

Summary
The mutated form of the Ca2+ channel CALHM1 (Ca2+ homeostasis modulator 1), P86L‐CALHM1, has been correlated with early onset of Alzheimer's disease (AD). P86L‐CALHM1 increases production of amyloid beta (Aβ) upon extracellular Ca2+ removal and its subsequent addback. The aim of this study was to investigate the effect of the overexpression of CALHM1 and P86L‐CALHM, upon Aβ treatment, on the following: (i) the intracellular Ca2+ signal pathway; (ii) cell survival proteins ERK1/2 and Ca2+/cAMP response element binding (CREB); and (iii) cell vulnerability after treatment with Aβ. Using aequorins to measure the effect of nuclear Ca2+ concentrations ([Ca2+]n) and cytosolic Ca2+ concentrations ([Ca2+]c) on Ca2+ entry conditions, we observed that baseline [Ca2+]n was higher in CALHM1 and P86L‐CALHM1 cells than in control cells. Moreover, exposure to Aβ affected [Ca2+]c levels in HeLa cells overexpressing CALHM1 and P86L‐CALHM1 compared with control cells. Treatment with Aβ elicited a significant decrease in the cell survival proteins p‐ERK and p‐CREB, an increase in the activity of caspases 3 and 7, and more frequent cell death by inducing early apoptosis in P86L‐CALHM1‐overexpressing cells than in CALHM1 or control cells. These results suggest that in the presence of Aβ, P86L‐CALHM1 shifts the balance between neurodegeneration and neuronal survival toward the stimulation of pro‐cytotoxic pathways, thus potentially contributing to its deleterious effects in AD.
Keywords: Alzheimer's disease, Ca2+ channel CALHM1, CREB, Ca2+ homeostasis, caspases, early apoptosis
Introduction
Alzheimer's disease (AD) is clinically characterized by progressive cognitive impairment that is believed to result from synaptic dysfunction and neurodegeneration initiated by the aggregated form of amyloid beta (Aβ) peptide (Hardy & Selkoe, 2002). Accumulated evidence suggests that AD is also linked to an imbalance of intracellular Ca2+ homeostasis (Bezprozvanny & Mattson, 2008; Green & LaFerla, 2008; Marambaud et al., 2009; Fernandez‐Morales et al., 2012), because Ca2+ plays a critical role in maintaining cell survival; for example, a mild elevation of [Ca2+]c promotes neuronal survival and plasticity, whereas more pronounced elevations can cause neurotoxicity (Berridge et al., 1998; Cano‐Abad et al., 2001). Thus, alterations in Ca2+ homeostatic mechanisms associated with aging, mutations in amyloid precursor protein (APP) and presenilins, and dysfunctional Ca2+ fluxes at the endoplasmic reticulum (ER) can promote neuronal cell death (Bezprozvanny & Mattson, 2008).
Although data from the literature indicate that neuronal death in AD is related to the action of Aβ on intracellular Ca2+ dyshomeostasis, little is known about the role of the novel Ca2+ channel, calcium homeostasis modulator 1 (CALHM1), in the disease. CALHM1 is expressed in all brain regions and neuronal cells, at the ER, and in the plasma membrane. CALHM1 generates Ca2+‐selective cation currents in the plasma membrane. It has also been shown to form a novel Ca2+‐permeable ion channel, whose gating is allosterically regulated by both membrane voltage and extracellular Ca2+ concentration; in addition, CALHM1 is insensitive to classic selective blockers of voltage‐gated Ca2+ channels, although it is inhibited by nonselective and inorganic Ca2+ channel blockers such as Co2+ (Dreses‐Werringloer et al., 2008; Moreno‐Ortega et al., 2010; Ma et al., 2012). But recently we described that CAHM1 is blocked by CGP37157 (Moreno‐Ortega et al., 2015).
A polymorphism of CALHM1, P86L‐CALHM1, which results in a proline to leucine substitution at codon 86, has been associated with early onset of sporadic AD (Dreses‐Werringloer et al., 2008); however, this association remains controversial. Thus, while some studies have shown a significant correlation (Boada et al., 2010; Cui et al., 2010), others have failed to find such an association (Bertram et al., 2008). While it is accepted that P86L‐CALHM1 is not a genetic risk factor for the development of AD, a meta‐analysis has shown that this polymorphism modulates the age of disease onset (Lambert et al., 2010). Transient expression of the P86L‐CALHM1 channel promotes accumulation of Aβ by altering membrane permeability to Ca2+ and, consequently, promotes an increase in [Ca2+]c (Dreses‐Werringloer et al., 2008). However, evidence implicating a role for Aβ‐induced disruption of Ca2+ homeostasis linked to CALHM1 or P86L‐CALHM1 and the activation of cell death signaling pathways has not been reported.
Selective neuronal vulnerability is a feature of a number of neurodegenerative diseases, but the processes that target specific neurons for death while allowing others to remain healthy are unclear. The differential activation of an internal death program in vulnerable neurons has been proposed as a mechanism to explain the selective death of neurons (Schreiber & Baudry, 1995). However, it is equally likely that specific neuronal populations contain an intrinsic survival mechanism. The presence and/or activity of such a pathway in various cell types could partly explain their varying sensitivities to detrimental brain insults. Several studies have recently implicated the transcription factor c‐AMP response element‐binding protein (CREB) as a possible regulator of a general survival program in neurons. CREB can be activated by various kinases in response to electrical activity, neurotransmitters, hormones, and neurotrophins, thus promoting the expression of many genes that contain cAMP response elements (Finkbeiner et al., 1997; Hardingham & Bading, 1998). CREB also plays a central role in memory formation (West et al., 2001). The transcriptional activation of CREB is crucially dependent on phosphorylation of Ser133 by kinases such as Ca2+/calmodulin kinase (CaMK), ras/mitogen‐activated protein kinase (MAPK), ERK1/2 (Wu et al., 2001), and protein kinases A and C (Hardingham et al., 1999). Extracellular signal‐regulated kinases (ERKs) are key genes in activating survival pathways (Roskoski, 2012), and their transient activation plays an important role in memory‐related processes (Costa & Silva, 2002).
As Ca2+ dyshomeostasis is found in AD and P86L‐CALHM1 is considered a risk factor for AD, we investigated how native CALHM1 and P86L‐CALHM1 could contribute to Ca2+ homeostasis, survival signaling pathways (namely, ERK and the transcription factor CREB), and cell survival at baseline or after treatment with Aβ. We used transfected HeLa cells with the empty vector (control) and cells transfected with vectors including CALHM1 and P86L‐CALHM1 to study the kinetics of the changes of [Ca2+]c and [Ca2+]n generated by reintroduction of Ca2+ and treatment with Aβ. We also analyzed ERK, CREB activation, and apoptosis pathways upon exposure to Aβ. Our results indicate that P86L‐CALHM1 could contribute to neuronal vulnerability by affecting cytosolic and nuclear Ca2+ homeostatic mechanisms and survival signaling pathways.
Results
Effect of CALHM1 and P86L‐CALHM1 overexpression on the nuclear concentration of Ca2+
Several authors have investigated the participation of CALHM1 expression in different Ca2+ compartments such as cytosol (Dreses‐Werringloer et al., 2008; Moreno‐Ortega et al., 2010; Ma et al., 2012), mitochondria ([Ca2+]mt) (Moreno‐Ortega et al., 2010), and ER ([Ca2+]ER) (Gallego‐Sandin et al., 2011). However, the regulation of nuclear Ca2+ homeostasis by CALHM1 and P86L‐CALHM1 has not yet been described.
Because CALHM1 is anchored to the ER membrane (Dreses‐Werringloer et al., 2008) and the ER membrane constitutes the nuclear envelope, we hypothesized that upon CALHM1 opening, and the channel could be releasing Ca2+ from the ER into the nucleus. Furthermore, variations in the [Ca2+]c can promote changes in [Ca2+]n that could regulate cellular functions ranging from proliferation to cell death (Alonso et al., 2011). Therefore, we used nuclear‐targeted aequorin (nu_AEQ) to explore whether CALHM1 or P86L‐CALHM1 overexpression could promote changes in [Ca2+]n upon reintroduction of Ca2+.
Cells transfected with nu_AEQ were initially perfused with a 0 Ca2+/EGTA solution for 2 min. This solution was then switched to another one containing 1 mm Ca2+. Figure 1A shows that the [Ca2+]n was stable at around 0.38 μm in control cells in 0 Ca2+/EGTA; in CALHM1 and P86L‐CALHM1 cells, [Ca2+]n was quite stable at 1.83 and 1.5 μm, respectively. Upon reintroduction of 1 mm Ca2+, [Ca2+]n rose to a peak at 0.97 ± 0.09 μm and then decayed to near baseline values, indicating inactivation of the constitutive capacitative Ca2+ entry channel of the control HeLa cells. The kinetics of the transient [Ca2+]n in CALHM1‐overexpressing cells were considerably different from those of the control; the activation rate was significantly slower and peaked at 2.79 ± 0.13 μm before slowly decaying to a stable plateau at around 1.5 μm. In P86L‐CALHM1‐overexpressing cells, the transient [Ca2+]n developed much more slowly, reaching a peak at 2.32 ± 0.16 μm and stabilizing as a plateau, with little decay.
Figure 1.
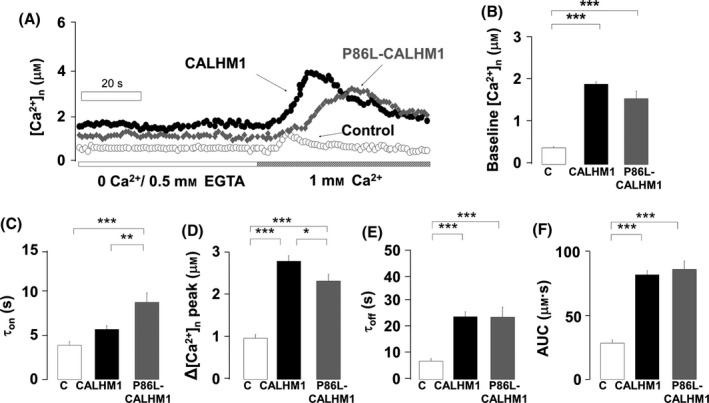
Kinetics of the nuclear Ca2+ transients ([Ca2+]n) measured using aequorin targeting the nucleus. (A) Typical traces of the time course of [Ca2+]n elevation elicited during the time period is indicated. Ca2+ was reintroduced as indicated on the bottom horizontal bar. Data are represented as follows: (B) baseline [Ca2+]n, (C) time constant for activation (τon), (D) peak [Ca2+]n transient amplitude, (E) time constant for inactivation (τoff), and (F) area under the curve (AUC) of the transients in cells overexpressing the empty vector (C), CALHM1, or P86L‐CALHM1. Bar graphs of B–F were computed with pooled data from 20 experiments (control), 34 experiments (CALHM1), and 29 experiments (P86L‐CALHM1) performed with cells from 10 different cultures and according to protocols such as those shown in A. Data are expressed as mean ± SEM. anova post hoc Bonferroni, *P < 0.05; **P < 0.01; ***P < 0.001.
Quantitative averaged data from 20, 34, and 29 experiments for control, CALHM1, and P86L‐CALHM1 cells, respectively, show a 4.86‐fold increase over baseline [Ca2+]n in CALHM1 cells and 3.98‐fold increase for P86L‐CALHM1 cells, with respect to the control cells (Fig. 1B). In addition, the kinetics of the [Ca2+]n transients differed between the three cell types. For instance, the time constant for the rate of the transient rise (τon) was 1.47‐fold and 2.27‐fold higher in CALHM1 and P86L‐CALHM1 cells, respectively, than in control cells (Fig. 1C), suggesting slower activation of the [Ca2+]n signal. Moreover, the peak heights were 2.86‐fold and 2.34‐fold greater (Fig. 1D). The rate of signal decay was considerably slower in CALHM1 cells (τoff 3.46‐fold higher) and in P86L‐CALHM1 cells (τoff 3.41‐fold higher), with respect to control cells (Fig. 1E). Finally, we calculated the area under the curve (AUC) of each transient as a reflection of the total [Ca2+]n, considering the net rise in [Ca2+]n from baseline for each cell type: we observed that it was 2.87‐fold higher in CALHM1 and 3.03‐fold higher in P86L‐CALHM1 cells than in controls (Fig. 1F).
CALHM1 and P86L‐CALHM1 overexpression and Ca2+ release at nucleoplasma regions
Activation of inositol 1,4,5‐trisphosphate receptors (InsP3R) is a key mechanism of Ca2+ entry into the nucleus. As the ER membrane forms part of the nuclear envelope and CALHM1 is anchored to the ER, we explored whether CALHM1 or P86L‐CALHM1 overexpression could affect the kinetics of the [Ca2+]n transients elicited by indirect InsP3R activation by histamine. We first perfused cells with a Ca2+ solution (1 mm) for 2 min and replaced this solution with another one containing 100 μm of histamine for 15 s. Figure 2A shows three superimposed typical traces on the [Ca2+]n variations elicited by histamine‐InsP3R stimulation. Once more, baseline [Ca2+]n was higher in CALHM1 and P86L‐CALHM1 cells than in control (Fig. 2B). As far as the histamine‐elicited transients was concerned, no significant changes were observed in the τon, peak [Ca2+]n, τoff, or AUC between the three cell types (Figs. 2C–F). One interpretation of these results could be that slow inactivation of InsP3R channels occurs upon Ca2+ leak through CALHM1 and P86L‐CALHM1, which would in turn slower Ca2+ release into the nucleus owing to InsP3R inactivation by Ca2+.
Figure 2.
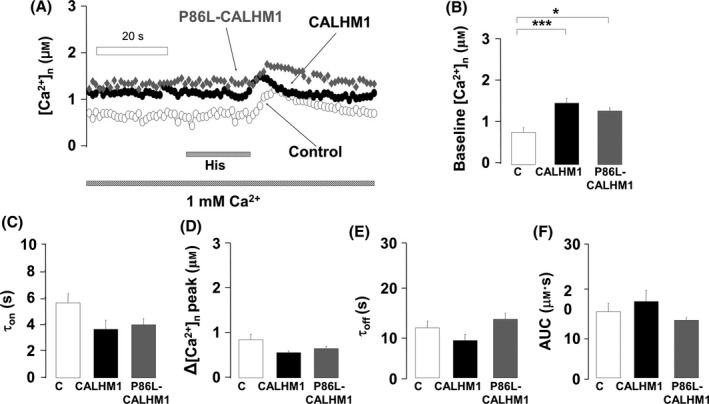
Kinetics of the [Ca2+]n transients elicited by histamine in control, CALHM1, and P86L‐CALHM1 cells. (A) Typical traces of the time course of [Ca2+]n elicited by 100 μm histamine, administered as indicated on the bottom bar. Data are analyzed as follows: (B) baseline [Ca2+]n, (C) time constant for activation (τon), (D) peak [Ca2+]n transient amplitude, (E) time constant for inactivation (τoff), and (F) area under the curve (AUC) of the transients in cells expressing the empty vector (C), CALHM1, or P86L‐CALHM1. Bar graphs of B–F were computed with pooled data from 16 experiments performed with cells from seven different cultures and according to the protocols shown in the A. Data are expressed as mean ± SEM. anova post hoc Bonferroni, *P < 0.05; ***P < 0.01.
Effects of Aβ on cytosolic Ca2+ signaling in cells overexpressing CALHM1 and P86L‐CALHM1
Previous results gave rise to the hypothesis that CALHM1 could behave as a leak channel regulating changes in the kinetics of nuclear [Ca2+]n changes and, in so doing regulates Aβ/APP ratio levels in a Ca2+‐dependent manner (Dreses‐Werringloer et al., 2008). However, the influence of extracellular Aβ on Ca2+ homeostasis in CALHM1‐ and P86L‐CALHM1‐overexpressing cells has not been investigated to date. To address this issue, we performed experiments to measure changes in [Ca2+]c occurring during acute Aβ treatment. To reveal possible changes in the rate of Ca2+ entry through CALHM1 or P86L‐CALHM1 channels, [Ca2+]c was measured under the channel activating form, that is removal of extracellular Ca2+ (0 Ca2+/EGTA) and its subsequent addback (1 mm Ca2+) in the absence or presence of Aβ.
In the absence of Aβ, the addback of Ca2+ elicited a significant increase in the [Ca2+]c, reaching 4.34 and 1.25 μm in CALHM1 and P86L‐CALHM1, respectively (Fig. 3A). In the presence of Aβ25‐35 (10 μm) and 1 mm extracellular Ca2+, slight oscillations in baseline [Ca2+]c in both CALHM1 and P86L‐CALHM1 cells were detected (data not shown). Extracellular Ca2+ was then withdrawn and this protocol repeated in the presence of Aβ; Ca2+ entry was significantly reduced in CALHM1‐ or P86L‐CALHM1‐overexpressing cells (Fig. 3B). Pooled data show that in CALHM1 cells, peak [Ca2+]c was reduced by 40.32%, from 4.34 μm (no Aβ) to 1.75 μm (plus Aβ), whereas in P86L‐CALHM1 cells, the peak was reduced by 44% from 1.25 to 0.55 μm. In control cells, [Ca2+]c changes were mild and similar in the presence or absence of Aβ (0.67 and 0.3 μm, respectively) (Fig. 3C). These modifications seem to be specific for the toxic form of Aβ since the scramble sequence of βA25–35 did not afford significant modifications in the [Ca2+]c (data not shown).
Figure 3.
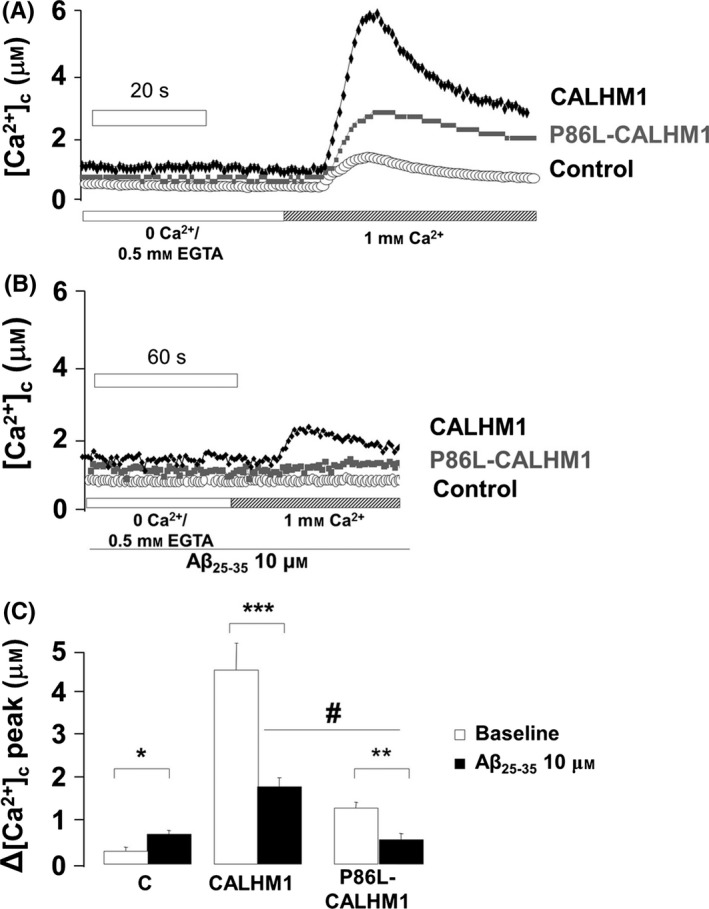
Signaling of cytosolic Ca2+ concentrations ([Ca2+]c) elicited by perfusion with Aβ in HeLa cells overexpressing control, CALHM1, and P86L‐CALHM1. (A) Example of typical traces of [Ca2+]c elicited by an addback Ca2+ protocol in HeLa cells transfected with the empty vector (control), CALHM1, or P86L‐CALHM1. (B) Experiment performed as in A, but in cells continuously perfused with amyloid β25–35 peptide (Aβ25–35) at 10 μm. (C) Δ[Ca2+]c of the peak of the transient amplitude of the Ca2+ entry elicited by the addback protocol. Pooled data are expressed as mean ± SEM from at least seven experiments performed with three different batches of cells. anova post hoc Dunnet, *P < 0.05; **P < 0.01; ***P < 0.001 compared with non‐Aβ control. One‐tail t‐test, # P < 0.05 compared CALHM1 versus P86L‐CALHM1.
Vulnerability of CALHM1‐ and P86L‐CALHM1‐overexpressing cells to different cytotoxic stimuli
No significant baseline cell death was observed in HeLa cells transiently expressing the empty vector (control), CALHM1, or P86L‐CALHM1 (Fig. 4A). When both types were incubated with oligomers of Aβ1–42, 5 μm for 24 h, only P86L‐CALHM1‐overexpressing cells showed significant cell toxicity (24% of cell death) compared with cells overexpressing the empty vector or the wild‐type channel (Fig. 4B).
Figure 4.
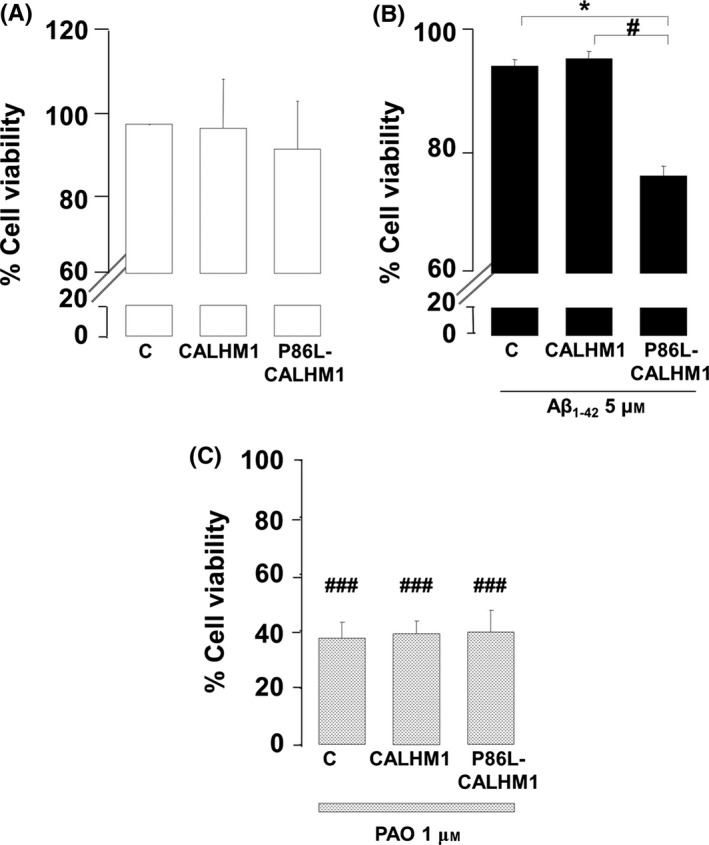
Cell vulnerability upon treatment with Aβ1–42 and phenylarsine oxide. The MTT assay was performed to test cell viability in control (C), CALHM1‐, or P86L‐CALHM1‐overexpressing HeLa cells. (A) Baseline viability. (B) Viability after 24 h of treatment with 5 μm of a mixture of protofibrils and oligomers of 1–42 of Aβ (Aβ1–42). (C) Viability after 24 h of treatment with 1 μm phenylarsine oxide (PAO). Triplicate measurements were obtained from four different cultures. Data are expressed as mean ± SEM. anova post hoc Bonferroni, *P < 0.05 compared with control cells treated with Aβ. anova post hoc Dunnet, # P < 0.01; ### P < 0.001, compared with untreated cells.
We also evaluated vulnerability to oxidative stress stimuli using phenylarsine oxide (PAO), which causes oxidative stress via a mitochondria‐dependent mechanism (Vay et al., 2009). PAO reduced cell viability in all three cell types (Fig. 4C). Therefore, cells expressing P86L‐CALHM1 did not show higher vulnerability to oxidative stress, in contrast to the observations with Aβ treatment.
Activation of apoptosis in CALHM1‐ and P86L‐CALHM1‐overexpressing cells upon Aβ exposure
To clarify the mechanism involved in the cell death observed in Fig 4B, the next reasonable step was to explore whether treatment with Aβ induced apoptosis in cells overexpressing CALHM1 or P86L‐CALHM1. To this end, we explored the different apoptosis stages in control, CALHM1, and P86L‐CALHM1 cells upon exposure to Aβ1–42 (5 μm) for 24 h. We observed a clear tendency toward early triggering of apoptosis only in cells overexpressing the mutated form P86L‐CALHM1 at 3 and 6 h (data not shown). Thus, we incubated the cells overexpressing CALHM1 and P86L‐CALHM1 for a longer time period to determine how long it would the apoptosis stage upon treatment with Aβ. After 24 h, only P86L‐CALHM1‐overexpressing cells activated the early apoptosis pathway (Fig. 5B). These results were independent of cell type, because the neuronal hippocampal cell line HT‐22 overexpressing CALHM1 and P86L‐CALHM1 were also vulnerable when treated with Aβ25–25 (50 μm) for 24 h. (Supplemental Results and figures).
Figure 5.
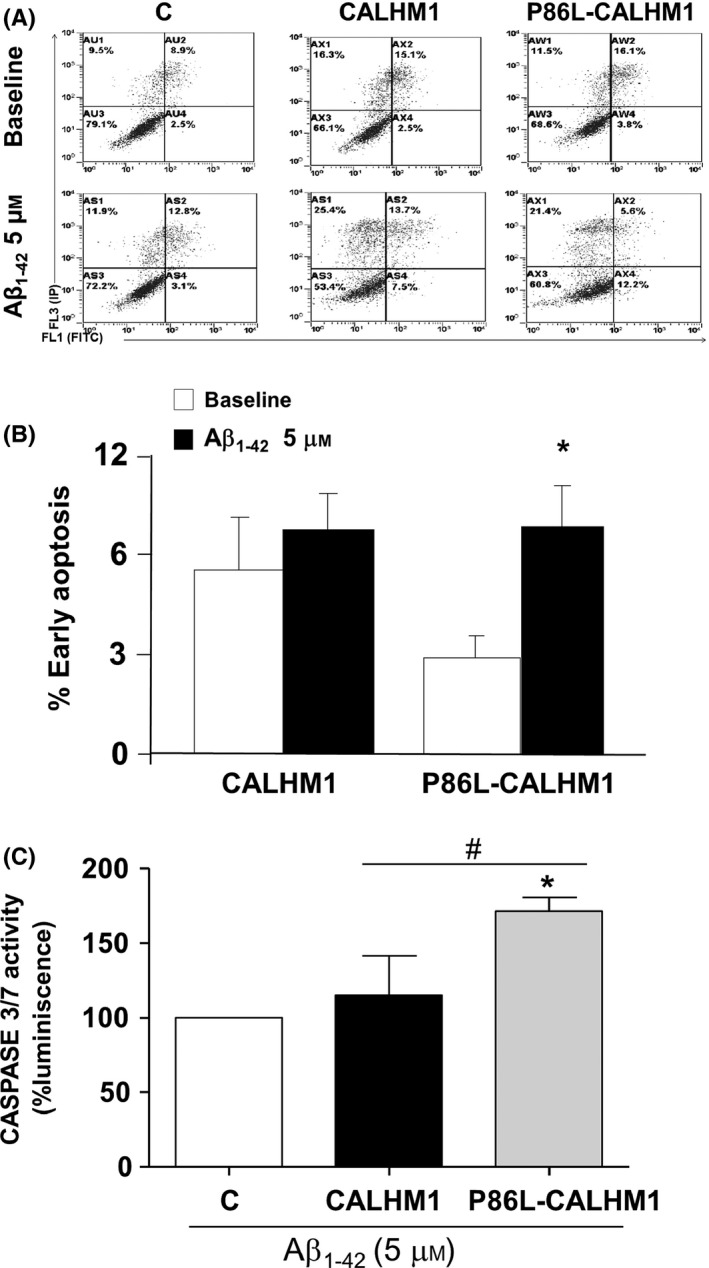
Apoptosis triggered by treatment with Aβ1–42 in CALHM1 and P86L‐CALHM1 cells. Determination of the different phases of apoptosis induced by 5 μm Aβ1–42 for 24 h in cells transfected with empty vector, CALHM1, and P86L‐CALHM1. (A), a typical flow cytometry; the upper pictures correspond to nontreated control, CALHM1, and P86L‐CALHM1 cells, and the bottom pictures to cells treated with 5 μm of a mixture of protofibrils and oligomers of Aβ1–42 for 24 h. (B) shows early apoptotic cells without treatment (white bars) or upon treatment with Aβ (black bars). Pooled data are expressed as mean ± SEM from at least 12 experiments performed with seven different cultures. One‐way t‐test, *P < 0.05 compared with nontreated cells. (C), Activation of caspases 3 and 7 induced by Aβ1–42. Measurement of activation of caspases 3 and 7 after 8 h of treatment with 5 μm Aβ1–42 in control (C), CALHM1‐, and P86L‐CALHM1‐expressing cells. The results are expressed as the difference in relative luminescence units per second after comparing cells in the absence of Aβ. Triplicate measurements were obtained from three different cultures. Data are expressed as mean ± SEM. anova post hoc Tukey, *P < 0.05 compared with control cells. T‐test, # P < 0.05 compared with CALHM1 versus P86L‐CALHM1.
To confirm that apoptosis was taking place (observed in Figs. 4B and 5B), we measured caspases 3 and 7. After exposure to Aβ1–42 5 μm for 8 h, the activity of caspases 3 and 7 rose significantly only in cells overexpressing P86L‐CALHM1 versus CALHM1 (Fig. 5C).
Regulation of ERK and CREB in CALHM1‐ and P86L‐CALHM1‐overexpressing cells
Ca2+ is critically involved in synaptic activity and memory formation by regulating specific signal transduction pathways that implicate key protein effectors, such as CAMK, MAPK/ERK, and CREB. Therefore, we performed experiments to clarify whether Aβ‐treated cells expressing CALHM1 or P86L‐CALHM1 could be regulating a key gene implicated in survival pathways such as ERK and CREB. No significant changes were detected in the expression of p‐ERK or t‐ERK between controls and CALHM1 cells that were untreated or treated with Aβ (5 μm of oligomers of Aβ1–42 for 1 h). However, treatment with Aβ significantly decreased both p‐ERK expression and t‐ERK expression in P86L‐CALHM1 cells (Fig. 6A and C).
Figure 6.
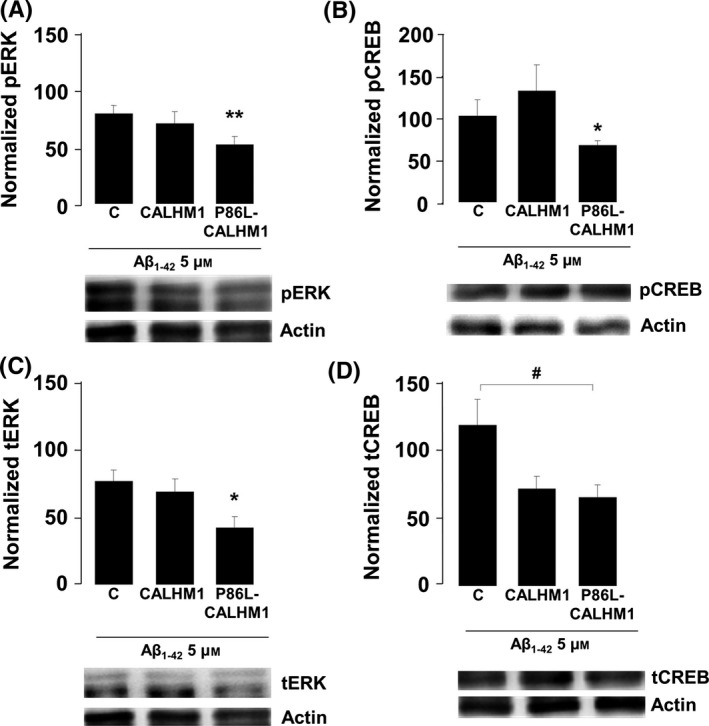
Aβ exposure reduces ERK and CREB signaling in P86L‐CALHM1‐expressing cells. Levels of protein expression of phosphorylated ERK (pERK) (A), total ERK (tERK) (B), phosphorylated CREB (pCREB) (C), and total CREB (tCREB) (D) after 1 h of treatment with 5 μm oligomers of the human fragment Aβ1–42 (Aβ1–42) in control (C), CALHM1‐ or P86L‐CALHM1‐expressing HeLa cells. Protein levels are normalized with respect to actin and to their respective baseline protein levels in cells without Aβ treatment. Data correspond to the mean ± SEM of five different cultures. anova post hoc Bonferroni, *P < 0.05, **P > 0.01 compared with levels in P86L‐CALHM1 cells not treated with Aβ; anova post hoc Dunnett, # P < 0.05 compared with control cells treated with Aβ1–42.
We also measured CREB, a transcriptional factor involved in memory and neuronal survival that can be activated via its phosphorylation at serine (Ser) 133 by several kinases, including ERK. In P86L‐CALHM1 cells treated with Aβ, we observed significantly lower expression of p‐CREB than P86L‐CALHM1 cells not treated with Aβ (Fig. 6B). As for t‐CREB, P86L‐CALHM1 cells treated with Aβ showed significantly lower expression levels than control cells exposed to Aβ; a trend toward lower expression levels was also observed in CALHM1‐expressing cells, although it was not statistically significant (Fig. 6D).
Discussion
We found alterations in [Ca2+]n signaling in HeLa cells transfected with the wild‐type CALHM1 Ca2+ channel and its mutated form P86L‐CALHM1. Baseline [Ca2+]n values in CALHM1 and P86L‐CALHM1 cells were twice those of controls both after perfusion with a 0 Ca2+/EGTA solution and under physiological conditions in 1 mm Ca2+ (Figs. 1B and 2B). Furthermore, upon reintroduction of Ca2+, the kinetics of the [Ca2+]n transients generated developed at a slower rate and decayed to a long‐lasting plateau in CALHM1 and P86L‐CALHM1 compared with control cells (Figs. 1A, C, E). Of interest was the fact that peak and total [Ca2+]n elevations (AUC) were somewhat higher in the cells expressing both forms of CALHM1; the differences detected are similar to those observed with [Ca2+]c under the same experimental conditions (Moreno‐Ortega et al., 2010), indicating that [Ca2+]n depends at least partially on [Ca2+]cit (Alonso & Garcia‐Sancho, 2011). Except for a slower decay in P86L‐CALHM1 cells, the transients were similar to those of CALHM1 cells. Slower and longer [Ca2+]n signals in CALHM1 and P86L‐CALHM1 can be altered because, upon transfection, these channels are preferentially expressed in ER membranes (Dreses‐Werringloer et al., 2008) and these membranes form the nuclear envelope; thus, the CALHM1 channel could be eliciting elevation of [Ca2+]n by simply acting as a leak channel pore or as an InsP3R pore. This last possibility seems unlikely because the [Ca2+]n transients generated by histamine were similar in control cells and in CALHM1 and P86L‐CALHM1 cells (Fig. 2). It therefore seems plausible that CALHM1 behaves as a leak Ca2+ channel and that the greater baseline [Ca2+]n levels in CALHM1 and P86L‐CALHM1 cells could be explained by Ca2+ leakage from the ER lumen into the nucleus. This interpretation is in line with the idea that the nucleus might have specific Ca2+ transients at specific functionally distinct subcompartments; in fact, the nuclear reticulum can generate localized nuclear Ca2+ gradients (Gerasimenko et al., 1995). The alternative hypothesis implies that cytosolic Ca2+ signals can generate nuclear Ca2+ signals by simple Ca2+ diffusion (Gerasimenko et al., 1995; Chamero et al., 2008; Alonso & Garcia‐Sancho, 2011).
Given that the mutated channel P86L‐CALHM1 causes accumulation of Aβ and thus increases Aβ levels in a Ca2+‐dependent manner (Dreses‐Werringloer et al., 2008), it could be responsible for cell vulnerability. In fact, we observed that expression of P86L‐CALHM1 significantly decreased cell viability (Fig. 4B) by initiating early apoptosis (Fig. 5B) in cells exposed to Aβ compared with treated cells overexpressing the empty vector or the CALHM1 channel. When control, CALHM1‐, or P86L‐CALHM1‐overexpressing cells were exposed to channel activation by Ca2+ addback, no significant vulnerability was observed in any of the cells; these results are in agreement with those reported by Dreses‐Werringloer and co‐workers (Dreses‐Werringloer et al., 2008). However, mitochondrial oxidative stressor, such as PAO, did not significantly increase the vulnerability of P86L‐CALHM1‐expressing cells compared with control or CALHM1‐expressing cells (Figs. 4C), indicating the presence of a different cell death mechanism between mitochondrial stressors and P86L‐CALHM1‐induced vulnerability. Additionally, the molecular mechanism of cell death implicated in P86L‐CALHM1 cells exposed to Aβ seems to be related to activation of early apoptosis (Fig. 5B); during this phase, phosphatidylserine is flipped to the outer side of the plasma membrane in a caspase‐dependent process (Bouchier‐Hayes et al., 2008); therefore, activation of caspases 3 and 7 was observed (see Fig. 5C). It is noteworthy that the cell death mechanism triggered by P86L‐CALHM1 in the presence of Aβ does not depend on the cell type, because the neuronal hippocampal cell line HT‐22 is also vulnerable to this stimulus, which triggers early apoptosis (Fig. S2). Moreover, other AD‐like insults such us okadaic acid (OKA) induced cell death in Hela P86L‐CALHM1‐overexpresing cells versus control and CALHM1, significantly (Fig S3). Taken together, these results could add evidence of the influence of P86L‐CALHM1 on the onset of AD and Aβ.
The enhanced vulnerability of P86L‐CALHM1‐overexpressing cells upon exposure to Aβ has been associated with the reduction observed in the expression of proteins related to cell survival signaling pathways such as ERK and CREB (Walton & Dragunow, 2000; Lonze & Ginty, 2002). In fact, in the presence of Aβ, P86L‐CALHM1‐expressing cells showed lower expression levels of p‐ERK and t‐ERK than controls (Fig. 6A, C). Changes in phosphorylation of ERK in CALHM1 and P86L‐CALHM1 were also recently described by Dreses‐Werringloer et al. (2013).
The transcriptional factor CREB has been related to neuronal survival, synaptic plasticity, and memory (Walton & Dragunow, 2000); phosphorylation of its Ser133 has been identified as the key event that must occur for CREB to function as a stimulus‐dependent transcriptional activator. After phosphorylation at Ser133, CREB recruits CREB‐binding protein to act as a transcriptional coactivator (Chrivia et al., 1993). A number of Ca2+‐dependent signaling pathways, such as MAPK/ERKs, have been implicated in the nuclear phosphorylation of CREB at Ser133. Reduction in t‐ERK and p‐ERK in cells expressing the mutated form of the channel could account for the reduction in p‐CREB detected in these cells (Fig. 6B). Curiously, cells overexpressing CALHM1 tended to reduce t‐CREB values, although this was not related to a diminution in the active form, p‐CREB, or the lack of alterations found in p‐ERK. Therefore, cell vulnerability was not increased.
Conclusion
We showed that P86L‐CALHM1 impairs plasma and nuclear membrane Ca2+ permeability, increases cytosolic and nuclear steady‐state Ca2+ levels, depresses the cell survival ERK/CREB pathway, and increases cell vulnerability to Aβ by triggering early apoptosis and activation of caspases 3 and 7. Therefore, in the presence of Aβ, P86L‐CALHM1 seems to shift the balance between neurodegeneration and neuronal survival toward the stimulation of pro‐cytotoxic pathways, which may in turn contribute to its deleterious effects in AD.
Experimental procedures
Chemicals
Metafectene® was purchased from Biontex (Munich, Germany). Wild‐type coelenterazine was purchased from Biotium (Hayward, CA, USA). 4′6‐diamidino‐2‐phenylindole (DAPI), Aβ25–35, anti‐β‐actin, histamine, human Aβ1–42, paraformaldehyde, propidium iodide (PI), thiazolyl blue tetrazolium bromide (MTT), TritonX‐100, and Tween 20 were purchased from Sigma (Madrid, Spain). The antibodies anti‐c‐Myc and anti‐CALHM1 and the PVDF membranes were from Millipore (Madrid, Spain), and Alexa 488 and Image iT‐FX signal enhancer were from Thermo Fisher Scientific (Madrid, Spain). The BCA Protein Assay Kit Reagent was from GE™ Healthcare, and the ECL Advance™ Western Blotting Detection Kit was from Fisher Scientific. The DAKO® mounting medium was purchased from DAKO (Barcelona, Spain). The antibodies anti‐CREB, anti‐P‐CREB, and anti‐P‐ERK were from Cell Signalling (Madrid, Spain); anti‐total ERK and secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, USA). We analyzed apoptosis using the FITC Apoptosis Detection Kit (Immunostep, Salamanca, Spain). Other general chemicals were purchased from Sigma (Madrid, Spain) or Panreac Química S.L.U. (Barcelona, Spain). The cDNA encoding for aequorins was a gift from Professor Tullio Pozzan (University of Padua). The cDNA encoding for CALHM1 and P86L‐CALHM1 was a gift from Professor Philippe Marambaud (Albert Einstein College of Medicine, New York, USA).
Culture of HeLa cells
HeLa cells were grown in plastic flasks in DMEM supplemented with 10% fetal bovine serum, 2 mm glutamine, 25 U mL−1 penicillin, and 25 μg mL−1 streptomycin (all products purchased from Lonza, Basel, Switzerland).
Measurements of [Ca2+]n and [Ca2+]c with aequorins
Cell experiments were performed with 8 × 104 cells seeded on 12‐mm‐diameter coverslips and grown to 60–70% confluence. Transfection with the genetically encoded photoprotein aequorins targeting the nucleus (nu_AEQ) or cytosol (cyt_AEQ) was achieved using Metafectene® as described elsewhere for cells (Moreno‐Ortega et al., 2010). Empty vector (control), or vectors containing CALHM1 or P86L‐CALHM1 were transiently co‐transfected with aequorins at a ratio of 1:1. Experiments to measure changes in [Ca2+]n or [Ca2+]c were performed 36 to 48 h after transfection. The two recombinant proteins were expressed in the same subset of cells (AEQ and the channel) (Brini et al., 1995).
HeLa cells expressing nu_AEQ or cyt_AEQ were reconstituted by adding 5 μm wild‐type coelenterazine for 1.5 h before the experiment. To ensure the total translocation of nu_AEQ to the nucleus, cells were incubated with dexamethasone 10 μm for 2 h immediately before the experiment was carried out (Brini et al., 1995). The cell monolayer was continuously superfused at room temperature (24 ± 2 °C) with Krebs–Hepes buffer (KHB) of the following composition: 125 mm NaCl, 5 mm KCl, 1 mm Na3PO4, 1 mm MgSO4, 5.5 mm glucose, and 20 mm HEPES (pH 7.4); the zero Ca2+ solution contained 0.5 mm ethylene glycol tetraacetic acid. To induce entry of Ca2+, KHB deprived of Ca2+ was switched to another solution containing 1 mm CaCl2, as specified in the figure legends. When used, 10 μm Aβ25–35 or 100 μm histamine was added to the KHB. Light emission was measured in a purpose‐built luminometer and calibrated in terms of [Ca2+], as described by (Rizzuto et al., 1992). At the end of the experiment, cells were lysed by superfusing them with KHB containing 10 mm CaCl2 and 100 μm digitonin to expose them to excess Ca2+ to burn out the aequorin remaining at the end of each experiment and to normalize the Ca2+ transients to the fraction of total aequorin consumed at each point during the experiment.
Cell treatment with a mixture of protofibrils and oligomers of Aβ1–42
HeLa cells were seeded on 24‐well plates and transfected as described above; 24 h after transfection, cells were treated with a mixture of protofibrils and oligomers of Aβ1–42 (Aβ1–42) (5 μm) for 1 h and then harvested and lysed to determine the expression of CREB, pSer133CREB, ERK1/2, and pERK1/2 (Western blot).
Aggregation of Aβ1–42 at 5 μm was achieved as previously described (Parodi et al., 2010). Briefly, human Aβ1–42 was dissolved with dimethylsulfoxide (DMSO) at 2.3 mm; an aliquot of the 2.3‐mm solution was then dissolved in PBS to a final concentration of 80 μm. Finally, this solution was incubated at 37 °C for 2 h under constant shaking.
Monitoring of cell viability
Cell viability was measured using an MTT assay as described elsewhere (Alonso et al., 2013). Briefly, 5 × 104 HeLa cells were seeded in 48‐well plates and transfected with 0.5 μg of empty vector, CALHM1, or P86L‐CALHM1. Twenty‐four hours after transfection, cells were treated with 5 μm of aggregated Aβ1–42, oligomycin (10 μm) plus rotenone (30 μm), or phenylarsine oxide (PAO, 1 μm) for 24 h. Cells were then incubated with MTT reagent for reducing for 30 min to form formazan crystal, which was then dissolved with DMSO. Optical density (OD) was read using an ELISA reader at 540 nm (Berthold Detection Systems, Sirius). Cell viability was expressed as a percentage of the control and calculated using the following equation: viability = [OD test/OD baseline] × 100.
Analysis of apoptosis phases
We analyzed the different phases of apoptosis using the FITC Apoptosis Detection Kit (Immunostep, Salamanca, Spain), which is based on the ability of annexin V to bind specifically to phosphatidylserine flipped into the outer layer of the plasma membrane and the ability of the nonvital dye propidium iodide (PI) to bind to DNA only in altered membrane cells. Thus, double staining enables discrimination between intact cells (annexin V–negative and PI‐negative), early apoptotic cells (annexin V–positive, PI‐negative), and late apoptotic cells (annexin V‐positive and PI‐positive) or necrotic cells (without the characteristic cell integrity).
In brief, HeLa (2 × 105) cells were seeded in 6‐well plates and transfected with 0.75 μg of empty vector and 2 μg of CALHM1 or P86L‐CALHM1. Twenty‐four hours after transfection, HeLa cells were treated for 24 h with 5 μm of a mixture of protofibrils and oligomers of Aβ1–42 and collected and incubated with fluorescent annexin V and PI. Apoptosis was determined by flow cytometry.
Measurement of caspase activation
We measured the activation of caspases 3 and 7 using a commercial kit based on luminescence (Caspase‐Glo (R) 3/7 Assay, Promega Biotech Ibérica S.L., Madrid, Spain) as described by (Alonso et al., 2013). In brief, 105 HeLa cells were seeded in 48‐well plates and transfected with 0.5 μg of empty vector, CALHM1, or P86L‐CALHM1. Twenty‐four hours after transfection, cells were treated with 5 μm of a mixture of protofibrils and oligomers of Aβ1–42 for 8 h. The readings were performed in black 96‐well plates with a plate reader (Glo‐Max Multi Detection System, Promega Biotech Ibérica S.L., Madrid, Spain). Activation of caspases 3 and 7 is expressed as a percentage of nontreated transfected cells.
Measurement of protein expression by Western blot
HeLa cells transfected with 0.5 μg of empty vector, CALHM1, or P86L‐CALHM1 and treated or not with 5 μm of a mixture of protofibrils and oligomers of Aβ1–42 for 24 h were lysed with 100 μL of cold lysis buffer containing 1% Nonidet P‐40, 10% glycerol, 137 mm NaCl, 20 mm Tris‐HCl pH 7.5, 1 mg mL−1 leupeptin, 1 mm phenylmethylsulfonyl fluoride, 20 mm NaF, 1 mm Na4P2O7, and 1 mm Na3PO4. Once the amount of protein was quantified using the BCA Protein Assay Kit reagent, electrophoresis was performed by running 30 μg of protein in polyacrylamide gels for 2 h at constant amperage. Proteins were transferred to PVDF membranes for 2 h at 70 mA. Membranes were then blocked for 2 h with Tween 20‐Tris Buffered Saline (TTBS) containing albumin 4% and incubated with anti‐p‐ERK, anti‐total ERK, anti‐P‐CREB, or anti‐total CREB and anti‐β actin for 2 h. After washing several times with TTBS, the corresponding secondary antibodies were added for 45 min. Finally, the membranes were revealed using the Western Blotting Detection Kit (Thermo Fisher Science) and analyzed and quantified using Scion‐Image software (Scion Corporation Informer Technologies Inc, Meyer Instruments Inc., Houston, EEUU).
Statistics
Values are given as mean ± SEM. The statistical differences between means were assessed using the t‐test or anova and Bonferroni's, Dunnett's, or Tukey's tests in a post hoc analysis. Differences between experimental groups were considered statistically significant at P < 0.05.
Funding
This work was partly supported by the following grants: Ministerio de Economía y Competitividad, FPU Program, Refs. AP2009/0343 (AJMO) and AP2010/1219 (IB). ARN: FIS PI10/01426. MGL: Ministerio de Economía y Competitividad, Ref. SAF2012‐23332. MFCA: Consolidación de grupos de investigación UAM‐CAM 1004040047. We also thank Fundación Teófilo Hernando, Madrid, Spain, for their continued support.
Conflict of interest
None declared.
Supporting information
Fig. S1 Cellular localization of CALHM1 and P86L‐CALHM1.
Fig. S2 Cell vulnerability after treatment with Aβ25–35 in HT‐22.
Fig. S3 Early apoptosis triggered by treatment with Aβ25–35 in CALHM1‐ and P86L‐CALHM1–expressing HT‐22 cells.
Fig. S4 Dose response curve of Okadaic Acid.
Data S1 Supplemental Material and Methods.
Acknowledgments
We are grateful to Dr. Philippe Marambaud (Albert Einstein College of Medicine, New York, USA) for the gifts of cDNAs for CALHM1 and P86L‐CALHM1. We thank Professor Tullio Pozzan (University of Padua, Italy) for cDNAs for cyt_AEQ and nu_AEQ. We thank Ms Francisca Molina for her excellent work in the confocal experiments and Laura Molero, PhD, for her help in the flow cytometry measurements.
References
- Alonso MT, Garcia‐Sancho J (2011) Nuclear Ca(2+) signalling. Cell Calcium 49, 280–289. [DOI] [PubMed] [Google Scholar]
- Alonso MT, Manjarres IM, Garcia‐Sancho J (2011) Privileged coupling between Ca(2+) entry through plasma membrane store‐operated Ca(2+) channels and the endoplasmic reticulum Ca(2+) pump. Mol. Cell. Endocrinol. 353, 37–44. [DOI] [PubMed] [Google Scholar]
- Alonso E, Cano‐Abad MF, Moreno‐Ortega AJ, Novalbos J, Milla J, Garcia AG, Ruiz‐Nuno A (2013) Nanomolar ouabain elicits apoptosis through a direct action on HeLa cell mitochondria. Steroids 78, 1110–1118. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD, Lipp P (1998) Calcium–a life and death signal. Nature 395, 645–648. [DOI] [PubMed] [Google Scholar]
- Bertram L, Schjeide BM, Hooli B, Mullin K, Hiltunen M, Soininen H, Ingelsson M, Lannfelt L, Blacker D, Tanzi RE (2008) No association between CALHM1 and Alzheimer's disease risk. Cell 135, 993–994; author reply 994–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezprozvanny I, Mattson MP (2008) Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci. 31, 454–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boada M, Antunez C, Lopez‐Arrieta J, Galan JJ, Moron FJ, Hernandez I, Marin J, Martinez‐Lage P, Alegret M, Carrasco JM, Moreno C, Real LM, Gonzalez‐Perez A, Tarraga L, Ruiz A (2010) CALHM1 P86L polymorphism is associated with late‐onset Alzheimer's disease in a recessive model. J. Alzheimers Dis. 20, 247–251. [DOI] [PubMed] [Google Scholar]
- Bouchier‐Hayes L, Munoz‐Pinedo C, Connell S, Green DR (2008) Measuring apoptosis at the single cell level. Methods 44, 222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brini M, Marsault R, Bastianutto C, Alvarez J, Pozzan T, Rizzuto R (1995) Transfected aequorin in the measurement of cytosolic Ca2+ concentration ([Ca2+]c) A critical evaluation. J. Biol. Chem. 270, 9896–9903. [DOI] [PubMed] [Google Scholar]
- Cano‐Abad MF, Villarroya M, Garcia AG, Gabilan NH, Lopez MG (2001) Calcium entry through L‐type calcium channels causes mitochondrial disruption and chromaffin cell death. J. Biol. Chem. 276, 39695–39704. [DOI] [PubMed] [Google Scholar]
- Chamero P, Manjarres IM, Garcia‐Verdugo JM, Villalobos C, Alonso MT, Garcia‐Sancho J (2008) Nuclear calcium signaling by inositol trisphosphate in GH3 pituitary cells. Cell Calcium 43, 205–214. [DOI] [PubMed] [Google Scholar]
- Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, Goodman RH (1993) Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature 365, 855–859. [DOI] [PubMed] [Google Scholar]
- Costa RM, Silva AJ (2002) Molecular and cellular mechanisms underlying the cognitive deficits associated with neurofibromatosis 1. J. Child Neurol. 17, 622–626; discussion 627‐629, 646–651. [DOI] [PubMed] [Google Scholar]
- Cui PJ, Zheng L, Cao L, Wang Y, Deng YL, Wang G, Xu W, Tang HD, Ma JF, Zhang T, Ding JQ, Cheng Q, Chen SD (2010) CALHM1 P86L polymorphism is a risk factor for Alzheimer's disease in the Chinese population. J. Alzheimers Dis. 19, 31–35. [DOI] [PubMed] [Google Scholar]
- Dreses‐Werringloer U, Lambert JC, Vingtdeux V, Zhao H, Vais H, Siebert A, Jain A, Koppel J, Rovelet‐Lecrux A, Hannequin D, Pasquier F, Galimberti D, Scarpini E, Mann D, Lendon C, Campion D, Amouyel P, Davies P, Foskett JK, Campagne F, Marambaud P (2008) A polymorphism in CALHM1 influences Ca2+ homeostasis, Abeta levels, and Alzheimer's disease risk. Cell 133, 1149–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreses‐Werringloer U, Vingtdeux V, Zhao H, Chandakkar P, Davies P, Marambaud P (2013) CALHM1 controls the Ca(2)(+)‐dependent MEK, ERK, RSK and MSK signaling cascade in neurons. J. Cell Sci. 126, 1199–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez‐Morales JC, Arranz‐Tagarro JA, Calvo‐Gallardo E, Maroto M, Padin JF, Garcia AG (2012) Stabilizers of neuronal and mitochondrial calcium cycling as a strategy for developing a medicine for Alzheimer's disease. ACS Chem. Neurosci. 3, 873–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME (1997) CREB: a major mediator of neuronal neurotrophin responses. Neuron 19, 1031–1047. [DOI] [PubMed] [Google Scholar]
- Gallego‐Sandin S, Alonso MT, Garcia‐Sancho J (2011) Calcium homoeostasis modulator 1 (CALHM1) reduces the calcium content of the endoplasmic reticulum (ER) and triggers ER stress. Biochem J. 437, 469–475. [DOI] [PubMed] [Google Scholar]
- Gerasimenko OV, Gerasimenko JV, Tepikin AV, Petersen OH (1995) ATP‐dependent accumulation and inositol trisphosphate‐ or cyclic ADP‐ribose‐mediated release of Ca2+ from the nuclear envelope. Cell 80, 439–444. [DOI] [PubMed] [Google Scholar]
- Green KN, LaFerla FM (2008) Linking calcium to Abeta and Alzheimer's disease. Neuron 59, 190–194. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H (1998) Nuclear calcium: a key regulator of gene expression. Biometals 11, 345–358. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Chawla S, Cruzalegui FH, Bading H (1999) Control of recruitment and transcription‐activating function of CBP determines gene regulation by NMDA receptors and L‐type calcium channels. Neuron 22, 789–798. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356. [DOI] [PubMed] [Google Scholar]
- Lambert JC, Sleegers K, Gonzalez‐Perez A, Ingelsson M, Beecham GW, Hiltunen M, Combarros O, Bullido MJ, Brouwers N, Bettens K, Berr C, Pasquier F, Richard F, Dekosky ST, Hannequin D, Haines JL, Tognoni G, Fievet N, Dartigues JF, Tzourio C, Engelborghs S, Arosio B, Coto E, De Deyn P, Del Zompo M, Mateo I, Boada M, Antunez C, Lopez‐Arrieta J, Epelbaum J, Schjeide BM, Frank‐Garcia A, Giedraitis V, Helisalmi S, Porcellini E, Pilotto A, Forti P, Ferri R, Delepine M, Zelenika D, Lathrop M, Scarpini E, Siciliano G, Solfrizzi V, Sorbi S, Spalletta G, Ravaglia G, Valdivieso F, Vepsalainen S, Alvarez V, Bosco P, Mancuso M, Panza F, Nacmias B, Bossu P, Hanon O, Piccardi P, Annoni G, Mann D, Marambaud P, Seripa D, Galimberti D, Tanzi RE, Bertram L, Lendon C, Lannfelt L, Licastro F, Campion D, Pericak‐Vance MA, Soininen H, Van Broeckhoven C, Alperovitch A, Ruiz A, Kamboh MI, Amouyel P (2010) The CALHM1 P86L polymorphism is a genetic modifier of age at onset in Alzheimer's disease: a meta‐analysis study. J. Alzheimers Dis. 22, 247–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD (2002) Function and regulation of CREB family transcription factors in the nervous system. Neuron 35, 605–623. [DOI] [PubMed] [Google Scholar]
- Ma Z, Siebert AP, Cheung KH, Lee RJ, Johnson B, Cohen AS, Vingtdeux V, Marambaud P, Foskett JK (2012) Calcium homeostasis modulator 1 (CALHM1) is the pore‐forming subunit of an ion channel that mediates extracellular Ca2+ regulation of neuronal excitability. Proc. Natl Acad. Sci. USA 109, E1963–E1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marambaud P, Dreses‐Werringloer U, Vingtdeux V (2009) Calcium signaling in neurodegeneration. Mol. Neurodegener. 4, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno‐Ortega AJ, Ruiz‐Nuno A, Garcia AG, Cano‐Abad MF (2010) Mitochondria sense with different kinetics the calcium entering into HeLa cells through calcium channels CALHM1 and mutated P86L‐CALHM1. Biochem. Biophys. Res. Commun. 391, 722–726. [DOI] [PubMed] [Google Scholar]
- Moreno‐Ortega AJ, Martínez‐Sanz FJ, Lajarín‐Cuesta R, de Los RC, Cano‐Abad MF (2015) Benzothiazepine CGP37157 and its 2′‐isopropyl analogue modulate Ca2+ entry through CALHM1. Neuropharmacology 95, 503–510. [DOI] [PubMed] [Google Scholar]
- Parodi J, Sepulveda FJ, Roa J, Opazo C, Inestrosa NC, Aguayo LG (2010) Beta‐amyloid causes depletion of synaptic vesicles leading to neurotransmission failure. J. Biol. Chem. 285, 2506–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, Simpson AW, Brini M, Pozzan T (1992) Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature 358, 325–327. [DOI] [PubMed] [Google Scholar]
- Roskoski R Jr (2012) ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol. Res. 66, 105–143. [DOI] [PubMed] [Google Scholar]
- Schreiber SS, Baudry M (1995) Selective neuronal vulnerability in the hippocampus–a role for gene expression? Trends Neurosci. 18, 446–451. [DOI] [PubMed] [Google Scholar]
- Vay L, Hernandez‐SanMiguel E, Lobaton CD, Moreno A, Montero M, Alvarez J (2009) Mitochondrial free [Ca2+] levels and the permeability transition. Cell Calcium 45, 243–250. [DOI] [PubMed] [Google Scholar]
- Walton MR, Dragunow I (2000) Is CREB a key to neuronal survival? Trends Neurosci. 23, 48–53. [DOI] [PubMed] [Google Scholar]
- West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, Greenberg ME (2001) Calcium regulation of neuronal gene expression. Proc. Natl Acad. Sci. USA 98, 11024–11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GY, Deisseroth K, Tsien RW (2001) Activity‐dependent CREB phosphorylation: convergence of a fast, sensitive calmodulin kinase pathway and a slow, less sensitive mitogen‐activated protein kinase pathway. Proc. Natl Acad. Sci. USA 98, 2808–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Cellular localization of CALHM1 and P86L‐CALHM1.
Fig. S2 Cell vulnerability after treatment with Aβ25–35 in HT‐22.
Fig. S3 Early apoptosis triggered by treatment with Aβ25–35 in CALHM1‐ and P86L‐CALHM1–expressing HT‐22 cells.
Fig. S4 Dose response curve of Okadaic Acid.
Data S1 Supplemental Material and Methods.