

Sir,
Hutchinson-Gilford progeria syndrome (HGPS; MIM: 176670) is a rare genetic premature aging disorder that affects the skin, bones, and cardiovascular system.[1] It is characterized by progeroid features from a very early age of life, short stature, low birth weight, sclerodermoid features, early loss of hair, marked loss of subcutaneous fat, prominent superficial veins, craniofacial disproportion, beaked nose, pigeon chest, and thin limbs that resemble an aged person.
The most frequent cause of death among individuals with HGPS is myocardial infarction early in life. Less than 144 cases of this interesting disorder have been in the world literature till date.[2] The present communication is intended to document yet another interesting case of this rare and unusual entity for academic interest.
A 6-year-old male child born to first-degree consanguineous parents was brought to our outpatient clinic with the complaints of short stature and large head with peculiar facies since birth, with thin atrophic, tight, shiny skin, characteristic absence of hair over the scalp, eyebrows, and eyelashes soon after birth. His developmental of milestones were delayed, with a low intelligence quotient (IQ).
General physical examination of the patient revealed short stature, thin build, and poor nourishment. The older sibling of the patient was normal.
Dermatological examination revealed characteristic progeroid facies with frontal bossing, macrocephaly, prominent dilated veins over scalp, beaked nose, sparse eyebrows and eyelashes, overcrowding of teeth, mottled pigmentation over the neck and flanks, and subcostal retraction of the chest with pectus excavatum [Figures 1a–c and 2a, b].
Figure 1.
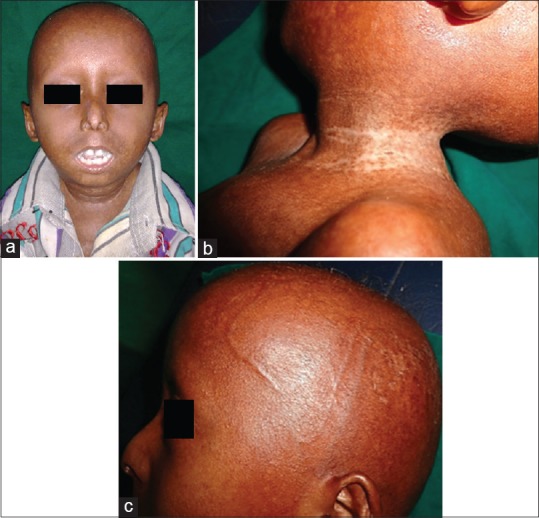
(a) Progeriod facies with frontal bossing, beaked nose. (b) mottled pigmentation around the neck. (c) Prominent dilated veins over the scalp
Figure 2.
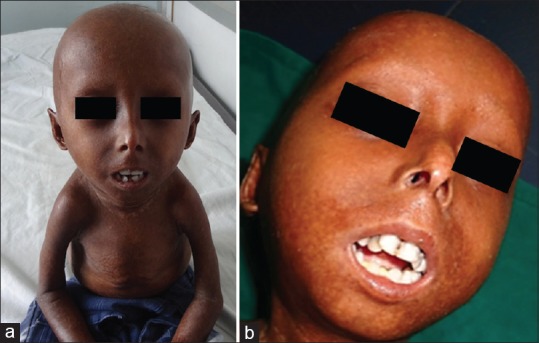
(a) Stunted growth, thin limbs. (b) Overcrowding of teeth
Routine laboratory investigations including hemogram, liver and kidney function tests, and lipid profile were within normal limits. Radiological features of skull showed incomplete fusion of anterior and posterior fontanelles [Figure 3].
Figure 3.
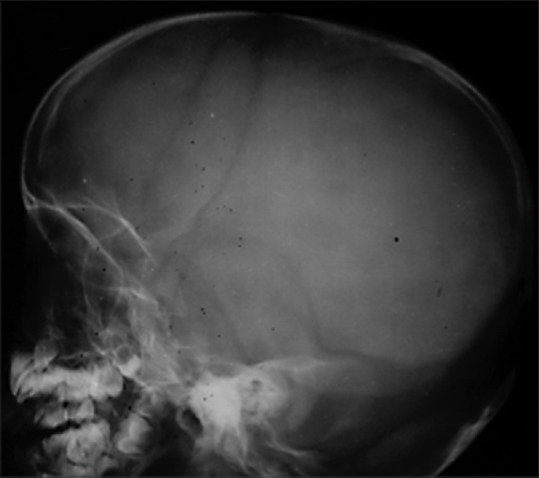
Radiography of skull showing incomplete fusion of anterior fontanelle
The skin biopsy of the sclerodermoid plaque from the flank revealed atrophy of the epidermis and reticular dermis and replacement of the subcutis with fibrocollagenous tissue. [Figure 4].
Figure 4.
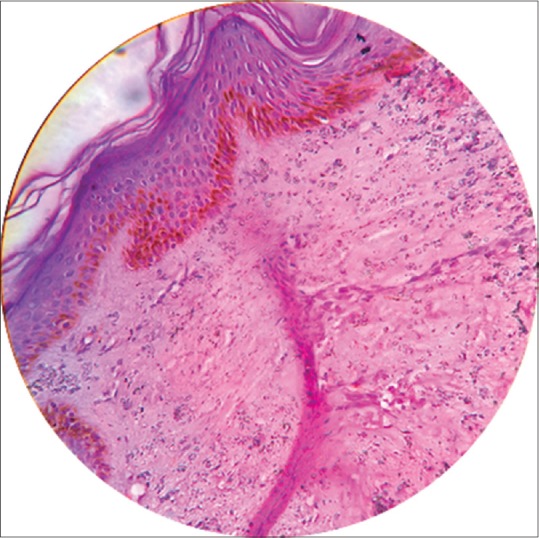
Atrophy of the epidermis and reticular dermis and replacement of the subcutis with fibrocollagenous tissue. (H and E, x40)
Progeria is a rare hereditary disorder that is characterized phenotypically by features of premature aging. The first case of this interesting disorder was described by Hutchinson in 1886, whereas the name progeria derived from Greek word “gerios” meaning old was coined by Gilford in 1904. De Buskin in 1972 redesignated this entity with the present nomenclature of “Hutchinson-Gilford progeria syndrome”.[3]
HGPS occurs sporadically with a strong racial susceptibility for whites who represent 97% of this condition. Males are more commonly affected. The mode of inheritance is not clear but is described to be either autosomal dominant or recessive or both in the literature.[4] The exact etiology of HGPS is unknown, but many cases occur de novo with autosomal mutation in the Laminin A (LMNA) gene located on band 1q21.1-q21.3.[5] LMNA encodes laminins A and C that are the main components of the intermediate filamentous lamina; they provide structural support, participate in DNA replication, and mRNA transcription.
Prelamin A is a protein located on the nuclear membrane of cells; it needs to be cleaved to form laminin A, a process that is defective in patients with progeria. Increased prelamin A causes nuclear blebbing and altered shape of the nucleus. However, the mechanism by which the altered shape of nucleus leads to symptoms of premature aging is not clear. The rate of aging is accelerated by about seven times. In progeria, cutaneous manifestations occur earlier, followed by skeletal and cardiovascular changes. HGPS children appear normal at birth but display the effects of accelerated aging within one year.
The clinical manifestations of HGPS include short stature, low birth weight, lipodystrophy, micrognathia, macrocephaly, prominent scalp veins, generalized alopecia, delayed dentition, pyriform thorax, thin limbs, decreased joint mobility, and sclerodermatous changes over the abdomen, neck folds, proximal thighs and buttocks, persistently patent anterior fontanelle, dystrophic nails, and beaked nose and protruding ears. Motor and mental development is normal.
Radiological findings include diffuse osteopenia and osteolysis. These children usually have severe atherosclerosis and death results mainly due to myocardial infarction generally between ages 5 and 20 years with a median lifespan of 13 years.
Differential diagnosis of HGPS includes mandibulo-acral dysplasia, Werner syndrome, Cockayne syndrome, and Hallerman-Strief syndrome.
Our case had the classical manifestations of HGPS as highlighted in the case report.
There is no specific treatment for HGPS and these children need a symptomatic approach, which includes early identification and prompt management of the complications. Farnesyl transferase inhibitors (FTIs) such as lonafarnib have shown some promise in reversing the structural abnormalities of the nucleus (prelamin A). The statin drug pravastatin normally used for lowering cholesterol and preventing cardiovascular disease, and the bisphosphonate drug zoledronic acid used for improving osteoporosis are the other drugs advocated for management of HGPS patients.[6,7] Proper counseling of the parents about this condition is important. Long-term follow-up is needed to observe the cardiovascular and skeletal complications in these patients.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Badame AJ. Progeria. Arch Dermatol. 1989;125:540–4. [PubMed] [Google Scholar]
- 2.Liag L, Zhang H, Gu X. Homozygous LMNA mutation R527C in atypical Hutchinson-Gilford progeria syndrome: Evidence for autosomal recessive inheritance. Acta Paediatr. 2009;98:1315–8. doi: 10.1111/j.1651-2227.2009.01324.x. [DOI] [PubMed] [Google Scholar]
- 3.DeBusk FL. The Hutchinson-Gilford progeria syndrome. Report of 4 cases and review of the literature. J Pediatr. 1972;80:697–724. doi: 10.1016/s0022-3476(72)80229-4. [DOI] [PubMed] [Google Scholar]
- 4.Beauregard S, Gilchrest BA. Syndromes of premature aging. Dermatol Clin. 1987;5:109–21. [PubMed] [Google Scholar]
- 5.Pollex RL, Hegele RA. Hutchinson-Gilford progeria syndrome. Clin Genet. 2004;66:375–81. doi: 10.1111/j.1399-0004.2004.00315.x. [DOI] [PubMed] [Google Scholar]
- 6.Varela I, Pereira S, Ugalde AP, Navarro CL, Suárez MF, Cau P, et al. Combined treatment with statins and aminobisphosphonates extends longevity in a mouse model of human premature aging. Nat Med. 2008;14:767–72. doi: 10.1038/nm1786. [DOI] [PubMed] [Google Scholar]
- 7.Gordon LB, Massaro J, D’Agostino RB, Sr, Campbell SE, Brazier J, Brown WT, et al. Progeria Clinical Trials Collaborative. Impact of farnesylation inhibitors on survival in Hutchinson-Gilford progeria syndrome. Circulation. 2014;130:27–34. doi: 10.1161/CIRCULATIONAHA.113.008285. [DOI] [PMC free article] [PubMed] [Google Scholar]