

Abstract
Naegeli–Franceschetti–Jadassohn Syndrome (NFJS) is a rare, autosomal dominant inherited form of ectodermal dysplasia, caused by mutation in the KRT14 gene. We report here a case of NFJS in a 27-year-old male who presented with reticulate hyperpigmentation over skin, dental changes, absence of dermatoglyphics, hypohidrosis, and hair changes.
Keywords: Dermatopathia pigmentosa reticularis, KRT 14 gene, Naegeli–Franceschetti–Jadassohn, reticulate hyperpigmentation
INTRODUCTION
Naegeli–Franceschetti–Jadassohn Syndrome (NFJS) is a rare form of ectodermal dysplasia inherited as an autosomal dominant disorder. NFJS and its closely related allelic disorder, dermatopathia pigmentosa reticularis (DPR), are characterized by the absence of dermatoglyphics, reticulate pattern of skin hyperpigmentation, palmoplantar keratoderma, abnormal sweating, and other subtle developmental anomalies of teeth, hair, and skin. Several mutations in the KRT 14 gene have been found to cause NFJS/DPR. NFJS and DPR were originally described as separate conditions; however, because of very few differences in the clinical presentation and being caused by mutations in the same gene, they are now often considered different forms of the same disorder.
CASE REPORT
A 27-year-old, unmarried male, clerk by occupation born out of non-consanguineous marriage, presented to the skin outpatient department with complaints of reticulate hyperpigmentation over body including the palms and soles, photophobia, hypohidrosis, complaint of heat intolerance especially during summers and nail dystrophy since birth. His family history was insignificant.
On dermatologic examination, the patient had reticulate hyperpigmentation involving the whole body [Figure 1], and hair showed mild pigmentary dilution with patchy golden brown discoloration [Figure 2]. Pigmentation was especially dense over the palms and soles, which showed rain drop pattern of pigmentation, whereas that on elbows, knees, and dorsal aspect of tongue exhibited reticulate pattern [Figure 3]. Teeth showed yellowish discoloration, abnormal dentition, and enamel defects [Figure 4]. The skin over dorsal aspect of hands and feet was atrophic, shiny wrinkled, and nails showed severe dystrophy involving all finger and toe nails [Figure 5]. There was absence of dermatoglyphics [Figure 6]. The skin was xerotic. On general physical examination, the patient was thinly built, and had pallor. Systemic examination was normal, except for ophthalmic examination that showed mild corneal degeneration. Other ophthalmic examination findings were within normal limits. Baseline investigations (complete blood counts, liver and renal function tests, urine analysis) were within normal limits except for hemoglobin level, which was 9 g/dL. Blood arsenic level of the patient was below detection limit.
Figure 1.
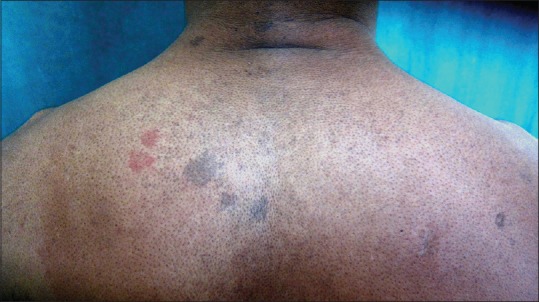
Reticulate and mottled hyperpigmentation over body
Figure 2.
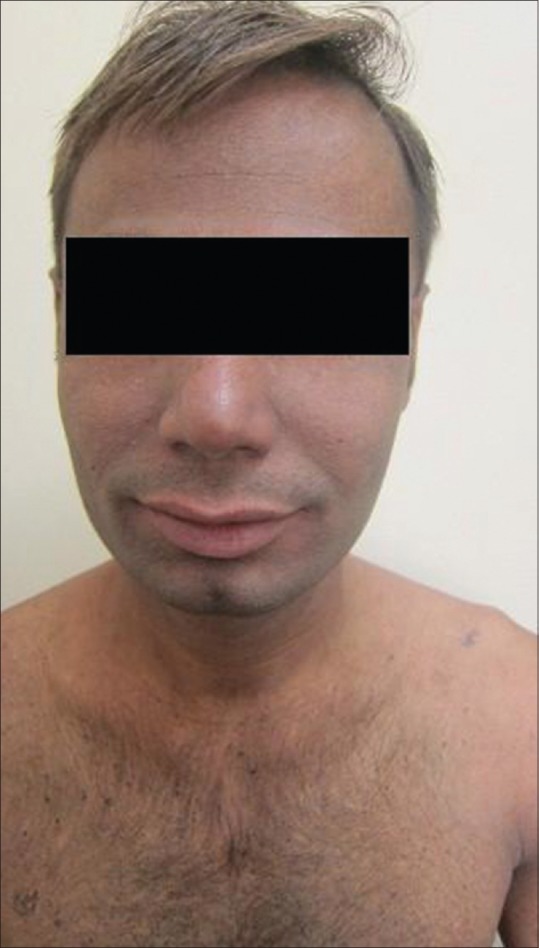
Pigmentary dilution of hair
Figure 3.
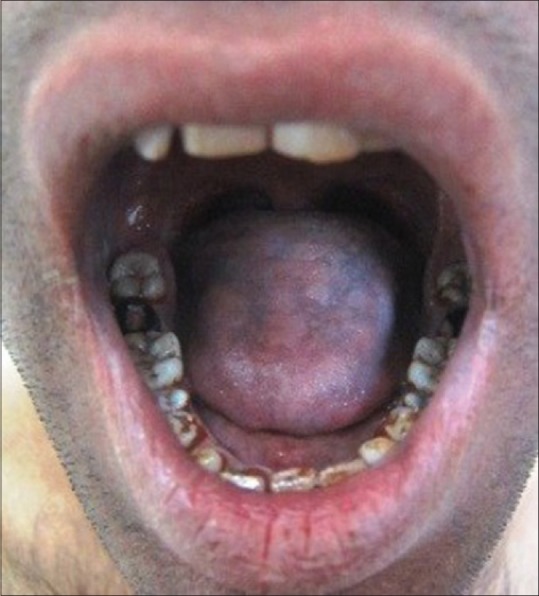
Reticulate hyperpigmentation over the dorsal aspect of tongue
Figure 4.
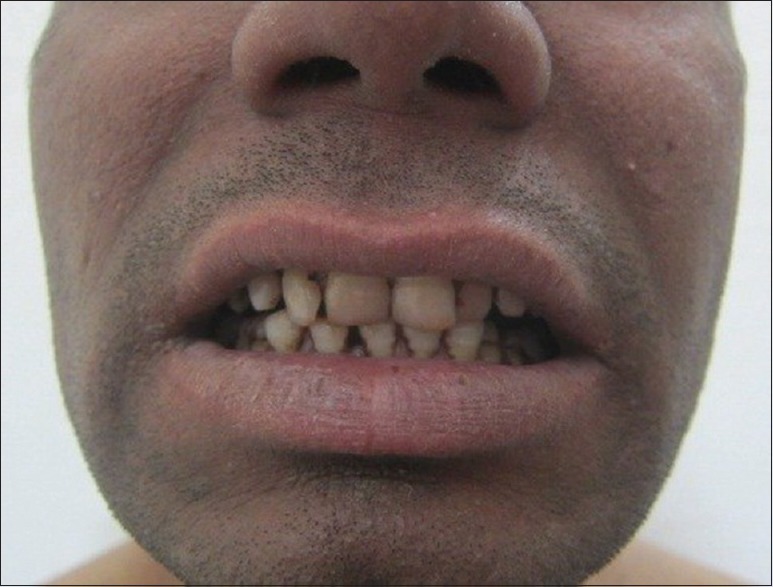
Yellowish discoloration of teeth
Figure 5.
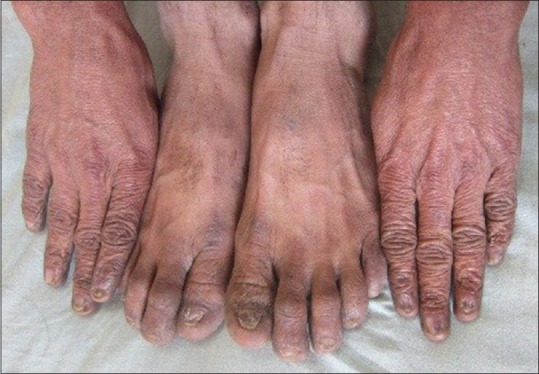
Dystrophic nails and pigmentation over dorsum of hand and feet
Figure 6.
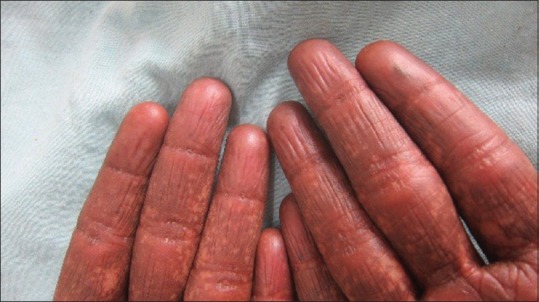
Loss of dermatoglyphics
A biopsy specimen from a hyperpigmented macule on the back showed findings of epidermal atrophy, vacuolar degeneration, hyperpigmentation of the basal layer, and pigmentary incontinence. The dermis showed perivascular chronic inflammation, increased dermal fibrosis, with presence of dermal melanophages.
Molecular analysis for KRT 14 gene mutation using genomic DNA from the blood of the patient was done by the method of PCR-bidirectional sequencing of exons 1, 4, and 6. It showed absence of mutation in the three exons (1, 4, and 6), which ruled out epidermolysis bullosa simplex (which is also caused by the mutation in this gene); however, possibility of NFJS/DPR cannot be ruled out on the basis of this study and it requires further exome sequencing study of KRT 14 gene.
He was advised strict photo protection and teeth care. Advice to avoid strenuous activity and to maintain adequate hydration was also given. He was given oral antioxidant once daily and topical emollients.
DISCUSSION
NFJS (MIM 161000) is a rare autosomal dominant form of ectodermal dysplasia that affects the skin, sweat glands, nails, and teeth.[1,2] Incidence is estimated to be one case in two to four million population. The syndrome is allelic to DPR[3] (MIM 125595).
NFJS/DPR results from mutations in the KRT14 gene located on chromosome 17q11.2–q21. This gene provides instructions for making the protein keratin 14. The KRT14 gene mutations that cause NFJS/DPR most likely reduce the amount of functional keratin 14 that is produced in cells. A shortage of this protein makes cells in the epidermis more likely to undergo apoptosis. The resulting loss of these cells alters the normal development and structure of ectodermal tissues, which underlies most of the skin and nail problems characteristic of NFJS/DPR.[4,5]
Among the most distinctive characteristics of this syndrome is the complete absence of the patterned ridges on the skin of the hands and feet, called dermatoglyphics that are the basis for each person's unique fingerprints.[6,7]
Brown or grey-brown reticulated hyperpigmentation develops by the age of 2 years, without a preceding inflammatory stage. The pigmentary changes are most often observed on the abdomen and periocular and perioral regions, with variable involvement of the neck, trunk, proximal extremities, axillae, and groin. Hyperpigmentation often fades after puberty and may eventually disappear.[8]
Some affected individuals also have blistering on their palms and soles. Nails may show onychodystrophy, onycholysis, subungual hyperkeratosis, and congenital malalignment of the great toe nails.[8]
Other features are hypohidrosis; heat intolerance; noncicatricial alopecia on the scalp, eyebrows, and underarms; enamel defects; palmoplantar keratoderma; and other dental anomalies.[8]
DPR shares key features with NFJS but has been distinguished from it by lifelong persistence of the skin hyperpigmentation, partial alopecia, and absence of dental anomalies. Compared with reticulate hyperpigmentation, which fades, hypohidrosis and palmoplantar keratoderma usually persist in NFJ syndrome.[1,8]
To date, all patients reported had normal intelligence and were usually in good health.[9]
NFJ syndrome is a reticulate pigmentary disorder. Other reticulate pigmentary diseases and differentials in this case include DPR, Dowling–Degos disease, reticulate acropigmentation of Kitamura, and dyskeratosis congenita.
The distribution of the reticulate pigmentation in Dowling–Degos disease, dyschromatosis symmetrica hereditaria, and reticulate acropigmentation of Kitamura makes these disorders easier to distinguish.[10]
DPR is similar to NFJS but is distinguished by reticulate pigmentation lasting till adulthood, alopecia, and the lack of dental abnormalities. Both NFJS and DPR are allelic disorders.[11]
Dyskeratosis congenita is X-linked recessive disorder and patients can have features similar to NFJS patients, but in addition patients can also have alopecia, mucosal leukoplakia, poikiloderma, and blood dyscrasias.[12]
In our patient we were able to observe all the clinical features of the NFJS, which are mentioned in the literature along with the additional finding of pigmentary dilution of hair, which has not been reported previously.
No treatment is effective for NFJS. As with other ectodermal dysplasias, exposure to heat should be limited and sufficient hydration[1] is recommended. Tooth care to prevent early caries is indicated. Doxycycline has been found to interfere with tumor necrosis factor-alfa–mediated signalling and apoptosis in vitro and may have a role in future treatment.[11] Strenuous activity should be avoided.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Lugassy J, Itin P, Ishida-Yamamoto A, Holland K, Huson S, Geiger D, et al. NaegeliFranceschetti-Jadassohn syndrome and dermatopathia pigmentosa reticularis: Two allelic ectodermal dysplasias caused by dominant mutations in KRT14. Am J Hum Genet. 2006;79:724–30. doi: 10.1086/507792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Itin P, Lautenschlager S, Meyer R, Mevorah B, Rufli T. Natural history of the Naegeli–Franceschetti–Jadassohn syndrome and further delineation of its clinical manifestations. J Am Acad Dermatol. 1993;28:942–50. doi: 10.1016/0190-9622(93)70135-g. [DOI] [PubMed] [Google Scholar]
- 3.Itin PH, Lautenschlager S. Genodermatosis with reticulate, patchy and mottled pigmentation of the neck--A clue to rare dermatologic disorders. Dermatology. 1998;197:281–90. doi: 10.1159/000018015. [DOI] [PubMed] [Google Scholar]
- 4.Hesse M, Zimek A, Weber K, Magin TM. Comprehensive analysis of keratin gene clusters in humans and rodents. Eur J Cell Biol. 2004;83:19–26. doi: 10.1078/0171-9335-00354. [DOI] [PubMed] [Google Scholar]
- 5.Irvine AD. Inherited defects in keratins. Clin Dermatol. 2005;23:6–14. doi: 10.1016/j.clindermatol.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 6.Babler WJ. Embryologic development of epidermal ridges and their configurations. Birth Defects Orig Artic Ser. 1991;27:95–112. [PubMed] [Google Scholar]
- 7.Baird HW., 3rd Kindred showing congenital absence of the dermal ridges (fingerprints) and associated anomalies. J Pediatr. 1964;64:621–31. doi: 10.1016/s0022-3476(64)80610-7. [DOI] [PubMed] [Google Scholar]
- 8.Chang MW. Disorders of hyperpigmentation. In: Bolognia JL, Jorizzo JL, Schaffer JV, editors. Dermatology. 3rd ed. Atlanta: Saunders; 2012. p. 1068. [Google Scholar]
- 9.Heimer WL, 2nd, Brauner G, James WD. Dermatopathia pigmentosa reticularis: A report of a family demonstrating autosomal dominant inheritance. J Am Acad Dermatol. 1992;26:298–301. doi: 10.1016/0190-9622(92)70039-i. [DOI] [PubMed] [Google Scholar]
- 10.Franceschetti A, Jadassohn W. On incontinentia pigmenti and differentiation of two syndromes appearing under the same name. Dermatologica. 1954;108:1–28. [PubMed] [Google Scholar]
- 11.Sparrow GP, Samman PD, Wells RS. Hyperpigmentation and hypohidrosis. (The Naegeli–Franceschetti–Jadassohn syndrome): report of a family and review of the literature. Clin Exp Dermatol. 1976;1:127–40. doi: 10.1111/j.1365-2230.1976.tb01408.x. [DOI] [PubMed] [Google Scholar]
- 12.Chang MW. Disorders of hyperpigmentation. In: Bolognia JL, Jorizzo JL, Schaffer JV, editors. Dermatology. 3rd ed. Atlanta: Saunders; 2012. p. 1066. [Google Scholar]