (16).
Cell culture and treatment, viability and proliferation assays, and plasmid constructs
T-ALL cell lines and tumors were virally transduced as described previously (53, 54). miRNA-cDNA expression constructs and miRNA sponges were cloned in murine stem cell virus (MSCV)–based retroviral vectors (4, 17), whereas miR-ZIP antagomirs were expressed from lentiviral vectors (System Biosciences), including MZIP29a-PA-1, MZIP150-PA-1, MZIP155-PA-1, MZIP200c-PA-1, and the scrambled control MZIP000-PA-1. miRNA-specific sponges were designed and tested as described previously (19).
In vitro screens of individual miRNAs or sponges
The T-ALL cell lines KoptK1, RPMI-8402, DND41, and TALL-1 were transduced with HIV-based lentiviral vectors that express both the miRNA (as indicated) and GFP. Seventy-two hours after infection, the percentage of GFP+ cells was determined by flow cytometric analysis. Cells were then monitored every 48 hours for the expression of GFP over a 14-day period. Similarly, MOHITO cells were transduced with retroviruses expressing miRNA-specific sponges and GFP and were then monitored for GFP enrichment or depletion at the population level by flow cytometry.
Generation of murine T-ALL and transplantation assays
The mouse T-ALL model has been described previously (17, 55). Briefly, fetal liver stem cells (from 13.5- to 14.5-day-old embryos) derived from either wild-type or miRNA knockout mice were virally transduced with retrovirus expressing ICN together with the indicated miRNA inhibitor or gene (cDNA encoding Myb or Hbp1) and then were transplanted into lethally radiated syngeneic C57BL/6 host mice. Survival data were analyzed in Kaplan-Meier format with the log-rank (Mantel-Cox) test for statistical significance. Analysis of cell surface markers was performed as described previously (55). For transplantation assays, primary murine leukemic cells derived from ICN and a dominant-negative form of IKAROS, IK6-expressing HPCs were transduced with MSCV-based vectors expressing miR-150, miR-155, miR-200c, a short-hairpin against Myb (shMyb-2), or empty vector control. Cells were then selected in culture medium containing puromycin and were injected into sublethally radiated syngeneic recipient mice (25,000 cells per mouse).
Western blotting and qRT-PCR analysis
Western blotting analysis was performed with whole-cell lysates as described previously (42). Antibodies used were specific for c-Myb (clone C-19, 1:500; Santa Cruz Biotechnology, sc-517), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Cell Signaling), and actin (1:5000; Sigma, AC-15). Total RNA was extracted from cells with the AllPrep kit (Qiagen). Synthesis of cDNA, qRT-PCR assays, and data analysis by the ΔΔCt method were performed as described previously (40). The following TaqMan Gene Expression Assays were used: c-Myb (Hs00920554_m1, Applied Biosystems) and Hbp1 (Mm00462715_m1, Applied Biosystems). The abundances of these mRNAs were normalized to those of housekeeping genes, including human β-actin or GusB and mouse actin or GAPDH (Applied Biosystems). To measure miRNA abundance, the following TaqMan assays were used: miR-29a (TM: 002112), miR-31 (TM: 002279), miR-150 (TM: 473), miR-155 (TM: 479), and miR-200c (TM: 002300). The abundances of these miRNAs were normalized to that of U6 small nuclear RNA (TM: 001093, Applied Biosystems).
Discriminative model with sparse logistic regression
To determine the shared targets of tumor suppressor miRNAs that were not also targeted by highly abundant miRNAs in T-ALL, we used logistic regression with a lasso constraint to discriminate between tumor suppressor miRNAs (+1 class) and highly abundant miRNAs (−1 class) on the basis of feature vectors of predicted target genes. We used prediction scores from miRSVR (http://miRNA.org) as features and restricted to a subset of genes that were statistically significantly increased in abundance in human T-ALL samples on the basis of two published data sets (22, 23) and our RNA sequencing analysis of eight T-ALL samples. The logistic regression model expresses the conditional probability of belonging to the tumor suppressor miRNA class as:
where xmiR,g is the mirSVR score of the miRNA for a given gene “g,” and wg is the regression coefficient for the gene g. For sparse logistic regression, the optimization problem consists of minimizing the negative log likelihood plus a lasso term:
We implemented sparse logistic regression with the glmnet R software package. Because we had a small set of six tumor suppressor miRNAs on which to train, we fixed our positive training examples and repeatedly sub-sampled nine highly abundant miRNAs to train slightly different regression models. After training a total of 100 different models, target genes were ranked according to how frequently they were included with positive regression coefficients in these models.
Target prediction
With the seed sequence (position 2:8) for each miRNA, we identified conserved mRNA targets by requiring the complementary sequence to occur in the 3′UTRs of both mouse and human orthologous mRNAs. We used the minimum number of seed matches from both UTRs as the conserved 7-mer count for each miRNA:mRNA pair. We used Biopython to extract 3′UTR sequences for mouse and human with RefSeq (Release 42). The longest 3′UTR was used when multiple RefSeq transcripts were present for a single gene.
Enrichment of miRNAs
The empirical P value for the enrichment of miRNAs in Table 1 was determined by selecting 10,000 random samples of genes (of size equal to the set of tumor suppressor genes) from the full list of human genes for which we had 3′UTRs and target predictions. We computed the sum of TargetScan context scores for each broadly conserved miRNA seed family relative to each of these random gene sets and ranked the miRNA families according to these scores. This procedure generated a randomized version of Table 1 for each randomly selected gene set. We then calculated enrichment scores for these rankings with a Wilcoxon rank sum statistic comparing the “positive” miRNAs (those that passed all screens) to all other miRNAs. Finally, to compute the empirical P value, we counted the number of times over the 10,000 randomly sampled gene sets that we obtained a Wilcoxon rank sum score as good as or better than the one we obtained with the tumor suppressor gene set. We found that 428 random samplings performed as well as or better than the tumor suppressor gene set, which corresponded to a statistically significant empirical P value (P < 0.043).
Table 1. Groups of miRNAs used in the lasso regression analysis.
The positive set includes tumor suppressor miRNAs, whereas the negative set includes highly abundant miRNAs.
| Positive set | Negative set |
|---|---|
| miR-29a/c, miR-31, miR-150, miR-155, miR-200 | miR-223, miR-222, miR-30c, miR-30a, miR-342-3p, miR-19b, miR-191, miR-181a, miR-376a, miR-92a, miR-20a, miR-19a, miR-106b, miR-17, miR-196a, miR-142-3p, miR-15b, miR-486-5p, miR-21, miR-662, miR-26a, miR-26b, miR-451, miR-30b, miR-617, miR-16, miR-140-5p, miR-181d, miR-93 |
Comparison of cytogenetic and mutational subgroups
Differential miRNA expression analysis was performed with t tests for groups of patient samples by cytogenetic criteria or by the presence and absence of Notch1 or Fbxw7 mutations (7). Cell lines were grouped by the indicated mutations and sensitivity to γ-secretase inhibitors, as reported previously (43). Benjamini-Hochberg correction was used for multiple hypotheses correction.
Luciferase assays
The 3′UTR of human Myb was a gift from J. Lieberman (Harvard Medical School). For the Myb 3′UTR luciferase assays, wild type refers to the complete 1191–base pair (bp) 3′UTR fragment that contains all of the miRNA-binding sites. For miR-150, miR-155, and miR-200c, site 1, site 2, and site 3 refer to mutation of the predicted miRNA-binding sites located in the positions indicated from left to right in fig. S5A. For miR-150, the positions of each site correspond to site 1 (base pairs 87 to 93), site 2 (base pairs 829 to 836), and site 3 (base pairs 904 to 911). For miR-155, the positions of each site correspond to site 1 (base pairs 15 to 21) and site 2 (base pairs 431 to 437). For miR-200c, the positions of each site correspond to site 1 (base pairs 319 to 325) and site 2 (base pairs 373 to 379). Site 1,2 or Site 1,2,3 refers to two or more mutated sites. The luciferase assays were performed as described previously (56). The primers used for mutagenesis of regions of the 3′UTR are listed in table S6.
Supplementary Material
Fig. S1. Changes in the abundances of miRNAs in T-ALL.
Fig. S2. Competition experiments testing the antileukemic potential of various miRNAs.
Fig. S3. Additional data concerning the in vivo tumor suppressor activity of various miRNAs.
Fig. S4. Validation data sets confirm Myb as a relevant target of tumor suppressor miRNAs.
Fig. S5. Myb is a target of three tumor suppressor miRNAs in T-ALL.
Fig. S6. HBP1 accelerates the rate of development of ICN-driven leukemia.
Table S1. Expression profiles of miRNAs in normal T cells, T-ALL patient samples, and T-ALL cell lines.
Table S2. miRNAs with decreased abundance in T-ALL samples as compared to normal T cell subsets based on subtype.
Table S3. Expression profiles of miRNAs for previously reported candidate tumor suppressor miRNAs.
Table S4. Identification of target genes.
Table S5. Full list of genes from the lasso regression model.
Table S6. Sequences of primers used for making mutations in the Myb 3′UTR.
Acknowledgments
We thank A. J. Capobianco, L. Beverly, A. A. Ferrando, J. Cools, W. Pear, J. Lieberman, and T. Bender for reagents. We thank the Memorial Sloan Kettering (MSK) animal facility and Research Animal Resource Center (RARC), Viale of the MSK Genomics Core, and K. Huberman of the Geoffrey Beene Translational Oncology Core Facility for supervising next-generation sequencing.
Funding: This work was supported by grants from the National Cancer Institute (R01-CA142798-01 to H.G.W.); a May & Samuel Rudin Foundation Award (H.G.W.); W. H. Goodwin and A. Goodwin and the Commonwealth Foundation for Cancer Research (H.G.W.); the Fund for Scientific Research (F.W.O.) Flanders Ph.D. grant to J.V.d.M., postdoctoral grants to P.R., T.T., and P.V.V., project grants G.0869.10N and G.0202.09N to F.S., and grant 3G002711 to T.T., and its Odysseus research program (grant to T.T.); the GOA-UGent (grant no. 12051203); Stichting tegen Kanker, FOD ALL action; the Belgian Program of Interuniversity Poles of Attraction; and the Belgian Foundation Against Cancer. H.G.W. is a Scholar of the Leukemia and Lymphoma Society.
Footnotes
Author contributions: V.R.S. and K.J.M. equally contributed to experimental work, study design, and data analysis; M.B., P.M., and A.L.W. performed experimental work; J.V.d.M. performed genomic analysis, including mutational analysis, arrayCGH, and miRNA profiling on T-ALL patient samples; M.C., C.S.L., and N.D.S. performed computational analyses; F.S. was involved in the design of genomic analyses for T-ALLs and miRNA profiling of thymocytes and supervised data mining and interpretation; T.T. performed experimental design and supervised the collection of thymocytes; P.V.V. and P.R. were responsible for experimental design, data mining, interpretation of thymocyte data,and T-ALL miRNA profiling; Y.B. performed the collection and clinical annotation of human T-ALL samples; and H.G.W. designed the study and wrote the paper.
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: RNA sequencing data have been deposited in the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/).
SUPPLEMENTARY MATERIALS
REFERENCES AND NOTES
- 1.Weng AP, Ferrando AA, Lee W, t Morris JP, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, Aster JC. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- 2.Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, Lu C, Chen SC, Wei L, Collins-Underwood JR, Ma J, Roberts KG, Pounds SB, Ulyanov A, Becksfort J, Gupta P, Huether R, Kriwacki RW, Parker M, McGoldrick DJ, Zhao D, Alford D, Espy S, Bobba KC, Song G, Pei D, Cheng C, Roberts S, Barbato MI, Campana D, Coustan-Smith E, Shurtleff SA, Raimondi SC, Kleppe M, Cools J, Shimano KA, Hermiston ML, Doulatov S, Eppert K, Laurenti E, Notta F, Dick JE, Basso G, Hunger SP, Loh ML, Devidas M, Wood B, Winter S, Dunsmore KP, Fulton RS, Fulton LL, Hong X, Harris CC, Dooling DJ, Ochoa K, Johnson KJ, Obenauer JC, Evans WE, Pui CH, Naeve CW, Ley TJ, Mardis ER, Wilson RK, Downing JR, Mullighan CG. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–163. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palomero T, Sulis ML, Cortina M, Real PJ, Barnes K, Ciofani M, Caparros E, Buteau J, Brown K, Perkins SL, Bhagat G, Agarwal AM, Basso G, Castillo M, Nagase S, Cordon-Cardo C, Parsons R, Zúñiga-Pflücker JC, Dominguez M, Ferrando AA. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med. 2007;13:1203–1210. doi: 10.1038/nm1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, Hammond SM. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mu P, Han YC, Betel D, Yao E, Squatrito M, Ogrodowski P, de Stanchina E, D’Andrea A, Sander C, Ventura A. Genetic dissection of the miR-17~92 cluster of microRNAs in Myc-induced B-cell lymphomas. Genes Dev. 2009;23:2806–2811. doi: 10.1101/gad.1872909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mavrakis KJ, Wolfe AL, Oricchio E, Palomero T, de Keersmaecker K, McJunkin K, Zuber J, James T, Khan AA, Leslie CS, Parker JS, Paddison PJ, Tam W, Ferrando A, Wendel HG. Genome-wide RNA-mediated interference screen identifies miR-19 targets in Notch-induced T-cell acute lymphoblastic leukaemia. Nat Cell Biol. 2010;12:372–379. doi: 10.1038/ncb2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mavrakis KJ, Van Der Meulen J, Wolfe AL, Liu X, Mets E, Taghon T, Khan AA, Setty M, Rondou P, Vandenberghe P, Delabesse E, Benoit Y, Socci NB, Leslie CS, Van Vlierberghe P, Speleman F, Wendel HG. A cooperative microRNA-tumor suppressor gene network in acute T-cell lymphoblastic leukemia (T-ALL) Nat Genet. 2011;43:673–678. doi: 10.1038/ng.858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsu PW, Huang HD, Hsu SD, Lin LZ, Tsou AP, Tseng CP, Stadler PF, Washietl S, Hofacker IL. miRNAMap: Genomic maps of microRNA genes and their target genes in mammalian genomes. Nucleic Acids Res. 2006;34:D135–D139. doi: 10.1093/nar/gkj135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Felekkis K, Voskarides K, Dweep H, Sticht C, Gretz N, Deltas C. Increased number of microRNA target sites in genes encoded in CNV regions. Evidence for an evolutionary genomic interaction. Mol Biol Evol. 2011;28:2421–2424. doi: 10.1093/molbev/msr078. [DOI] [PubMed] [Google Scholar]
- 10.Miska EA, Alvarez-Saavedra E, Abbott AL, Lau NC, Hellman AB, McGonagle SM, Bartel DP, Ambros VR, Horvitz HR. Most Caenorhabditis elegans microRNAs are individually not essential for development or viability. PLOS Genet. 2007;3:e215. doi: 10.1371/journal.pgen.0030215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park CY, Jeker LT, Carver-Moore K, Oh A, Liu HJ, Cameron R, Richards H, Li Z, Adler D, Yoshinaga Y, Martinez M, Nefadov M, Abbas AK, Weiss A, Lanier LL, de Jong PJ, Bluestone JA, Srivastava D, McManus MT. A resource for the conditional ablation of microRNAs in the mouse. Cell Rep. 2012;1:385–391. doi: 10.1016/j.celrep.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ebert MS, Sharp PA. Roles for microRNAs in conferring robustness to biological processes. Cell. 2012;149:515–524. doi: 10.1016/j.cell.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K, Rassenti L, Kipps T, Negrini M, Bullrich F, Croce CM. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2002;99:15524–15529. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raveche ES, Salerno E, Scaglione BJ, Manohar V, Abbasi F, Lin YC, Fredrickson T, Landgraf P, Ramachandra S, Huppi K, Toro JR, Zenger VE, Metcalf RA, Marti GE. Abnormal microRNA-16 locus with synteny to human 13q14 linked to CLL in NZB mice. Blood. 2007;109:5079–5086. doi: 10.1182/blood-2007-02-071225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boldin MP, Taganov KD, Rao DS, Yang L, Zhao JL, Kalwani M, Garcia-Flores Y, Luong M, Devrekanli A, Xu J, Sun G, Tay J, Linsley PS, Baltimore D. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J Exp Med. 2011;208:1189–1201. doi: 10.1084/jem.20101823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mestdagh P, Van Vlierberghe P, De Weer A, Muth D, Westermann F, Speleman F, Vandesompele J. A novel and universal method for microRNA RT-qPCR data normalization. Genome Biol. 2009;10:R64. doi: 10.1186/gb-2009-10-6-r64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pear WS, Aster JC, Scott ML, Hasserjian RP, Soffer B, Sklar J, Baltimore D. Exclusive development of T cell neoplasms in mice transplanted with bone marrow expressing activated Notch alleles. J Exp Med. 1996;183:2283–2291. doi: 10.1084/jem.183.5.2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mavrakis KJ, Wendel HG. TargetScreen: An unbiased approach to identify functionally important microRNA targets. Cell Cycle. 2010;9:2080–2084. doi: 10.4161/cc.9.11.11807. [DOI] [PubMed] [Google Scholar]
- 19.Kluiver J, Gibcus JH, Hettinga C, Adema A, Richter MK, Halsema N, Slezak-Prochazka I, Ding Y, Kroesen BJ, van den Berg A. Rapid generation of microRNA sponges for microRNA inhibition. PLOS One. 2012;7:e29275. doi: 10.1371/journal.pone.0029275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scherr M, Venturini L, Battmer K, Schaller-Schoenitz M, Schaefer D, Dallmann I, Ganser A, Eder M. Lentivirus-mediated antagomir expression for specific inhibition of miRNA function. Nucleic Acids Res. 2007;35:e149. doi: 10.1093/nar/gkm971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Betel D, Koppal A, Agius P, Sander C, Leslie C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010;11:R90. doi: 10.1186/gb-2010-11-8-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Vlierberghe P, Ambesi-Impiombato A, Perez-Garcia A, Haydu JE, Rigo I, Hadler M, Tosello V, Della Gatta G, Paietta E, Racevskis J, Wiernik PH, Luger SM, Rowe JM, Rue M, Ferrando AA. ETV6 mutations in early immature human T cell leukemias. J Exp Med. 2011;208:2571–2579. doi: 10.1084/jem.20112239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Homminga I, Pieters R, Langerak AW, de Rooi JJ, Stubbs A, Verstegen M, Vuerhard M, Buijs-Gladdines J, Kooi C, Klous P, van Vlierberghe P, Ferrando AA, Cayuela JM, Verhaaf B, Beverloo HB, Horstmann M, de Haas V, Wiekmeijer AS, Pike-Overzet K, Staal FJ, de Laat W, Soulier J, Sigaux F, Meijerink JP. Integrated transcript and genome analyses reveal NKX2-1 and MEF2C as potential oncogenes in T cell acute lymphoblastic leukemia. Cancer Cell. 2011;19:484–497. doi: 10.1016/j.ccr.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 24.Xiao C, Calado DP, Galler G, Thai TH, Patterson HC, Wang J, Rajewsky N, Bender TP, Rajewsky K. MiR-150 controls B cell differentiation by targeting the transcription factor c-Myb. Cell. 2007;131:146–159. doi: 10.1016/j.cell.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 25.Gartel AL, Goufman E, Tevosian SG, Shih H, Yee AS, Tyner AL. Activation and repression of p21WAF1/CIP1 transcription by RB binding proteins. Oncogene. 1998;17:3463–3469. doi: 10.1038/sj.onc.1202240. [DOI] [PubMed] [Google Scholar]
- 26.Zhuma T, Tyrrell R, Sekkali B, Skavdis G, Saveliev A, Tolaini M, Roderick K, Norton T, Smerdon S, Sedgwick S, Festenstein R, Kioussis D. Human HMG box transcription factor HBP1: A role in hCD2 LCR function. EMBO J. 1999;18:6396–6406. doi: 10.1093/emboj/18.22.6396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sekkali B, Szabat E, Ktistaki E, Tolaini M, Roderick K, Harker N, Patel A, Williams K, Norton T, Kioussis D. Human high mobility group box transcription factor 1 affects thymocyte development and transgene variegation. J Immunol. 2005;175:5203–5212. doi: 10.4049/jimmunol.175.8.5203. [DOI] [PubMed] [Google Scholar]
- 28.Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM, Dang CV, Thomas-Tikhonenko A, Mendell JT. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet. 2008;40:43–50. doi: 10.1038/ng.2007.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klinakis A, Szabolcs M, Politi K, Kiaris H, Artavanis-Tsakonas S, Efstratiadis A. Myc is a Notch1 transcriptional target and a requisite for Notch1-induced mammary tumorigenesis in mice. Proc Natl Acad Sci USA. 2006;103:9262–9267. doi: 10.1073/pnas.0603371103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palomero T, Lim WK, Odom DT, Sulis ML, Real PJ, Margolin A, Barnes KC, O’Neil J, Neuberg D, Weng AP, Aster JC, Sigaux F, Soulier J, Look AT, Young RA, Califano A, Ferrando AA. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci USA. 2006;103:18261–18266. doi: 10.1073/pnas.0606108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, Karnezis AN, Swigart LB, Nasi S, Evan GI. Modelling Myc inhibition as a cancer therapy. Nature. 2008;455:679–683. doi: 10.1038/nature07260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soucek L, Helmer-Citterich M, Sacco A, Jucker R, Cesareni G, Nasi S. Design and properties of a Myc derivative that efficiently homodimerizes. Oncogene. 1998;17:2463–2472. doi: 10.1038/sj.onc.1202199. [DOI] [PubMed] [Google Scholar]
- 33.Costinean S, Zanesi N, Pekarsky Y, Tili E, Volinia S, Heerema N, Croce CM. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in Eμ-miR155 transgenic mice. Proc Natl Acad Sci USA. 2006;103:7024–7029. doi: 10.1073/pnas.0602266103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eis PS, Tam W, Sun L, Chadburn A, Li Z, Gomez MF, Lund E, Dahlberg JE. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci USA. 2005;102:3627–3632. doi: 10.1073/pnas.0500613102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pekarsky Y, Santanam U, Cimmino A, Palamarchuk A, Efanov A, Maximov V, Volinia S, Alder H, Liu CG, Rassenti L, Calin GA, Hagan JP, Kipps T, Croce CM. Tcl1 expression in chronic lymphocytic leukemia is regulated by miR-29 and miR-181. Cancer Res. 2006;66:11590–11593. doi: 10.1158/0008-5472.CAN-06-3613. [DOI] [PubMed] [Google Scholar]
- 36.Santanam U, Zanesi N, Efanov A, Costinean S, Palamarchuk A, Hagan JP, Volinia S, Alder H, Rassenti L, Kipps T, Croce CM, Pekarsky Y. Chronic lymphocytic leukemia modeled in mouse by targeted miR-29 expression. Proc Natl Acad Sci USA. 2010;107:12210–12215. doi: 10.1073/pnas.1007186107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tam W, Dahlberg JE. miR-155/BIC as an oncogenic microRNA. Genes Chromosomes Cancer. 2006;45:211–212. doi: 10.1002/gcc.20282. [DOI] [PubMed] [Google Scholar]
- 38.Lahortiga I, De Keersmaecker K, Van Vlierberghe P, Graux C, Cauwelier B, Lambert F, Mentens N, Beverloo HB, Pieters R, Speleman F, Odero MD, Bauters M, Froyen G, Marynen P, Vandenberghe P, Wlodarska I, Meijerink JP, Cools J. Duplication of the MYB oncogene in T cell acute lymphoblastic leukemia. Nat Genet. 2007;39:593–595. doi: 10.1038/ng2025. [DOI] [PubMed] [Google Scholar]
- 39.Clappier E, Cuccuini W, Kalota A, Crinquette A, Cayuela JM, Dik WA, Langerak AW, Montpellier B, Nadel B, Walrafen P, Delattre O, Aurias A, Leblanc T, Dombret H, Gewirtz AM, Baruchel A, Sigaux F, Soulier J. The C-MYB locus is involved in chromosomal translocation and genomic duplications in human T-cell acute leukemia (T-ALL), the translocation defining a new T-ALL subtype in very young children. Blood. 2007;110:1251–1261. doi: 10.1182/blood-2006-12-064683. [DOI] [PubMed] [Google Scholar]
- 40.Biedenkapp H, Borgmeyer U, Sippel AE, Klempnauer KH. Viral myb oncogene encodes a sequence-specific DNA-binding activity. Nature. 1988;335:1628–1640. doi: 10.1038/335835a0. [DOI] [PubMed] [Google Scholar]
- 41.Zuber J, Rappaport AR, Luo W, Wang E, Chen C, Vaseva AV, Shi J, Weissmueller S, Fellmann C, Taylor MJ, Weissenboeck M, Graeber TG, Kogan SC, Vakoc CR, Lowe SW. An integrated approach to dissecting oncogene addiction implicates a Myb-coordinated self-renewal program as essential for leukemia maintenance. Genes Dev. 2011;25:1628–1640. doi: 10.1101/gad.17269211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thompson BJ, Buonamici S, Sulis ML, Palomero T, Vilimas T, Basso G, Ferrando A, Aifantis I. The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J Exp Med. 2007;204:1825–1835. doi: 10.1084/jem.20070872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O’Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, Hardwick J, Welcker M, Meijerink JP, Pieters R, Draetta G, Sears R, Clurman BE, Look AT. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to γ-secretase inhibitors. J Exp Med. 2007;204:1813–1824. doi: 10.1084/jem.20070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mansour MR, Sanda T, Lawton LN, Li X, Kreslavsky T, Novina CD, Brand M, Gutierrez A, Kelliher MA, Jamieson CH, von Boehmer H, Young RA, Look AT. The TAL1 complex targets the FBXW7 tumor suppressor by activating miR-223 in human T cell acute lymphoblastic leukemia. J Exp Med. 2013;210:1545–1557. doi: 10.1084/jem.20122516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sanda T, Lawton LN, Barrasa MI, Fan ZP, Kohlhammer H, Gutierrez A, Ma W, Tatarek J, Ahn Y, Kelliher MA, Jameson CH, Staudt LM, Young RA, Look AT. Core transcriptional regulatory circuit controlled by the TAL1 complex in human T cell acute lymphoblastic leukemia. Cancer Cell. 2012;22:209–221. doi: 10.1016/j.ccr.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wolfe AL, Singh K, Zhong Y, Drewe P, Rajasekhar VK, Sanghvi VR, Mavrakis KJ, Jiang M, Van Der Meulen J, Schatz JH, Rodrigo CM, Zhao C, Rondue P, deStanchina E, Teruya-Feldstein J, Kelliher MA, Speleman F, Porco JA, Jr, Pelletier J, Rätsch G, Wendel HG. RNA G-quadruplexes cause eIF4A-dependent oncogene translation in cancer. Nature. 2014;513:65–70. doi: 10.1038/nature13485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Richon VM, Zhou X, Secrist JP, Cordon-Cardo C, Kelly WK, Drobnjak M, Marks PA. Histone deacetylase inhibitors: Assays to assess effectiveness in vitro and in vivo. Methods Enzymol. 2004;376:199–205. doi: 10.1016/S0076-6879(03)76013-3. [DOI] [PubMed] [Google Scholar]
- 48.Bernstein E, Kim SY, Carmell MA, Murchison EP, Alcorn H, Li MZ, Mills AA, Elledge SJ, Anderson KV, Hannon GJ. Dicer is essential for mouse development. Nat Genet. 2003;35:215–217. doi: 10.1038/ng1253. [DOI] [PubMed] [Google Scholar]
- 49.Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet. 2007;39:673–677. doi: 10.1038/ng2003. [DOI] [PubMed] [Google Scholar]
- 50.Van de Walle I, De Smet G, De Smedt M, Vandekerckhove B, Leclercq G, Plum J, Taghon T. An early decrease in Notch activation is required for human TCR-αβ lineage differentiation at the expense of TCR-γδ T cells. Blood. 2009;113:2988–2998. doi: 10.1182/blood-2008-06-164871. [DOI] [PubMed] [Google Scholar]
- 51.Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, Lao KQ, Livak KJ, Guegler KJ. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005;33:e179. doi: 10.1093/nar/gni178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mestdagh P, Feys T, Bernard N, Guenther S, Chen C, Speleman F, Vandesompele J. High-throughput stem-loop RT-qPCR miRNA expression profiling using minute amounts of input RNA. Nucleic Acids Res. 2008;36:e143. doi: 10.1093/nar/gkn725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mavrakis KJ, Zhu H, Silva RL, Mills JR, Teruya-Feldstein J, Lowe SW, Tam W, Pelletier J, Wendel HG. Tumorigenic activity and therapeutic inhibition of Rheb GTPase. Genes Dev. 2008;22:2178–2188. doi: 10.1101/gad.1690808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Plas DR, Talapatra S, Edinger AL, Rathmell JC, Thompson CB. Akt and Bcl-xL promote growth factor-independent survival through distinct effects on mitochondrial physiology. J Biol Chem. 2001;276:12041–12048. doi: 10.1074/jbc.M010551200. [DOI] [PubMed] [Google Scholar]
- 55.Wendel HG, De Stanchina E, Fridman JS, Malina A, Ray S, Kogan S, Cordon-Cardo C, Pelletier J, Lowe SW. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature. 2004;428:332–337. doi: 10.1038/nature02369. [DOI] [PubMed] [Google Scholar]
- 56.Xiao C, Srinivasan L, Calado DP, Patterson HC, Zhang B, Wang J, Henderson JM, Kutok JL, Rajewsky K. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol. 2008;9:405–414. doi: 10.1038/ni1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Changes in the abundances of miRNAs in T-ALL.
Fig. S2. Competition experiments testing the antileukemic potential of various miRNAs.
Fig. S3. Additional data concerning the in vivo tumor suppressor activity of various miRNAs.
Fig. S4. Validation data sets confirm Myb as a relevant target of tumor suppressor miRNAs.
Fig. S5. Myb is a target of three tumor suppressor miRNAs in T-ALL.
Fig. S6. HBP1 accelerates the rate of development of ICN-driven leukemia.
Table S1. Expression profiles of miRNAs in normal T cells, T-ALL patient samples, and T-ALL cell lines.
Table S2. miRNAs with decreased abundance in T-ALL samples as compared to normal T cell subsets based on subtype.
Table S3. Expression profiles of miRNAs for previously reported candidate tumor suppressor miRNAs.
Table S4. Identification of target genes.
Table S5. Full list of genes from the lasso regression model.
Table S6. Sequences of primers used for making mutations in the Myb 3′UTR.