

Summary
Aging is associated with marked deficiency in circulating IGF‐1, which has been shown to contribute to age‐related cognitive decline. Impairment of moment‐to‐moment adjustment of cerebral blood flow (CBF) via neurovascular coupling is thought to play a critical role in the genesis of age‐related cognitive impairment. To establish the link between IGF‐1 deficiency and cerebromicrovascular impairment, neurovascular coupling mechanisms were studied in a novel mouse model of IGF‐1 deficiency (Igf1 f/f‐TBG‐Cre‐AAV8) and accelerated vascular aging. We found that IGF‐1‐deficient mice exhibit neurovascular uncoupling and show a deficit in hippocampal‐dependent spatial memory test, mimicking the aging phenotype. IGF‐1 deficiency significantly impaired cerebromicrovascular endothelial function decreasing NO mediation of neurovascular coupling. IGF‐1 deficiency also impaired glutamate‐mediated CBF responses, likely due to dysregulation of astrocytic expression of metabotropic glutamate receptors and impairing mediation of CBF responses by eicosanoid gliotransmitters. Collectively, we demonstrate that IGF‐1 deficiency promotes cerebromicrovascular dysfunction and neurovascular uncoupling mimicking the aging phenotype, which are likely to contribute to cognitive impairment.
Keywords: arachidonic acid metabolites, astrocyte, endothelial dysfunction, functional hyperemia, Insulin‐like growth factor‐1, neurovascular uncoupling, nitric oxide, somatomedin C, vascular aging, vascular cognitive impairment
Introduction
Vascular cognitive impairment (VCI) in the aging population poses a serious challenge to developed countries around the world. With the expansion of the aging population, understanding potentially reversible and preventable vascular contributions to age‐related cognitive impairment and dementia is of critical importance.
There is increasing evidence that in addition to pathologies affecting the larger cerebral arteries (e.g. atherosclerosis), pathophysiological alterations of the cerebral microcirculation have a critical role in age‐related decline in brain function (Gorelick et al., 2011). Among them, age‐related functional changes in the neurovascular unit that have the potential to impair local regulation of cerebral blood flow are of great importance (Iadecola et al., 2009). The energy requirements of neurons are high. Yet, the brain contains little energy reserves and, during neuronal activation, there is a need for rapid increases in oxygen and glucose delivery. This is ensured by neurovascular coupling, a vital feed‐forward control mechanism involving neuronal signaling via neurotransmitters, which adjusts local cerebral blood flow (CBF) to the energy requirements of activated neurons. The resulting functional hyperemia is responsible for maintenance of an optimal local microenvironment in the cerebral tissue by increasing delivery of oxygen and glucose and removal of potentially deleterious by‐products of cerebral metabolism. Aging is associated with significant impairment of functional hyperemia (termed ‘neurovascular uncoupling’), and the ensuing disruption of the cerebral microenvironment likely contributes to impairment of higher cerebral function in elderly patients and aged laboratory animals (Zaletel et al., 2005; Park et al., 2007; Topcuoglu et al., 2009; Fabiani et al., 2013; Sorond et al., 2013; Stefanova et al., 2013; Toth et al., 2014a). Yet, the specific age‐related mechanisms responsible for neurovascular uncoupling are not yet understood.
There is increasing evidence suggesting that neuroendocrine mechanisms have an important role in age‐related vascular alterations (Ungvari & Csiszar, 2012; Sonntag et al., 2013). In particular, the age‐related decline in circulating insulin‐like growth factor‐1 (IGF‐1) levels appears to contribute significantly to age‐related microvascular changes and cognitive decline (reviewed recently in Sonntag et al. (2013)). Neurovascular coupling depends on an intact functional network of neurons, vascular endothelial cells, and astrocytes (Attwell et al., 2010; Chen et al., 2014). Although these cell types abundantly express IGF‐1 receptors and are known targets of IGF‐1 (Sonntag et al., 2013), the role of IGF‐1 in the regulation of functional hyperemia in response to neuronal activation is not well understood. The cellular mechanisms underlying neurovascular coupling include endothelial production of nitric oxide (NO) (Ma et al., 1996; Stobart et al., 2013; Chen et al., 2014) as well as astrocytic production of vasodilator metabolites of arachidonic acid, including epoxygenase‐derived epoxyeicosatrienoic acids (EETs) and cyclooxygenase‐derived prostaglandins (Peng et al., 2002; Zonta et al., 2003; Takano et al., 2006). Importantly, previous studies demonstrate that IGF‐1 deficiency leads to endothelial dysfunction and impaired bioavailability of NO in the peripheral circulation (reviewed in Ungvari & Csiszar (2012)). IGF‐1 was also shown to regulate cellular arachidonic acid metabolism (Tahara et al., 1991; Berenbaum et al., 1994; Damke et al., 1994; Sharma et al., 1997). Despite these advances, the effects of IGF‐1 deficiency on the cerebral microcirculation and mediation of neurovascular coupling by NO, EETs, and eicosanoid gliotransmitters remain elusive.
This study was designed to test the hypotheses that IGF‐1 regulates synthesis/release of NO and vasodilator eicosanoid gliotransmitters in the cerebral microcirculation and that low circulating IGF‐1 levels impair neurovascular coupling in the brain, mimicking the aging phenotype. To test our hypotheses, we used a novel mouse model of adult‐onset, isolated endocrine IGF‐1 deficiency induced by adeno‐associated viral knockdown of IGF‐1 specifically in the liver of postpubertal mice using Cre‐lox technology (Igf1 f/f + TBG‐Cre‐AAV8) (Toth et al., 2014b). Neurovascular coupling, synthesis of eicosanoid gliotransmitters, astrocytic gene expression, and cerebromicrovascular endothelial function were tested. To substantiate our findings, behavioral studies known to be sensitive for neurovascular uncoupling (tests indicative for learning and memory) were conducted.
Results
IGF‐1 deficiency impairs neurovascular coupling and cognitive function
Figure 1A shows that mice receiving TBG‐Cre‐AAV8 had significantly lower serum IGF‐1 levels compared with control mice receiving TBG‐eGFP‐AAV8. Both groups had similar serum IGF‐1 levels prior to the administration of liver‐targeted viruses (data not shown). Consistent with the concept that circulating IGF‐1 contributes to the maintenance of IGF‐1 levels in the brain (Nishijima et al., 2010), we found that mice receiving TBG‐Cre‐AAV8 also had significantly lower tissue IGF‐1 levels in the cerebral cortex compared with control mice receiving TBG‐eGFP‐AAV8 (4.8 ± 1.7 and 11.4 ± 2.4 pg mg−1 of tissue, respectively; P = 0.03).
Figure 1.
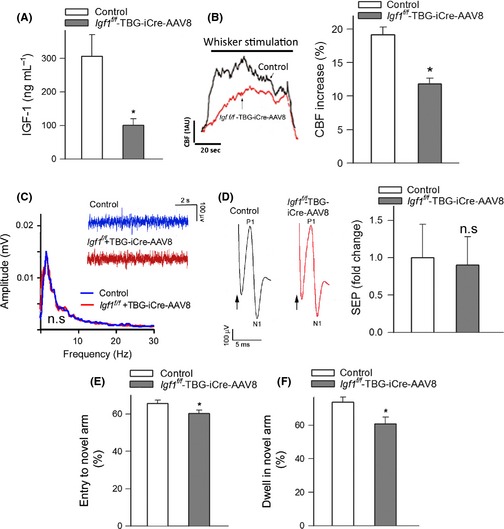
IGF‐1 deficiency impairs neurovascular coupling and cognitive function. Panel A shows that adeno‐associated viral knockdown of hepatic Igf1 (Igf1 f/f + TBG‐Cre‐AAV8) decreases significantly the level of circulating IGF‐1 compared to control animals (Igf1 f/f + TBG‐eGFP‐AAV8) (*P < 0.05 vs. control). (B) Representative traces of cerebral blood flow (CBF) measured with a laser Doppler probe above the whisker barrel cortex during contralateral whisker stimulation (5 Hz) in control and IGF‐1‐deficient mice. 1 AU corresponds to ~5% increase in CBF from baseline. Right panel depicts the summary data of the CBF responses (ΔAUC as % of baseline; n = 12, *P < 0.001 vs. control). (C–D) Spontaneous and evoked neural activity is not altered in IGF‐1‐deficient mice. (C) The amplitude and frequency distribution of neocortical electrical activity are nearly identical in control and IGF‐1‐deficient mice (inlet shows original recording of electrocorticograms, n = 6, P = 0.4). (D) The somatosensory evoked potential (SEP) responses in the somatosensory cortex evoked by contralateral whisker pad stimulation are comparable in control and IGF‐1‐deficient mice. The arrow indicates the application of the stimulus. The amplitude of the negative wave of the field potentials (N1) does not differ between control and IGF‐1‐deficient mice (n = 6, P = 0.6). (E–F) Spatial memory testing of mice in Y‐maze. The IGF‐1‐deficient animals (Igf1 f/f + TBG‐Cre‐AAV8) exhibited impaired spatial memory as shown by the decreased number of entries in novel arm (E; * P = 0.001 vs. control) and shorter exploratory time spent in novel arm of the Y‐maze during retrieval trial (F; and P = 0.01 vs. control). Data are mean ± S.E.M., n = 20 in each group.
Changes in CBF in the whisker barrel cortex in response to contralateral whisker stimulation were significantly attenuated in IGF‐1‐deficient mice (Fig. 1B), indicating that IGF‐1 deficiency leads to neurovascular uncoupling, mimicking the aging phenotype (Toth et al., 2014a). IGF‐1 deficiency could reduce functional hyperemia by impairing neural activity evoked by whisker stimulation. To examine this possibility, we recorded spontaneous and evoked neural activity in control and IGF‐1‐deficient mice. We found that the amplitude and frequency distribution of the electrocorticogram and the amplitude of the somatosensory field potentials produced by the activation of the whisker pad do not differ between control and IGF‐1‐deficient mice (Fig. 1C–D). Therefore, IGF‐1 deficiency is unlikely to contribute to impaired functional hyperemia by modulating the neural activity evoked by whisker stimulation.
Our recent studies demonstrate that selective experimental disruption of neurovascular coupling responses is associated with significant impairment of cognitive function, recapitulating neurological symptoms and signs observed in brain aging (Tarantini, Ungvari and Toth, manuscript in preparation 2015). In this study, for the hippocampal‐dependent spatial memory test, the control mice entered the novel arm more often than the IGF‐1‐deficient mice following the intertrial interval (Fig. 1C). The control mice also spent significantly more time in the novel arm than the previously visited arms, whereas the IGF‐1‐deficient mice spent significantly less time in the novel arm (Fig. 1D), indicating that neurovascular uncoupling induced by IGF‐1 deficiency is also associated with impaired spatial working memory and novelty‐seeking behavior. Previous studies using the Morris water maze (Trejo et al., 2007) and the Barnes maze (Sonntag and Csiszar, unpublished data, 2012) also yielded similar results, showing that IGF‐1‐deficient mice exhibit impaired spatial working memory. Learning and/or memory deficits were also observed in GH/IGF‐1‐deficient Lewis dwarf rats (Nieves‐Martinez et al., 2010), spontaneously dwarf SD rats (Li et al., 2011), and in Ames dwarf mice in some (Derenne et al., 2011), but not all (Sharma et al., 2010), studies.
IGF‐1 deficiency impairs cerebromicrovascular endothelial function: role in neurovascular uncoupling
We found that in control animals, the administration of the NO synthase inhibitor L‐NAME significantly decreased CBF responses in the barrel cortex elicited by contralateral whisker stimulation (Fig. 2A). In IGF‐1‐deficient animals, the effect of L‐NAME was blunted (Fig. 2A), suggesting that IGF‐1 deficiency impairs NO mediation, which contributes to neurovascular uncoupling. Topical application of the endothelium‐dependent vasodilator agent acetylcholine (ACh; 10−5 mol L−1) resulted in a significant increase in CBF in the barrel cortex of control mice (Fig. 2B). ACh‐induced CBF responses were significantly attenuated in IGF‐1‐deficient mice (Fig. 2B), supporting the concept that IGF‐1 deficiency impairs cerebromicrovascular endothelial function, mimicking the aging phenotype (Toth et al., 2014a). Previously, it has been found that in models of vascular aging in the periphery, IGF‐1 decreases vascular oxidative stress and improves endothelial function (Ungvari & Csiszar, 2012), whereas it does not significantly affect endothelium‐independent vasodilation elicited by NO donors (Bailey‐Downs et al., 2012). We found that in IGF‐1‐deficient mice, 3‐nitrotyrosine content in the cerebral cortex is significantly elevated (Fig. 2C) consistent with increased oxidative/nitrosative stress in the brain, which mimics the aging phenotype (Toth et al., 2014a). IGF‐1 deficiency was associated with decreased expression of Nos3 (Fig. 2D). In IGF‐1‐deficient mice, the expression of Nox1 and Nox2 subunits of the NADPH oxidase tended to increase; however, the differences did not reach statistical significance (Fig. 2D).
Figure 2.
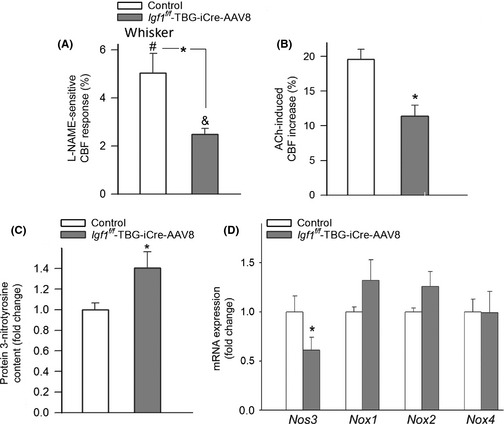
IGF‐1 deficiency impairs cerebromicrovascular endothelial function: role in neurovascular uncoupling. (A) L‐NAME‐sensitive, NO‐mediated portion of the CBF response (calculated based on the percentage decline in CBF in the presence of L‐NAME) measured above the barrel field of the primary somatosensory cortex in response to whisker stimulation in control and IGF‐1‐deficient (Igf1 f/f + TBG‐Cre‐AAV8) mice (n = 6, * P < 0.05 vs. control; # P < 0.05 vs. control w/o drug; & P < 0.05 vs. Igf1 f/f + TBG‐Cre‐AAV8 w/o drug). (B) CBF responses elicited by topical administration of acetylcholine to the barrel field of control and IGF‐1‐deficient mice (n = 6, *P < 0.05 vs. control). (C) Protein 3‐nitrotyrosine content, a biomarker of increased ONOO‐ formation, in cortical tissue of IGF‐1‐deficient and control mice (n = 5, *P < 0.05 vs. control). (D) qPCR data showing mRNA expression of the endothelial nitric oxide synthase (Nos3) and the NADPH oxidase subunits Nox1, Nox2, and Nox4 in cortical samples of IGF‐1‐deficient and control mice. Data are mean ± S.E.M. (n = 5, *P < 0.05 vs. control).
IGF‐1 deficiency impairs glutamate‐mediated CBF responses: role in neurovascular uncoupling
Astrocytes were suggested to sense neuronal‐derived glutamate through metabotropic glutamate receptors (mGluR) and NMDA receptors, leading to increased production of vasodilator gliotransmitters that contribute to neurovascular coupling (Petzold & Murthy, 2011). In support of this concept, we found that in control mice, the metabotropic Glu receptor blocker MPEP and NMDA receptor blocker D‐APV significantly decreased CBF responses in the barrel cortex elicited by contralateral whisker stimulation (Fig. 3A). In IGF‐1‐deficient animals, the effect of MPEP plus D‐APV was significantly decreased (Fig. 3A), suggesting that IGF‐1 deficiency impairs glutamate‐mediated neurovascular coupling. Further support for this concept is provided by the findings that in IGF‐1‐deficient mice, glutamate‐induced CBF changes were also significantly impaired (Fig. 3B). IGF‐1 deficiency did not alter glutamate release induced by neuronal activation (Fig. 3C–D), whereas it dysregulated the astrocytic expression of metabotropic glutamate receptors (Grm2, Grm4, Grm5) and NMDA receptors (Grin1 and Grin2; Fig. 3E).
Figure 3.
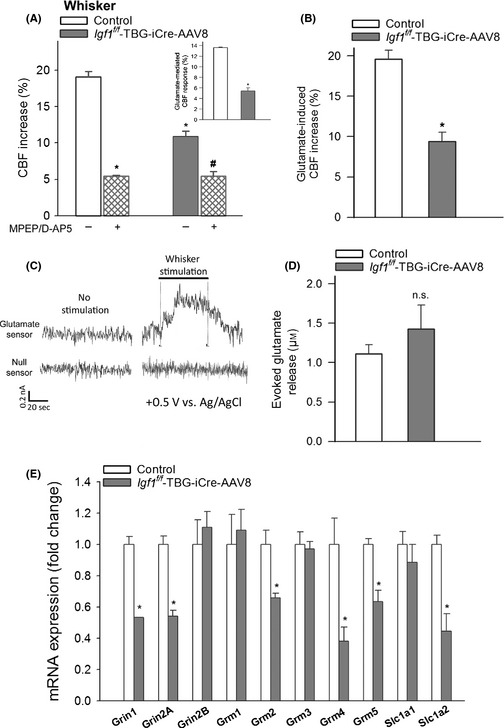
IGF‐1 deficiency impairs glutamate‐mediated CBF responses: role in neurovascular uncoupling. (A) Effects of treatment with antagonists of metabotropic glutamate receptors (MPEP, 5 × 10−5 mol L−1) and NMDA receptors (N‐methyl‐D‐aspartate, D‐APV, 5 × 10−5 mol L−1) on cerebral blood flow (CBF) responses measured above the barrel field of the primary somatosensory cortex in response to whisker stimulation in control and IGF‐1‐deficient mice (Igf1 f/f + TBG‐Cre‐AAV8). The inlet shows the glutamate‐mediated part of the neurovascular response in each group (n = 6 in each group, *P < 0.05 vs. control; # P < 0.05 vs. Igf1 f/f + TBG‐Cre‐AAV8). (B) CBF responses measured above the barrel field of the primary somatosensory cortex elicited by topical administration of L‐glutamate (500 μmol L−1) in control and IGF‐1‐deficient mice (n = 6 in each group, *P < 0.05 vs. control). Panel C Original recordings of changes in extracellular glutamate in response to whisker stimulation (5 Hz, 2 min) measured by amperometry using a glutamate biosensor inserted into the barrel cortex of mice (see Methods for details). ‘Null sensor’ indicates a biosensor constructed the same way as the glutamate sensors but without any enzymes for biosensing. Summary data are shown in Panel D. No significant differences (P = 0.4) were observed between cortical glutamate signals induced by whisker stimulation in control (n = 5) and IGF‐1‐deficient mice (Igf1 f/f + TBG‐Cre‐AAV8, n = 7). (E) qPCR data showing mRNA expression of NMDA receptors (Grin1, Grin2A, Grin2B), metabotropic glutamate receptors (Grm1, Grm2, Grm3, Grm4, Grm5), and glutamate transporters (Slc1a1, Slc1a2) on astrocytes isolated from control and IGF‐1‐deficient animals (n = 5). *P < 0.05 vs. control. Data are mean ± S.E.M. for every panel of the figure.
IGF‐1 deficiency impairs mediation of CBF responses by eicosanoid gliotransmitters: role in neurovascular uncoupling
Upon activation by neuronal‐derived glutamate, astrocytes were shown to convert arachidonic acid by cyclooxygenases to vasodilator prostaglandins and by P450 epoxygenase to vasodilator EETs (Petzold & Murthy, 2011). Consistent with this concept, we found that in control animals, the administration of the cyclooxygenase inhibitor indomethacin (Fig. 4A) and P450 epoxygenase inhibitor MS‐PPOH (Fig. 4B) significantly decreased CBF responses in the barrel cortex elicited by contralateral whisker stimulation. In IGF‐1‐deficient animals, the effects of indomethacin (Fig. 4A) and MS‐PPOH (Fig. 4B) were significantly decreased, suggesting that IGF‐1 deficiency impairs the mediation of neurovascular coupling by prostaglandins and EETs. The astrocyte‐derived vasoconstrictor eicosanoid 20‐HETE can also negatively impact neurovascular coupling. Using the cytochrome P450 ω‐hydroxylase inhibitor HET0016, we found that IGF‐1 deficiency tended to increase the HET0016‐sensitive component of the CBF response, indicating that a 20‐HETE‐dependent constrictor response is present in IGF‐1‐deficient mice (Fig. 4C). LC/MS/MS measurements demonstrated that IGF‐1 deficiency resulted in a diminished cerebral production of the potent vasodilator 14,15‐EET (which is the most prevalent among the four EET regioisomers in the murine brain) in response to glutamate stimulation of brain slices (Fig. 4D). IGF‐1 deficiency also tended to increase the cerebral production of 20‐HETE (Fig. 4E). Among the investigated factors known to be involved in the synthesis of eicosanoid gliotransmitters, the expression of Cyp2c55 decreased, whereas the expression of the 20‐HETE‐producing ω‐hydroxylase Cyp4a10 increased in IGF‐1‐deficient mice (Fig. 4F).
Figure 4.
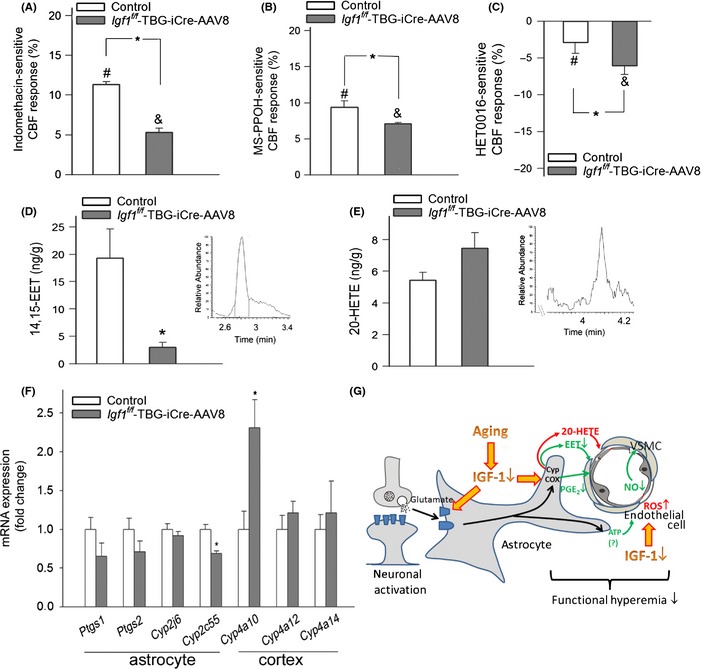
IGF‐1 deficiency impairs mediation of CBF responses by eicosanoid gliotransmitters: role in neurovascular uncoupling. (A) Indomethacin‐sensitive, prostaglandin‐mediated portion of the CBF response (calculated based on the percentage decline in CBF in the presence of INDO) measured above the barrel field of the primary somatosensory cortex in response to whisker stimulation in control and IGF‐1‐deficient (Igf1 f/f + TBG‐Cre‐AAV8) mice (n = 6 in each group, *P < 0.05 vs. control; # P < 0.05 vs. control w/o INDO; & P < 0.05 vs. Igf1 f/f + TBG‐Cre‐AAV8 w/o INDO). (B) MS‐PPOH‐sensitive, EET‐mediated portion of the CBF response (calculated based on the percentage decline in CBF in the presence of MS‐PPOH) measured above the barrel field of the primary somatosensory cortex in response to whisker stimulation in control and IGF‐1‐deficient (Igf1 f/f + TBG‐Cre‐AAV8) mice (n = 6 in each group, *P < 0.05 vs. control; # P < 0.05 vs. control w/o MS‐PPOH; & P < 0.05 vs. Igf1 f/f + TBG‐Cre‐AAV8 w/o MS‐PPOH). (C) HET0016‐sensitive, 20‐HETE‐mediated portion of the CBF response (calculated based on the percentage decline in CBF in the presence of the cytochrome P450 ω‐hydroxylase inhibitor HET0016) measured above the barrel field of the primary somatosensory cortex in response to whisker stimulation in control and IGF‐1‐deficient (Igf1 f/f + TBG‐Cre‐AAV8) mice (n = 6 in each group, *P < 0.05 vs. control; # P < 0.05 vs. control w/o HET0016; & P < 0.05 vs. Igf1 f/f + TBG‐Cre‐AAV8 w/o HET0016). (D–E) Production of 14,15 EET (D) and 20‐HETE (E) in glutamate‐activated brain slices from control and Igf1 f/f + TBG‐Cre‐AAV8 mice as measured by liquid chromatography/mass spectrometry (LC/MS) (n = 6 in each group; *P < 0.05 vs. control; see Methods). (F) qPCR data showing mRNA expression of cyclooxygenase‐1 and cyclooxygenase‐2 (Ptgs1, Ptgs2) and EET‐producing epoxygenases (Cyp2j6, Cyp2c55) in isolated astrocytes, and 20‐HETE‐producing ω‐hydroxylases (Cyp4a10, Cyp4a12, Cyp4a14) in cortical samples of control and IGF‐1‐deficient mice (n = 5 in each group). *P < 0.05 vs. control. Data are mean ± S.E.M. (G) Proposed mechanisms by which age‐related IGF‐1 deficiency may impair neurovascular coupling responses (see Discussion). The model predicts that IGF‐1 deficiency both alters astrocytic production of eicosanoid gliotransmitters and impairs cerebromicrovascular endothelial function.
Discussion
Circulating IGF‐1 concentrations decrease significantly with age, due to decreases in GH levels, both in humans and in laboratory animals (Sonntag et al., 2005, 2013). There is substantial evidence that circulating IGF‐1 is an important vascular protective factor and that the age‐related decline in IGF‐1 levels contributes to vascular aging, promoting atherogenesis and development of cardiovascular disease and stroke (reviewed recently in Ungvari & Csiszar (2012) and Sonntag et al. (2013)). Here, we show for the first time that circulating IGF‐1 deficiency also leads to profound neurovascular dysregulation, characterized by impaired CBF responses induced by synaptic activity (Fig. 1), which mimics the cerebromicrovascular aging phenotype (Toth et al., 2014a). Impairment of a key homeostatic mechanism matching energy supply with the needs of active neuronal tissue is predicted to have deleterious effects on brain function. Indeed, there is strong evidence that in elderly patients, impaired neurovascular coupling (Zaletel et al., 2005; Topcuoglu et al., 2009; Stefanova et al., 2013) associates with decline in higher cortical functions including cognition. Importantly, neurovascular uncoupling in IGF‐1‐deficient mice also associates with impaired cognitive function (Fig. 1), mimicking the aging phenotype (Csiszar et al., 2013).
Although IGF‐1 deficiency may impact multiple aspects of neuronal function (Sonntag et al., 2013), our recent studies strongly suggest that a direct mechanistic link exists between neurovascular uncoupling and cognitive decline. Accordingly, recently we found that experimentally induced acute neurovascular uncoupling in mice, in the absence of alterations in synaptic function, leads to impaired performance in tests relevant for hippocampal‐ and cortical‐dependent tasks of learning and memory (Tarantini, Ungvari and Toth, manuscript in preparation 2015). In humans, IGF‐1 deficiency is associated with progressive cognitive dysfunction that can be reversed by increasing circulating IGF‐1 levels (reviewed in Sonntag et al. (2013)). Rodents have a similar decrease in circulating IGF‐1 levels with age and treatment of aged F344xBN rats with IGF‐1 was shown to improve cognitive function (reviewed in Sonntag et al. (2013)). Thus, further studies are warranted to determine whether in the aforementioned models treatment with IGF‐1 rescues neurovascular coupling and to establish a causal link between IGF‐1‐dependent changes in functional hyperemia and cognitive function. In addition to exerting protective effects on neurovascular coupling responses, IGF‐1 may also protect brain function by additional mechanisms, including exerting trophic effects on central glutamatergic synapses (Trejo et al., 2007) and/or preventing blood brain barrier disruption and neuroinflammation (Toth et al., 2014b).
The effects of IGF‐1 deficiency on the cellular mechanisms involved in neurovascular coupling are likely multifaceted. Microvascular endothelial cells are directly exposed to circulating IGF‐1 and are known to abundantly express IGF‐1 receptors (Ungvari & Csiszar, 2012). Importantly, there is growing experimental evidence that NO production by cerebromicrovascular endothelial cells has an important role in functional hyperemia (Girouard et al., 2007; Longden & Nelson, 2011; Stobart et al., 2013). This concept is supported by our observation that inhibition of NO synthesis significantly reduces neurovascular coupling in control animals (Fig. 2). It is significant that impaired endothelial NO production was shown to contribute to age‐related neurovascular uncoupling in mice (Park et al., 2007; Toth et al., 2014a). The findings that in IGF‐1‐deficient mice (Fig. 2), the L‐NAME‐sensitive, NO‐mediated portion of the neurovascular coupling response was decreased suggest that cerebromicrovascular endothelial dysfunction also contributes to neurovascular uncoupling in IGF‐1 deficiency (Park et al., 2007), mimicking the aging phenotype (Park et al., 2007; Toth et al., 2014a). The mechanisms by which IGF‐1 deficiency impairs cerebromicrovascular endothelial function likely involve an increased breakdown of NO by elevated levels of ROS. Several lines of evidence support this concept. First, IGF‐1‐deficient mice, similar to aged mice (Park et al., 2007; Toth et al., 2014a), exhibit increased production of ROS (Csiszar et al., 2008). Second, the treatment of primary endothelial cells with IGF‐1 significantly attenuates cellular ROS generation (Csiszar et al., 2008). Third, in the microcirculation, endothelium‐derived NO was shown to react with increased O2 .− forming ONOO−, thus decreasing the bioavailability of NO (Csiszar et al., 2002; Pacher et al., 2007). The brains of IGF‐1‐deficient mice, similar to aged mice (Toth et al., 2014a), exhibit an increased 3‐nitrotyrosine content (Fig. 2), a biomarker of increased ONOO− formation, indicating that increased scavenging of vasodilator NO contributes to impaired endothelial mediation of cerebromicrovascular dilation in IGF‐1 deficiency. Previous studies suggest that increased activity/expression of NADPH oxidases and increased ROS production by mitochondrial sources contribute significantly to aging‐induced microvascular oxidative stress (Park et al., 2007; Ungvari & Csiszar, 2012; Toth et al., 2014a). Accordingly, the findings that treatment with a pharmacological inhibitor of cellular ROS production is able to improve functional hyperemia provide direct evidence for the role of increased oxidative stress in neurovascular uncoupling both in IGF‐1‐deficient mice and in aged mice (Toth et al., 2014a). Importantly, impaired endothelial function and increased oxidative stress are also manifest in the peripheral circulation in IGF‐1 deficiency (Gong et al., 2014) (reviewed in Ungvari & Csiszar (2012)). In addition to its vasodilator action, NO also confers multifaceted endothelial protective effects, including pro‐angiogenic, anti‐apoptotic, and anti‐inflammatory effects (Ungvari & Csiszar, 2012; Sonntag et al., 2013). Thus, it is likely that impaired endothelial function associated with IGF‐1 deficiency has complex phenotypic consequences in the cerebral microcirculation (e.g., alterations in microvascular density (Ungvari & Csiszar, 2012)), which should be elucidated by future studies.
In addition to the microvascular endothelial cells, the activation of astrocytic production of vasodilator arachidonic acid metabolites (EETs, prostaglandins) by neuronal‐derived glutamate also has an important role in neurovascular coupling (Petzold & Murthy, 2011). Here, we provide the first evidence that IGF‐1 deficiency alters the phenotype of astrocytes, impairing the astrocyte‐mediated portion of neurovascular coupling. We found that IGF‐1 deficiency impairs glutamate‐mediated gliovascular coupling responses (Fig. 3A), but it does not affect glutamate release from neurons (Fig. 3C), suggesting a primary astrocytic deficit. Our findings indicate that the likely mechanisms by which IGF‐1 deficiency impairs glutamate‐mediated gliovascular coupling responses include decreased expression of astrocytic glutamate receptors (Fig. 3D) and dysregulation of astrocytic synthesis of eicosanoid gliotransmitters, likely due to altered expression of cytochrome P‐450 enzymes in the astrocytes (Fig. 4). On the basis of the aforementioned findings, one would predict that restoration of circulating IGF‐1 levels in aging would positively impact astrocyte function, improving gliovascular coupling. This hypothesis needs to be experimentally tested in future studies. It should be noted that NO per se is likely to regulate the metabolism of arachidonic acid by cytochrome P‐450 enzymes in the vasculature, inhibiting the production of vasoconstrictor 20‐HETE. Thus, future studies should also determine whether decreased bioavailability of NO contributes to increased production of 20‐HETE in the brains of IGF‐1‐deficient mice.
In conclusion, our results add to the growing evidence that IGF‐1 exerts an important cerebromicrovascular protective effect (Toth et al., 2014b), which likely supports multiple aspects of brain health. The findings that isolated circulating IGF1 deficiency results in functional and phenotypic alterations in endothelial cells and astrocytes and leads to neurovascular uncoupling have important clinical relevance for cognitive impairment associated with both aging and genetic IGF‐1 deficiency (e.g., Laron dwarfism). Our findings, taken together with the results of earlier studies (reviewed in Sonntag et al. (2013)), point to potential benefits of interventions preventing age‐related IGF‐1 deficiency and promoting microvascular health for the prevention of cognitive decline in the elderly.
Experimental procedures
All procedures were approved by and followed the guidelines of the Institutional Animal Care and Use Committee of OUHSC in accordance with the ARRIVE guidelines.
Postdevelopmental liver‐specific knockdown of Igf1 in mice
Male mice homozygous for a floxed exon 4 of the Igf1 gene (Igf1 f/f) in a C57BL/6 background were used (Toth et al., 2014b). These mice have the entirety of exon 4 of the Igf1 gene flanked by loxP sites, which allows for genomic excision of this exon when exposed to Cre recombinase. Transcripts of the altered Igf1 gene yield a protein upon translation that fails to bind the IGF receptor. Animals were housed in the Rodent Barrier Facility at OUHSC, on a 12‐h light/12‐h dark cycle, and given access to standard rodent chow (Purina Mills, Richmond, IN, USA) and water ad libitum. To target hepatocytes, adeno‐associated viruses (AAVs) were purchased from the University of Pennsylvania Vector Core (Philadelphia, PA, USA). At 2 months of age, approximately 1.3 × 1010 viral particles (as assayed by genome content at the University of Pennsylvania) of AAV8.TBG.PI.Cre.rBG or AAV8.TBG.PI.eGFP.WPRE.bGH were administered to Igf1 f/f mice to knockdown IGF‐1 or as a control, respectively, as described (Toth et al., 2014b). While AAV8 is effective at transducing multiple tissues after i.v. delivery, including liver, the thyroxine‐binding globulin (TBG) promoter restricts the expression solely to hepatocytes, as described (Toth et al., 2014b). Experiments were conducted 3 months postknockdown of Igf1.
Measurement of circulating and tissue IGF‐1 levels
Venous blood was collected from the submandibular veins of animals from both groups (Medipoint, Mineola, NY, USA). Whole blood was centrifuged at 2500 g for 20 min at 4°C to collect serum, which was then stored at −80°C. IGF‐1 levels in sera and cortical tissue samples were measured by ELISA (R&D Systems, Minneapolis, MN, USA) according to the manufacturer's protocol and are reported in ng mL−1 and pg mg−1 tissue, respectively.
Spatial memory testing of mice in Y‐maze
Three months after IGF‐1 knockdown, animals were tested for spatial working memory in the Y‐maze as described (Csiszar et al., 2013). In brief, a Y‐maze apparatus, made up of three enclosed transparent Plexiglas arms (40 cm length × 9 cm width × 16 cm height) with extra‐maze visual cues around the maze, was used to assess hippocampal‐dependent spatial recognition memory. The test consisted of two trials separated by an intertrial interval (4 h). All mice were transported to the behavioral testing room in their home cages at least 1 h before testing. In the first training (acquisition) trial, mice were placed in the maze facing the end of a pseudorandomly chosen start arm and allowed to explore the maze for 5 min with one of the arms closed (novel arm). Mice were returned to their home cage until the second (retrieval) trial, during which they could explore freely all three arms of the maze. The time spent in each arm and number of entries were measured and analyzed from video recordings (Ethovision, Noldus Information Technology Inc., Leesburg, VA, USA). Mice were required to enter an arm with all four paws in order for it to be counted as an entry. The results are expressed as number of entries (entry) and time spent (dwell) in the novel arm as % of all entries and time spent in novel and other arm (Sarnyai et al., 2000). Entering the novel arm more frequently and for longer periods of time indicates intact memory and novelty‐seeking behavior because of the innate tendency of mice to explore. The maze was cleaned with 70% ethanol between the trials.
Surgical procedures
Mice in each group were anesthetized with α‐chloralose (50 mg kg−1, i.p.) and urethane (750 mg kg−1, i.p.), endotracheally intubated, and ventilated (MousVent G500; Kent Scientific Co, Torrington, CT, USA). Rectal temperature was maintained at 37°C using a thermostatic heating pad (Kent Scientific Co). End‐tidal CO2 (including dead space) was maintained between 3.2% and 3.7% to keep blood gas values within the physiological range (PaCO2: 36.18 ± 1.8 mmHg, PaO2: 109.8 ±3.1 mmHg). Mice were immobilized, placed on a stereotaxic frame (Leica Microsystems Inc, Buffalo Grove, IL, USA), the scalp and periosteum were pulled aside and the skull was removed over the barrel cortex, and the dura was gently removed. The cranial window was superfused with artificial cerebrospinal fluid (ACSF, composition: NaCl 119 mm, NaHCO3 26.2 mm, KCl 2.5 mm, NaH2PO4 1 mm, MgCl2 1.3 mm, glucose 10 mm, CaCl2 2.5 mm, pH = 7.3, 37°C). The right femoral artery was cannulated for arterial blood pressure measurement (Living Systems Instrumentations, Burlington, VT, USA). The blood pressure was within the physiological range throughout the experiments (90–110 mmHg).
Cerebral blood flow responses to whisker stimulation and pharmacological studies
To assess neurovascular coupling, a laser Doppler probe (Transonic Systems Inc., Ithaca, NY, USA) was placed above the barrel cortex (1–1.5 mm posterior and 3–3.5 mm lateral to bregma), and the contralateral whiskers were stimulated for 1 min at 5 Hz from side to side. Changes in CBF (n = 7–8 mic in each group) were assessed in three trials (5‐ to 10‐min intervals). CBF responses to whisker stimulation were repeated in the presence of the following inhibitors administered topically onto the brain surface of separate groups of animals: HET0016 (inhibitor of 20‐hydroxyeicosatrienoic acid (20‐HETE) production, 10−6 mol L−1 for 30 min; Cayman Chemicals, Ann Arbor, MI, USA) (Liu et al., 2008), MS‐PPOH (inhibitor of EET production, 20 × 10−6 mol L−1 for 30 min; Cayman Chemicals) (Shi et al., 2008), L‐NAME (Nω‐Nitro‐L‐arginine methyl ester, inhibitor of nitric oxide synthase, 10−4 mol L−1 for 20 min; Sigma‐Aldrich, St. Louis, MO, U.S.A.), apocynin (inhibitor of NADPH oxidases, 3 × 10−4 mol L−1 for 30 min; Cayman Chemicals), fluoroacetate sodium (inhibitor of the tricarboxylic acid cycle predominantly in glial cells, 10−4 mol L−1 min; Sigma‐Aldrich, St. Louis, MO, U.S.A.) (Fonnum et al., 1997; Lecrux et al., 2012), indomethacin (cyclooxygenase inhibitor, 5 × 10−4 mol L−1; Sigma‐Aldrich, St. Louis, MO, U.S.A.) (Kitaura et al., 2007), MPEP (6‐Methyl‐2‐(phenylethynyl)pyridine hydrochloride, group I metabotropic glutamate receptors (mGluR) subtype 5 antagonist, 5 × 10−5 mol L−1) (Zonta et al., 2003), and the NMDA (N‐methyl‐D‐aspartate) receptor antagonist D‐APV (D‐2‐Amino‐5‐Phosphonovaleric acid, 5 × 10−5 mol L−1; Cayman Chemicals) (Stobart et al., 2013). In a separate series of experiments (n = 8 in each group), CBF responses to topical administration of L‐glutamate (500 μmol L−1) (Hall et al., 2014) were determined in the absence and presence of MPEP (5 × 10−5 mol L−1) and D‐APV (5 × 10−5 mol L−1) (Stobart et al., 2013). CBF responses to acetylcholine (ACh; 10−5 mol L−1) were also obtained to assess maximal endothelial NO‐mediated responses. Changes in CBF are expressed as percent (%) changes from baseline.
Spontaneous neuronal activity and evoked field potentials (SEP) in the primary somatosensory cortex
The animals were surgically prepared and ventilated as described above, and a glass‐insulated tungsten microelectrode (impedance, 2–3 MΩ; Kation Scientific, LLC, Minneapolis, MN, USA) was inserted into the left barrel cortex (1–1.5 mm posterior and 3–3.5 mm lateral to bregma) through the ACSF‐perfused open cranial window for recording local field potentials. An Ag/AgCl electrode inserted in the neck muscles served as reference. After basal activity was recorded, the right whisker pad was stimulated by a bipolar stimulating electrode placed to the ramus infraorbitalis of the trigeminal nerve and into the masticatory muscles. The stimulation protocol used to investigate neurovascular coupling and somatosensory evoked field potentials consisted of 10 stimulation presentation trials with an intertrial interval of 70 s, each delivering a 15‐s train of electrical pulses (2 Hz, 0.2 mA, intensity, and 0.3 ms pulse width) after a 10‐s prestimulation baseline period. The signal was amplified with an AC/DC differential amplifier (high pass at 1 Hz, low pass at 1 kHz) (Model 3000; A‐M Systems, Inc. Carlsborg, WA, USA) and digitalized by the PowerLab/Labchart data acquisition system (ADInstruments, Colorado Springs, CO, USA) with a sampling rate of 40 kHz. Basal activity was analyzed as distribution of wave amplitude as a function of frequency (Park et al., 2008), and the negative amplitude in the somatosensory evoked field potential response was considered as the excitatory postsynaptic potential (fEPSP) (Lind et al., 2013). Analyses were performed on the average of 10 stimulation trials.
Cerebral glutamate release to whisker stimulation
In a separate cohort of animals, we assessed changes in extracellular glutamate signal in response to whisker stimulation. The cranial window was superfused continuously with ACSF. The glutamate sensor is a platinum electrode encapsulated in a biolayer containing glutamate oxidase and protected against the interference with ascorbate, urate, dopamine, and 5‐hydroxytriptamine. Glutamate is oxidized into hydrogen peroxide which is sensed by the electrode. In the morning of the experiments, the working electrode (Sarissa GLU Biosensor, 25 μm tip; Sarissa Biomedical, Coventry, UK) was calibrated according to the manufacturer's guidelines in vitro, and then, it was inserted 1.5 mm caudal and 3 mm lateral from bregma into the cerebral tissue about 500 μm deep. Glutamate null sensors (lack of any enzymes for biosensing) were us ed as controls. The reference electrode (Ag/AgCl) was inserted into the cerebral tissue elsewhere, and the auxiliary electrode (Ag/AgCl) was placed between the scalp and the skull. The potential was set at 0.5 V vs. Ag/AgCl. A 3‐electrode potentiostat (Quadstat) with an eDAQ data acquisition system (eDAQ Pty Ltd., Colorado Springs, CO, USA) was used for constant potential amperometry. Following the insertion of the electrodes, we waited about 2‐3 min until a stable baseline developed. Then, we stimulated the right whiskers for 1 min at 5 Hz, in three consecutive trials divided by 5‐ to 10‐min intervals. The response was recorded in nA and converted to μM of glutamate using the calibration curve. Basal glutamate measurements preceding the evoked glutamate signal were included only, and amplitudes were calculated by obtaining the maximum increase from baseline (Onifer et al., 2012).
Astrocyte isolation
Astrocytes were immunopurified from the cortex of the experimental animals by targeting the extracellular epitope of Glast, a glutamate transporter specifically found on astrocytes. Cortices were isolated in ice‐cold HBSS, enzymatically digested in L15 media with 0.05% trypsin for 20 min at 37°C, titurated, and filtered through 0.7‐μm mesh cell strainer. The remaining cells were pelleted, resuspended in 100 μls of HBSS with 0.5% BSA, and incubated with anti‐Glast‐PE (1:10; Miltenyi Biotech, Bergisch Gladbach, Germany) for 10 min at 4°C. Anti‐PE microbeads (Miltenyi Biotech) were then added and incubated for an additional 10 min at 4°C. Cells were pelleted and washed three times with PBS with 0.5% BSA before magnetic bead column purification (Miltenyi Biotech).
Quantitative real‐time RT–PCR
A quantitative real‐time RT–PCR technique was used to analyze mRNA expression of nitric oxide synthase, NADPH oxidases, NMDA receptors, metabotropic glutamate receptors, cyclooxygenases, epoxygenases, and ω‐hydroxylases in cortical samples and isolated astrocytes from each experimental group using a Strategen MX3000 platform, as previously reported (Toth et al., 2013b). In brief, total RNA was isolated with a Mini RNA Isolation Kit (Zymo Research, Orange, CA, USA) and was reverse‐transcribed using Superscript III RT (Invitrogen, Burlington, ON, Canada) (Toth et al., 2013a,b). Amplification efficiencies were determined using a dilution series of a standard vascular sample. Quantification was performed using the efficiency‐corrected ΔΔCq method. The relative quantities of the reference genes Hprt, Ywhaz, B2 m, and Actb were determined, and a normalization factor was calculated based on the geometric mean for internal normalization. Fidelity of the PCR was determined by melting temperature analysis and visualization of the product on a 2% agarose gel.
Measurement of the production of arachidonic acid metabolites in brain slices
To determine how IGF‐1 deficiency affects the synthesis of eicosanoid gliotransmitters, horizontal hippocampal slices of 400 μm thickness from mice in each cohort were prepared in ice‐cold solution containing (in mmol L−1) sucrose 110, NaCl 60, KCl 3, NaH2PO4 1.25, NaHCO3 28, sodium ascorbate acid 0.6, glucose 5, MgCl2 7, and CaCl2 0.5 using a HM650V vibrating microtome (Thermo Scientific, Burlington, ON, Canada). Slices were then transferred to a custom‐made chamber which contained oxygenated artificial cerebrospinal fluid (aCSF) of the following composition (in mm): NaCl 126, KCl 2.5, NaH2PO4 1.25, MgCl2 2, CaCl2 2, NaHCO3 26, glucose 10, pyruvic acid 2, and ascorbic acid 0.4. To stimulate astrocytes, glutamate (3 × 10−4 mol L−1) was added to the chamber. The samples were weighed and snap‐frozen for further analysis.
The samples were homogenized in ice‐cold phosphate buffer (pH 6.8). Thousand units of E. coli β‐glucuronidase was added to the tissue extract to release 20‐HETE from conjugation with glucuronide. After incubation at 37°C for 2 h, the pH in the solution was adjusted to 3 by the addition of acetic acid. [2H4]‐20‐HETE (10 ng) was added, and the sample was extracted with acidified CHCl3/CH3OH (2:1) and purified by silica solid‐phase extraction. Different samples were placed into ice‐cold 0.15M KCl. After homogenization, synthetic [2H11]‐labeled 14,15‐DHET (5 ng) was added as internal standard. The EETs and DHETs were extracted from the tissue homogenates with acidified CHCl3/CH3OH (2:1) and purified by silica solid‐phase extraction, separating EETs and DHETs.
Quantification was performed by LC/MS/MS using Acquity BEH C18 columns (1.0 × 100 mm; 1.7 μm) connected to a TSQ‐Quantum Vantage triple quadrupole spectrometer (ThermoScientific) with a linear solvent gradient that went from 70% 15 mm aqueous ammonium acetate (pH 8.5), 30% acetonitrile to 40% 15 mm aqueous ammonium acetate (pH 8.5), 60% acetonitrile in 6 min and at a flow of 0.18 mL/min. For 20‐HETE analysis, we utilized collision‐induced fragmentation of 20‐HETE at m/z 319 and the [2H4]‐20‐HETE internal standard at m/z 325. The ratio of the area of the 20‐HETE peak compared to the area of the corresponding deuterated 20‐HETE was used for quantification. For EET analysis, the EETs were converted to the corresponding DHETs by treatment with acetic acid overnight. Then, we utilized collision‐induced fragmentation of the DHETs at m/z 337 and the [2H11]–DHET internal standards at m/z 448. Diagnostic selective product ion analysis was performed at m/z 206 for 14,15‐DHET. These same product ions were also used for the deuterated internal standards. Quantifications were performed using the ratio of the area of the DHET peaks compared to the area of the corresponding deuterated DHET peaks (Capdevila et al., 1991; Nakagawa et al., 2006).
Determination of cerebral oxidative stress
To characterize the effect of IGF‐1 deficiency on cerebral oxidative stress, the marker for peroxynitrate action, 3‐nitrotyrosine (3‐NT), was assessed in homogenates of cortical samples using OxiSelect Protein Nitrotyrosine ELISA Kits (Cell Biolabs, San Diego, CA, U.S.A.) following the manufacturer's guidelines, as previously described (Toth et al., 2013b).
Statistical analysis
Statistical analysis was carried out by unpaired t‐test or two‐way ANOVA for repeated measures followed by Bonferroni multiple comparison test, as appropriate, using Prism 5.0 for Windows (Graphpad Software, La Jolla, CA, USA). A P value <0.05 was considered statistically significant. Data are expressed as mean ± S.E.M.
Funding
No funding information provided.
Conflict of interest
The authors declare no competing financial interests.
Author contribution
PT, ST, NMA, MNVA, GLM, WES, AC, and ZU designed research and revised the manuscript; PT, ST, NMA, MNVA, GLM, AM, AC, and ZU performed experiments; PT, ST, AC, GLM, NMA, and ZU analyzed data; and PT, AC, and ZU wrote the manuscript.
Acknowledgment
This work was supported by grants from the American Heart Association (to PT, AC, ZT, ST, and ZU), the National Center for Complementary and Alternative Medicine (R01‐AT006526 to ZU), the National Institute on Aging (R01‐AG047879 to AC, R01‐AG038747 to WES, R01‐NS056218 to AC and WES), the Arkansas Claude Pepper Older Americans Independence Center at University of Arkansas Medical Center (to ZU; P30 AG028718), the Oklahoma Center for the Advancement of Science and Technology (to AC, ZU, WES), and the Bolyai János Research Scholarship of the Hungarian Academy of Sciences (BO/00327/14/5 to EF).
References
- Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA (2010) Glial and neuronal control of brain blood flow. Nature 468, 232–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey‐Downs LC, Mitschelen M, Sosnowska D, Toth P, Pinto JT, Ballabh P, Valcarcel‐Ares MN, Farley J, Koller A, Henthorn JC, Bass C, Sonntag WE, Ungvari Z, Csiszar A (2012) Liver‐specific knockdown of IGF‐1 decreases vascular oxidative stress resistance by impairing the Nrf2‐dependent antioxidant response: a novel model of vascular aging. J. Gerontol. Biol. Med. Sci. 67, 313–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berenbaum F, Thomas G, Poiraudeau S, Bereziat G, Corvol MT, Masliah J (1994) Insulin‐like growth factors counteract the effect of interleukin 1 beta on type II phospholipase A2 expression and arachidonic acid release by rabbit articular chondrocytes. FEBS Lett. 340, 51–55. [DOI] [PubMed] [Google Scholar]
- Capdevila JH, Dishman E, Karara A, Falck JR (1991) Cytochrome P450 arachidonic acid epoxygenase: stereochemical characterization of epoxyeicosatrienoic acids. Methods Enzymol. 206, 441–453. [DOI] [PubMed] [Google Scholar]
- Chen BR, Kozberg MG, Bouchard MB, Shaik MA, Hillman EM (2014) A critical role for the vascular endothelium in functional neurovascular coupling in the brain. J. Am. Heart Assoc. 3, e000787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csiszar A, Ungvari Z, Edwards JG, Kaminski PM, Wolin MS, Koller A, Kaley G (2002) Aging‐induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ. Res. 90, 1159–1166. [DOI] [PubMed] [Google Scholar]
- Csiszar A, Labinskyy N, Perez V, Recchia FA, Podlutsky A, Mukhopadhyay P, Losonczy G, Pacher P, Austad SN, Bartke A, Ungvari Z (2008) Endothelial function and vascular oxidative stress in long‐lived GH/IGF‐deficient Ames dwarf mice. Am. J. Physiol. Heart Circ. Physiol. 295, H1882–H1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csiszar A, Tucsek Z, Toth P, Sosnowska D, Gautam T, Koller A, Deak F, Sonntag WE, Ungvari Z (2013) Synergistic effects of hypertension and aging on cognitive function and hippocampal expression of genes involved in beta‐amyloid generation and Alzheimer's disease. Am. J. Physiol. Heart Circ. Physiol. 305, H1120–H1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damke H, Bouterfa H, Braulke T (1994) Effects of insulin‐like growth factor II on the generation of inositol trisphosphate, diacylglycerol and cAMP in human fibroblasts. Mol. Cell. Endocrinol. 99, R25–R29. [DOI] [PubMed] [Google Scholar]
- Derenne A, Brown‐Borg H, Feltman K, Corbett G, Lackman S (2011) Acquisition of steady‐state operant behavior in long‐living Ames Dwarf mice. Physiol. Behav. 104, 1048–1052. [DOI] [PubMed] [Google Scholar]
- Fabiani M, Gordon BA, Maclin EL, Pearson MA, Brumback‐Peltz CR, Low KA, McAuley E, Sutton BP, Kramer AF, Gratton G (2013) Neurovascular coupling in normal aging: a combined optical, ERP and fMRI study. NeuroImage 85, 592–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonnum F, Johnsen A, Hassel B (1997) Use of fluorocitrate and fluoroacetate in the study of brain metabolism. Glia 21, 106–113. [PubMed] [Google Scholar]
- Girouard H, Park L, Anrather J, Zhou P, Iadecola C (2007) Cerebrovascular nitrosative stress mediates neurovascular and endothelial dysfunction induced by angiotensin II. Arterioscler. Thromb. Vasc. Biol. 27, 303–309. [DOI] [PubMed] [Google Scholar]
- Gong Z, Kennedy O, Sun H, Wu Y, Williams GA, Klein L, Cardoso L, Matheny RW Jr, Hubbard GB, Ikeno Y, Farrar RP, Schaffler MB, Adamo ML, Muzumdar RH, Yakar S (2014) Reductions in serum IGF‐1 during aging impair health span. Aging Cell 13, 408–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelick PB, Scuteri A, Black SE, Decarli C, Greenberg SM, Iadecola C, Launer LJ, Laurent S, Lopez OL, Nyenhuis D, Petersen RC, Schneider JA, Tzourio C, Arnett DK, Bennett DA, Chui HC, Higashida RT, Lindquist R, Nilsson PM, Roman GC, Sellke FW, Seshadri S, American Heart Association Stroke Council CoE, Prevention CoCNCoCR, Intervention, Council on Cardiovascular S, Anesthesia (2011) Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the american heart association/american stroke association. Stroke 42, 2672–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland BA, O'Farrell FM, Buchan AM, Lauritzen M, Attwell D (2014) Capillary pericytes regulate cerebral blood flow in health and disease. Nature 508, 55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Park L, Capone C (2009) Threats to the mind: aging, amyloid, and hypertension. Stroke 40, S40–S44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitaura H, Uozumi N, Tohmi M, Yamazaki M, Sakimura K, Kudoh M, Shimizu T, Shibuki K (2007) Roles of nitric oxide as a vasodilator in neurovascular coupling of mouse somatosensory cortex. Neurosci. Res. 59, 160–171. [DOI] [PubMed] [Google Scholar]
- Lecrux C, Kocharyan A, Sandoe CH, Tong XK, Hamel E (2012) Pyramidal cells and cytochrome P450 epoxygenase products in the neurovascular coupling response to basal forebrain cholinergic input. J. Cereb. Blood Flow Metab. 32, 896–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E, Kim DH, Cai M, Lee S, Kim Y, Lim E, Hoon Ryu J, Unterman TG, Park S (2011) Hippocampus‐dependent spatial learning and memory are impaired in growth hormone‐deficient spontaneous dwarf rats. Endocr. J. 58, 257–267. [DOI] [PubMed] [Google Scholar]
- Lind BL, Brazhe AR, Jessen SB, Tan FC, Lauritzen MJ (2013) Rapid stimulus‐evoked astrocyte Ca2+ elevations and hemodynamic responses in mouse somatosensory cortex in vivo. Proc. Natl Acad. Sci. USA 110, E4678–E4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Li C, Falck JR, Roman RJ, Harder DR, Koehler RC (2008) Interaction of nitric oxide, 20‐HETE, and EETs during functional hyperemia in whisker barrel cortex. Am. J. Physiol. Heart Circ. Physiol. 295, H619–H631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longden T, Nelson M (2011) Recruitment of the vascular endothelium into neurovascular coupling. Proc. Br. Pharmacol. Soc. 9, 062P http://www.pA2online.org/abstracts/Vol9Issue3abst062P.pdf. [Google Scholar]
- Ma J, Ayata C, Huang PL, Fishman MC, Moskowitz MA (1996) Regional cerebral blood flow response to vibrissal stimulation in mice lacking type I NOS gene expression. Am. J. Physiol. 270, H1085–H1090. [DOI] [PubMed] [Google Scholar]
- Nakagawa K, Holla VR, Wei Y, Wang WH, Gatica A, Wei S, Mei S, Miller CM, Cha DR, Price E Jr, Zent R, Pozzi A, Breyer MD, Guan Y, Falck JR, Waterman MR, Capdevila JH (2006) Salt‐sensitive hypertension is associated with dysfunctional Cyp4a10 gene and kidney epithelial sodium channel. J. Clin. Invest. 116, 1696–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieves‐Martinez E, Sonntag WE, Wilson A, Donahue A, Molina DP, Brunso‐Bechtold J, Nicolle MM (2010) Early‐onset GH deficiency results in spatial memory impairment in mid‐life and is prevented by GH supplementation. J. Endocrinol. 204, 31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishijima T, Piriz J, Duflot S, Fernandez AM, Gaitan G, Gomez‐Pinedo U, Verdugo JM, Leroy F, Soya H, Nunez A, Torres‐Aleman I (2010) Neuronal activity drives localized blood‐brain‐barrier transport of serum insulin‐like growth factor‐I into the CNS. Neuron 67, 834–846. [DOI] [PubMed] [Google Scholar]
- Onifer SM, Quintero JE, Gerhardt GA (2012) Cutaneous and electrically evoked glutamate signaling in the adult rat somatosensory system. J. Neurosci. Methods 208, 146–154. [DOI] [PubMed] [Google Scholar]
- Pacher P, Beckman JS, Liaudet L (2007) Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 87, 315–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park L, Anrather J, Girouard H, Zhou P, Iadecola C (2007) Nox2‐derived reactive oxygen species mediate neurovascular dysregulation in the aging mouse brain. J. Cereb. Blood Flow Metab. 27, 1908–1918. [DOI] [PubMed] [Google Scholar]
- Park L, Gallo EF, Anrather J, Wang G, Norris EH, Paul J, Strickland S, Iadecola C (2008) Key role of tissue plasminogen activator in neurovascular coupling. Proc. Natl Acad. Sci. USA 105, 1073–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X, Carhuapoma JR, Bhardwaj A, Alkayed NJ, Falck JR, Harder DR, Traystman RJ, Koehler RC (2002) Suppression of cortical functional hyperemia to vibrissal stimulation in the rat by epoxygenase inhibitors. Am. J. Physiol. Heart Circ. Physiol. 283, H2029–H2037. [DOI] [PubMed] [Google Scholar]
- Petzold GC, Murthy VN (2011) Role of astrocytes in neurovascular coupling. Neuron 71, 782–797. [DOI] [PubMed] [Google Scholar]
- Sarnyai Z, Sibille EL, Pavlides C, Fenster RJ, McEwen BS, Toth M (2000) Impaired hippocampal‐dependent learning and functional abnormalities in the hippocampus in mice lacking serotonin(1A) receptors. Proc. Natl Acad. Sci. USA 97, 14731–14736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma HS, Nyberg F, Gordh T, Alm P, Westman J (1997) Topical application of insulin like growth factor‐1 reduces edema and upregulation of neuronal nitric oxide synthase following trauma to the rat spinal cord. Acta Neurochir. Suppl. 70, 130–133. [DOI] [PubMed] [Google Scholar]
- Sharma S, Haselton J, Rakoczy S, Branshaw S, Brown‐Borg HM (2010) Spatial memory is enhanced in long‐living Ames dwarf mice and maintained following kainic acid induced neurodegeneration. Mech. Ageing Dev. 131, 422–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Liu X, Gebremedhin D, Falck JR, Harder DR, Koehler RC (2008) Interaction of mechanisms involving epoxyeicosatrienoic acids, adenosine receptors, and metabotropic glutamate receptors in neurovascular coupling in rat whisker barrel cortex. J. Cereb. Blood Flow Metab. 28, 111–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonntag WE, Ramsey M, Carter CS (2005) Growth hormone and insulin‐like growth factor‐1 (IGF‐1) and their influence on cognitive aging. Ageing Res. Rev. 4, 195–212. [DOI] [PubMed] [Google Scholar]
- Sonntag WE, Deak F, Ashpole N, Toth P, Csiszar A, Freeman W, Ungvari Z (2013) Insulin‐like growth factor‐1 in CNS and cerebrovascular aging. Front. Aging Neurosci. 5, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorond FA, Hurwitz S, Salat DH, Greve DN, Fisher ND (2013) Neurovascular coupling, cerebral white matter integrity, and response to cocoa in older people. Neurology 81, 904–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanova I, Stephan T, Becker‐Bense S, Dera T, Brandt T, Dieterich M (2013) Age‐related changes of blood‐oxygen‐level‐dependent signal dynamics during optokinetic stimulation. Neurobiol. Aging 34, 2277–2286. [DOI] [PubMed] [Google Scholar]
- Stobart JL, Lu L, Anderson HD, Mori H, Anderson CM (2013) Astrocyte‐induced cortical vasodilation is mediated by D‐serine and endothelial nitric oxide synthase. Proc. Natl Acad. Sci. USA 110, 3149–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahara K, Grollman EF, Saji M, Kohn LD (1991) Regulation of prostaglandin synthesis by thyrotropin, insulin or insulin‐like growth factor‐I, and serum in FRTL‐5 rat thyroid cells. J. Biol. Chem. 266, 440–448. [PubMed] [Google Scholar]
- Takano T, Tian GF, Peng W, Lou N, Libionka W, Han X, Nedergaard M (2006) Astrocyte‐mediated control of cerebral blood flow. Nat. Neurosci. 9, 260–267. [DOI] [PubMed] [Google Scholar]
- Topcuoglu MA, Aydin H, Saka E (2009) Occipital cortex activation studied with simultaneous recordings of functional transcranial Doppler ultrasound (fTCD) and visual evoked potential (VEP) in cognitively normal human subjects: effect of healthy aging. Neurosci. Lett. 452, 17–22. [DOI] [PubMed] [Google Scholar]
- Toth P, Csiszar A, Tucsek Z, Sosnowska D, Gautam T, Koller A, Laniado Schwartzman M, Sonntag WE, Ungvari ZI (2013a) Role of 20‐HETE,TRP channels & BKCa in dysregulation of pressure‐induced Ca2+ signaling and myogenic constriction of cerebral arteries in aged hypertensive mice. Am. J. Physiol. Heart Circ. Physiol. 305, 698–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth P, Tucsek Z, Sosnowska D, Gautam T, Mitschelen M, Tarantini S, Deak F, Koller A, Sonntag WE, Csiszar A, Ungvari Z (2013b) Age‐related autoregulatory dysfunction and cerebromicrovascular injury in mice with angiotensin II‐induced hypertension. J. Cereb. Blood Flow Metab. 33, 1732–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth P, Tarantini S, Tucsek Z, Ashpole NM, Sosnowska D, Gautam T, Ballabh P, Koller A, Sonntag WE, Csiszar A, Ungvari ZI (2014a) Resveratrol treatment rescues neurovascular coupling in aged mice: role of improved cerebromicrovascular endothelial function and down‐regulation of NADPH oxidase. Am. J. Physiol. Heart Circ. Physiol. 306, H299–H308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth P, Tucsek Z, Tarantini S, Sosnowska D, Gautam T, Mitschelen M, Koller A, Sonntag WE, Csiszar A, Ungvari Z (2014b) IGF‐1 deficiency impairs cerebral myogenic autoregulation in hypertensive mice. J. Cereb. Blood Flow Metab. 34, 1887–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trejo JL, Piriz J, Llorens‐Martin MV, Fernandez AM, Bolos M, LeRoith D, Nunez A, Torres‐Aleman I (2007) Central actions of liver‐derived insulin‐like growth factor I underlying its pro‐cognitive effects. Mol. Psychiatry 12, 1118–1128. [DOI] [PubMed] [Google Scholar]
- Ungvari Z, Csiszar A (2012) The emerging role of IGF‐1 deficiency in cardiovascular aging: recent advances. J. Gerontol. A Biol. Sci. Med. Sci. 67, 599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaletel M, Strucl M, Pretnar‐Oblak J, Zvan B (2005) Age‐related changes in the relationship between visual evoked potentials and visually evoked cerebral blood flow velocity response. Funct. Neurol. 20, 115–120. [PubMed] [Google Scholar]
- Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann KA, Pozzan T, Carmignoto G (2003) Neuron‐to‐astrocyte signaling is central to the dynamic control of brain microcirculation. Nat. Neurosci. 6, 43–50. [DOI] [PubMed] [Google Scholar]