

Summary
Aging confers increased susceptibility to common pathogens including influenza A virus. Despite shared vulnerability to infection with advancing age in humans and rodents, the relatively long time required for immune senescence to take hold practically restricts the use of naturally aged mice to investigate aging‐induced immunological shifts. Here, we show accelerated aging Lmna Dhe mice with spontaneous mutation in the nuclear scaffolding protein, lamin A, replicate infection susceptibility, and substantial immune cell shifts that occur with advancing age. Naturally aged (≥20 month) and 2‐ to 3‐month‐old Lmna Dhe mice share near identically increased influenza A susceptibility compared with age‐matched Lmna WT control mice. Increased mortality and higher viral burden after influenza infection in Lmna Dhe mice parallel reduced accumulation of lung alveolar macrophage cells, systemic expansion of immune suppressive Foxp3+ regulatory T cells, and skewed immune dominance among viral‐specific CD8+ T cells similar to the immunological phenotype of naturally aged mice. Thus, aging‐induced infection susceptibility and immune senescence are replicated in accelerated aging Lmna Dhe mice.
Keywords: accelerated aging, infection susceptibility, models of immune senescence, progeria syndrome
Aging parallels an inevitable decline in protective immunity causing susceptibility to infection. This is highlighted by an estimated 40‐fold increased mortality among individuals 65 years or older after infection with ubiquitous pathogens such as influenza A virus (Thompson et al., 2003). This definitive indicator of human immune senescence is shared and proceeds with accelerated tempo in animals with shorter lifespans. In turn, aged mice are increasingly used to characterize immunological changes associated with advancing age. However, the expense related to maintenance and limited availability of aged mice on transgenic strain backgrounds still practically hinders mechanistic dissection of immune senescence. Herein, we propose accelerated aging phenotype mice can be exploited to bypass these limitations.
Nuclear scaffolding lamin proteins are essential for cellular homeostasis including DNA repair and chromatin structure maintenance. Reciprocally, aberrant accumulation of lamin A variants occurs in natural aging as well as accelerated aging progeria disorders (Gonzalez et al., 2011). In Hutchinson–Gilford syndrome, rapidly progressive loss of subcutaneous fat, joint contractures, and arteriosclerosis beginning in early childhood occurs in association with spontaneous mutations in the LMNA gene encoding lamin A (Hennekam, 2006; Merideth et al., 2008). Mice with spontaneous Lmna defects have similar accelerated aging phenotypes (Mounkes et al., 2003; Varela et al., 2005). For example, ‘disheveled’ Lmna Dhe mice have small stature, reduced adipose tissue, and bone mineral density caused by a point mutation in exon 1 of the murine LMNA homologue (Odgren et al., 2010). Considering the need for more practical models of immune senescence, we evaluated whether infection susceptibility and immunological shifts that occur with aging are replicated in accelerated aging Lmna Dhe mice.
In agreement with prior studies showing advancing age impairs immunity against influenza (Po et al., 2002; Yager et al., 2008), naturally aged (≥20 month) compared with young adult (2–3 month) C57Bl/6 mice were markedly more susceptible to infection. Aged mice uniformly succumbed within the first 10 days following influenza A intranasal inoculation with increased viral titers in the lung compared with young adult mice infected with either an identical or weight‐adjusted reduced inoculum (Fig. 1A,B). Susceptibility to influenza with aging was recapitulated in 2‐ to 3‐month‐old accelerated aging Lmna Dhe mice. Compared to age‐matched Lmna WT littermate controls, Lmna Dhe mice harbored significantly increased viral titers and succumbed to infection with kinetics similar to ≥20‐month‐old mice (Fig. 1A,B). Thus, aging‐induced susceptibility to influenza infection is reproduced in accelerated aging Lmna Dhe mice.
Figure 1.
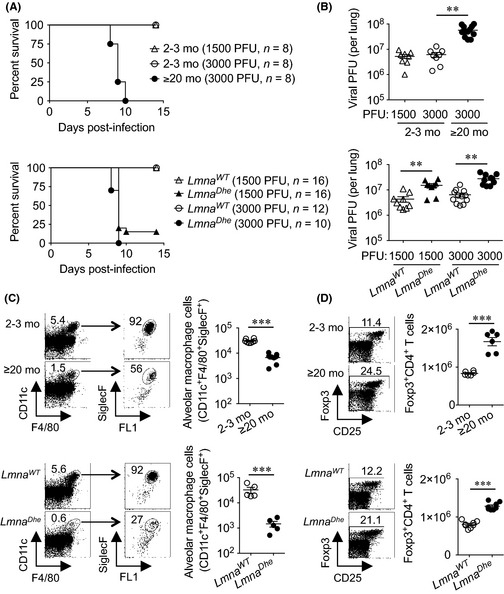
Influenza susceptibility together with skewed alveolar macrophage and regulatory T‐cell accumulation with aging are replicated in accelerated aging Lmna Dhe mice. (A) Survival of naturally aged (≥20 month) compared with 2‐ to 3‐month‐old mice (top), and 2‐ to 3‐month‐old Lmna Dhe mice compared with age‐matched Lmna WT control mice (bottom) after intranasal influenza infection. (B) Recoverable influenza viral titers day 8 postinfection for the mice described in panel A. (C) Cell gating scheme, and composite analysis of lung alveolar macrophage cells of ≥20‐month compared with 2‐ to 3‐month‐old mice (top), and 2‐ to 3‐month‐old Lmna Dhe mice compared with age‐matched Lmna WT control mice (bottom). (D) Representative FACS plots and composite analysis of Foxp3+ CD4+ splenocytes in ≥20‐month compared with 2‐ to 3‐month‐old mice (top), and 2‐ to 3‐month‐old Lmna Dhe mice compared with littermate Lmna WT control mice (bottom). These data are representative of at least two independent experiments each with similar results. Bar, mean ± 1 SE. **, P < 0.01; ***, P < 0.001.
Related experiments addressed whether immunological shifts implicated in causing influenza susceptibility are reiterated among naturally aged and Lmna Dhe mice. Given recent studies highlighting lung alveolar macrophage cells in innate protection early after influenza infection (Purnama et al., 2014; Schneider et al., 2014), the possibility that infection susceptibility with aging reflects reduced accumulation of these protective cells was evaluated. Sharply reduced accumulation of CD11c+F4/80+SiglecF+ alveolar macrophage cells was found in ≥20‐month compared with 2‐ to 3‐month‐old mice (Fig. 1C). This decline in alveolar macrophage cells with aging was reproduced, and even more prominent in Lmna Dhe compared to Lmna WT control mice (Fig. 1C). Comparatively, lung interstitial macrophage cells (CD11c−F4/80+) and dendritic cells (CD11c+F4/80−) that play non‐essential roles in protection against influenza did not differ significantly in ≥20‐month or 2‐ to 3‐month‐old Lmna Dhe mice compared with Lmna WT mice (Fig. S1). Thus, the natural decline in lung alveolar macrophage cells with aging is reproduced in accelerated aging Lmna Dhe mice, reinforcing the importance of these cells in innate antiviral immunity.
Expansion of immune suppressive regulatory T cells (Tregs) has also been causatively linked with immune senescence (Lages et al., 2008; Garg et al., 2014). In agreement, CD4+ splenocytes that co‐express the Treg defining marker, Foxp3, were expanded in ≥20‐month compared with 2–3‐month‐old mice (Fig. 1D). Aging‐induced accumulation of Tregs was recapitulated in Lmna Dhe mice that contain significantly increased frequency and number of Foxp3+ CD4+ T cells compared with Lmna WT controls (Fig. 1D). Foxp3+ cells in aged compared with Lmna Dhe mice also showed similar shifts in expression of some (e.g., CD103, ICOS), but not all Treg cell‐intrinsic functional markers (Vignali et al., 2008) (Fig. S2). Further analysis of Foxp3+ CD4+ T cells derived from Lmna Dhe donor bone marrow progenitors shows reconstitution in Lmna WT recipient mice efficiently eliminates quantitative expansion and qualitative shifts for Lmna Dhe Tregs (Fig. S3). Together with T‐cell‐extrinsic factors driving severe lymphopenia when lamin A is completely eliminated in Lmna‐deficient mice (Hale et al., 2010), these results indicate nonessential cell‐intrinsic roles for lamin A in peripheral CD4+ T‐cell homeostasis.
To investigate changes in other adaptive immune components associated with advancing age, CD8+ T‐cell dynamics prior to and expansion after influenza infection were evaluated in Lmna Dhe mice. Here, declining numbers of peripheral CD8+ T cells, their ratio to CD4+ T cells, and qualitative shifts favoring memory and exhaustion phenotypes (e.g., CD44hi, PD‐1hi) previously reported with aging were reproduced in our analysis of ≥20‐month compared with 2‐ to 3‐month‐old mice (Fig. S4) (Decman et al., 2012). Interestingly, however, with the exception of modestly increased CD44 and PD‐1 expression, these CD8+ T‐cell‐intrinsic shifts were not observed in Lmna Dhe compared with littermate Lmna WT control mice (Fig. S4).
Extending this analysis to mediastinal lymph node cells after influenza infection did not show significant differences in IFN‐γ production by CD8+ T cells of ≥20‐month compared to 2‐ to 3‐month‐old mice in response to NP336–374 peptide stimulation (Fig. 2A). Comparatively, IFN‐γ production by CD8+ T cells from aged mice and the ratio of IFN‐γ‐producing CD8+ T cells between aged compared with young adult mice were significantly increased after PA224–233 peptide stimulation (Fig. 2A and S5). Importantly, regardless of stimulation with NP336–374 or PA224–233 peptide under our experimental conditions, the accumulation and ratio of IFN‐γ‐producing influenza‐specific CD8+ T cells from Lmna Dhe compared with Lmna WT controls directly parallel results for aged compared to young adult mice (Figs 2A and S5).
Figure 2.
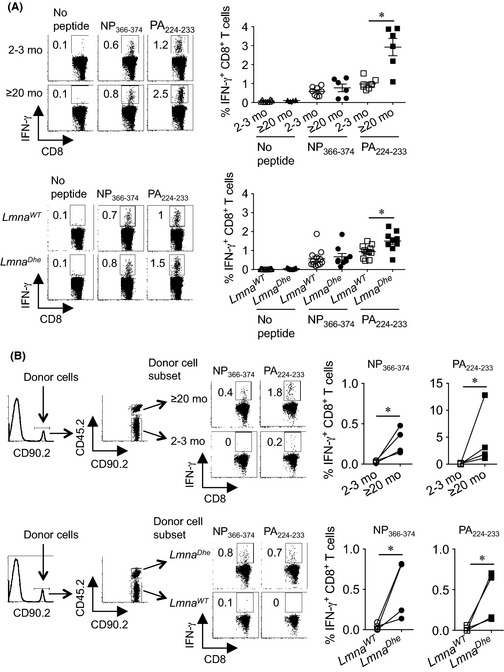
Skewed immune dominance of influenza virus‐specific CD8+ T cells with aging is replicated in accelerated aging Lmna Dhe mice. (A) Representative FACS plots and composite analysis of percent IFN‐γ‐producing mediastinal lymph node CD8+ T cell after stimulation with each influenza‐specific MHC class I peptide compared with no stimulation controls day 8 after influenza A infection (3000 PFUs) in ≥20‐month compared with 2‐ to 3‐month‐old mice (top), or 2‐ to 3‐month‐old Lmna Dhe mice compared with littermate Lmna WT control mice (bottom). (B) Cell gating scheme, and composite analysis of percent IFN‐γ‐producing CD8+ splenocytes after stimulation with PA 224–233 or NP 366–374 peptides for ≥20‐month compared with 2‐ to 3‐month‐old donor mice (top), or 2‐ to 3‐month‐old Lmna Dhe mice compared with Lmna WT donor mice (bottom) day 8 after influenza A infection (3000 PFUs). These data are representative of results from at least two independent experiments with similar results. Bar, mean ± 1 SE. *, P < 0.05.
To dissociate whether more robust PA224–233‐specific CD8+ T‐cell expansion in aged and Lmna Dhe mice reflects increased viral burden (Figs 1B) or cell‐intrinsic changes associated with aging (Yager et al., 2008; Decman et al., 2012), accumulation of IFN‐γ‐producing CD8+ T cells from each group of donor mice was compared after adoptive transfer into congenically discordant naive recipients (Fig. 2B). Remarkably, this analysis showed consistently increased expansion of PA224–233 as well as NP336–374‐specific CD8+ T cells from ≥20‐month and Lmna Dhe mice, each in comparison with cotransferred donor CD8+ T cells from 2‐ to 3‐month‐old Lmna WT mice (Fig. 2B). Thus, cell‐extrinsic factors shared by naturally aged and Lmna Dhe mice selectively restrict expansion of influenza NP366–374‐specific CD8+ T cells promoting skewed immune dominance with aging that favors expansion of PA224–233 to NP366–374 influenza‐specific CD8+ T cells (Po et al., 2002; Yager et al., 2008; Jiang et al., 2009).
Despite an increasing assortment of immunological shifts recognized to occur with aging (Dorshkind & Swain, 2009), the specific changes responsible for functional immune senescence with ensuing infection susceptibility remain largely undefined. A prohibitive roadblock using mouse infection models stems from long‐time intervals required for declining immunity to take hold (≥15–20 months) (Lages et al., 2008; Decman et al., 2012). To circumvent this limitation, we reasoned accelerated aging mouse models can be used to more efficiently unravel the key aging‐induced immunological shifts that control infection susceptibility. Although a variety of mice that display premature aging caused by spontaneous or engineered genetic defects have been described (Liao & Kennedy, 2014), this is the first reported use of any accelerated aging strain to probe aging‐induced infection susceptibility. We show influenza susceptibility that occurs with natural aging is faithfully reproduced in progeroid Lmna Dhe mice. Shifts in many innate and adaptive immune components implicated in causing influenza A susceptibility or diminished immunity with advancing age are also faithfully reproduced in Lmna Dhe mice. In particular, conserved decline in alveolar macrophage cells and expansion of immune suppressive Foxp3+ Tregs that parallel influenza susceptibility in naturally aged and Lmna Dhe mice suggest these immune cell shifts play dominant roles impairing host defense with advancing age. On the other hand, the discordance in CD8+ T‐cell numerical decline, along with expression of memory and exhaustion markers by these cells, between aged and Lmna Dhe mice suggests shifts in these immune components play less pivotal roles promoting influenza susceptibility. Additional studies are required to investigate whether these findings extend to aging‐induced susceptibility to pathogens that cause more persistent infection (Lages et al., 2008), and the use of accelerated aging mice to dissect the immune pathogenesis of aging increasingly recognized to parallel latent infection with other viral pathogens (Lages et al., 2008; Nikolich‐Zugich, 2014). Nonetheless, the fidelity whereby progeroid Lmna Dhe mice reproduce influenza susceptibility that occurs with advancing age highlights accelerated aging mice as important complementary tools for further dissecting the fundamental biology of aging‐induced immune senescence.
Materials and methods
Mice
C57Bl/6 (B6) mice (2–3 months old), congenic B6.SJL‐Ptprc a (CD45.1+), and B6.PL‐Thy1 a (CD90.1+) were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Naturally aged (≥20 month old) B6 mice were provided by The National Institute on Aging (Bethesda, MD, USA). Lmna Dhe and Lmna WT littermate control mice on B6 background were purchased from The Jackson Laboratory, bred in our specific pathogen free animal facility, and analyzed at 2–3 month of age. For generating mixed bone marrow chimeric mice, bone marrow progenitors cells (1 × 106) from congenically discordant Lmna Dhe (CD45.2+CD90.2+) and Lmna WT (CD45.2+CD90.1+) mice were combined at a 1:1 ratio, intravenously transferred into sublethally irradiated twice‐daily (550 rads) CD45.1+ recipients, and analyzed after 8–10 weeks of reconstitution. For adoptive transfer, mononuclear cells from the spleen plus peripheral lymph nodes of ≥20‐month‐aged (CD90.2+CD45.2+) and 2‐ to 3‐month‐old (CD90.2+CD45.2−) mice were combined at a 1:1 ratio (108 cells from each donor) and intravenously transferred into CD90.2−CD90.1+ naive young adult recipient mice one day prior to influenza infection. All experiments were performed under Cincinnati Children's Hospital Institutional Animal Care and Use Committee approved protocols.
Influenza infection
The PR8 strain of influenza A virus (A/Puerto Rico/34, H1N1) was provided by Dr. Monica M. McNeal (Cincinnati Children's Hospital). For infection, mice were anaesthetized intraperitoneally with ketamine (100 mg kg−1) and xylazine (10 mg kg−1) and intranasally inoculated with 1500 or 3000 plaque‐forming units (PFUs) of PR8 virus suspended in 30 μL endotoxin‐free saline. For enumerating recoverable influenza PFUs after infection, the lungs of each group of mice were sterilely dissected, homogenized in saline, and tittered in triplicate onto Madin–Darby canine kidney cell monolayers. After incubation with the lung homogenate for 18 hours, viral plaques were detected using a monoclonal mouse anti‐influenza A antibody (M4090913; Fitzgerald, Acton, MA, USA) followed by staining with FTIC‐conjugated goat anti‐mouse IgG antibody (AQ303F; Millipore, Billerica, MA, USA).
Flow cytometry
The following antibodies for cell surface, intracellular, or intranuclear staining were purchased from eBioscience (San Diego, CA, USA) or BD Biosciences (San Jose, CA, USA): anti‐CD4 (GK1.5), anti‐CD8a (53–6.7), anti‐CD25 (PC61), anti‐CD45 (30‐F11), anti‐CD11c (N418), anti‐SiglecF (E50‐2440), anti‐F4/80 (BM8), anti‐CTLA‐4 (UC10‐4B9), anti‐GITR (DTA‐1), anti‐ICOS (7E.17G9), anti‐OX40 (OX86), anti‐CD103 (2E7), anti‐Helios (22F6), anti‐Foxp3 (FJK‐16s), anti‐CD44 (IM7), anti‐CD62L (MEL‐14), anti‐CD69 (H1.2‐F3), anti‐CD127 (A7R34), anti‐KLRG1 (2F1), anti‐LAG3 (ebioC9B7W), anti‐PD‐1 (J43), CD90.2 (30‐H12), anti‐CD45.2 (104), anti‐IFN‐γ (XMG1.2). For intranuclear Foxp3 staining, surface stained cells were fixed and permeabilized using commercially available reagents (eBioscience) and stained with anti‐Foxp3 antibody. For cytokine production, single‐cell suspensions of mediastinal lymph node cells were stimulated with 10 μm of influenza A‐specific peptides NP366–374 (ASNENMETM) or PA224–233 (SSLENFRAYV) for 6 hours in the presence of GlogiPlug™ (BD Biosciences) compared with no stimulation controls and subjected to intracellular cytokine staining using commercially available reagents (BD Biosciences) and anti‐IFN‐γ. Flow cytometry samples were acquired on a BD FACSCanto™ cytometer and analyzed using FlowJo software (Tree Star, Ashland, OR, USA).
Isolation of lung tissue cells
The lungs were first cut into small pieces (0.1–0.2 cm) and then incubated in medium supplemented with 10% FBS, 500 μg mL−1 collagenase D, 50 μg mL−1 DNase I, and 500 μg mL−1 dispase for 60 min with gentle agitation. Thereafter, the lung homogenate was passed through a mesh filter (100 μm) and centrifuged on Percoll gradient (40% and 70%) for 20 min (530 g). Thereafter, each subset of cells (CD11c+F4/40+SiglecF+ alveolar macrophage cells, CD11c−F4/40+ interstitial macrophage cells, and CD11c+F4/40− dendritic cells) were identified among total mononuclear CD45+ leukocyte cells.
Statistics
The differences in survival after infection between groups of mice were analyzed using the log‐rank (Mantel–Cox) test. The differences in recoverable viral PFUs and cell number between groups of mice were analyzed using the Student's t‐test (two groups) or anova with Dunnett correction for multiple comparisons (more than two groups), with P < 0.05 taken as statistical significance.
Conflict of interest
The authors have no conflicts of interest to declare.
Supporting information
Fig. S1 Similar numbers of interstitial macrophage and dendritic cells in the lungs of naturally aged and accelerated aging Lmna Dhe mice.
Fig. S2 Expression of regulatory T cell‐intrinsic phenotypic markers in naturally aged and Lmna Dhe mice.
Fig. S3 Cell‐extrinsic factors drive Foxp3+ T cell shifts in Lmna Dhe mice.
Fig. S4 Discordant accumulation and phenotypical shifts for CD8+ T cells between naturally aged and Lmna Dhe mice.
Fig. S5 Selectively increased expansion of influenza PA224–233 compared with NP366–374‐specific CD8+ T cell from naturally aged and accelerated aging Lmna Dhe mice.
Acknowledgments and funding
We are grateful to Dr. Claire Chougnet and Hesham Shehata for providing lung tissue from aged mice through support from NIH grant R01‐AG0333057 and The National Institute on Aging for providing naturally aged mice. SSW holds an Investigator in the Pathogenesis of Infectious Disease award from the Burroughs Wellcome Fund.
References
- Decman V, Laidlaw BJ, Doering TA, Leng J, Ertl HC, Goldstein DR, Wherry EJ (2012) Defective CD8 T cell responses in aged mice are due to quantitative and qualitative changes in virus‐specific precursors. J. Immunol. 188, 1933–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorshkind K, Swain S (2009) Age‐associated declines in immune system development and function: causes, consequences, and reversal. Curr. Opin. Immunol. 21, 404–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg SK, Delaney C, Toubai T, Ghosh A, Reddy P, Banerjee R, Yung R (2014) Aging is associated with increased regulatory T‐cell function. Aging Cell 13, 441–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez JM, Pla D, Perez‐Sala D, Andres V (2011) A‐type lamins and Hutchinson‐Gilford progeria syndrome: pathogenesis and therapy. Front. Biosci. 3, 1133–1146. [DOI] [PubMed] [Google Scholar]
- Hale JS, Frock RL, Mamman SA, Fink PJ, Kennedy BK (2010) Cell‐extrinsic defective lymphocyte development in Lmna(‐/‐) mice. PLoS One 5, e10127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennekam RC (2006) Hutchinson‐Gilford progeria syndrome: review of the phenotype. Am. J. Med. Genet. A 140, 2603–2624. [DOI] [PubMed] [Google Scholar]
- Jiang J, Bennett AJ, Fisher E, Williams‐Bey Y, Shen H, Murasko DM (2009) Limited expansion of virus‐specific CD8 T cells in the aged environment. Mech. Ageing Dev. 130, 713–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lages CS, Suffia I, Velilla PA, Huang B, Warshaw G, Hildeman DA, Belkaid Y, Chougnet C (2008) Functional regulatory T cells accumulate in aged hosts and promote chronic infectious disease reactivation. J. Immunol. 181, 1835–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao CY, Kennedy BK (2014) Mouse models and aging: longevity and progeria. Curr. Top. Dev. Biol. 109, 249–285. [DOI] [PubMed] [Google Scholar]
- Merideth MA, Gordon LB, Clauss S, Sachdev V, Smith AC, Perry MB, Brewer CC, Zalewski C, Kim HJ, Solomon B, Brooks BP, Gerber LH, Turner ML, Domingo DL, Hart TC, Graf J, Reynolds JC, Gropman A, Yanovski JA, Gerhard‐Herman M, Collins FS, Nabel EG, Cannon RO 3rd, Gahl WA, Introne WJ (2008) Phenotype and course of Hutchinson‐Gilford progeria syndrome. N. Engl. J. Med. 358, 592–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mounkes LC, Kozlov S, Hernandez L, Sullivan T, Stewart CL (2003) A progeroid syndrome in mice is caused by defects in A‐type lamins. Nature 423, 298–301. [DOI] [PubMed] [Google Scholar]
- Nikolich‐Zugich J (2014) Aging of the T cell compartment in mice and humans: from no naive expectations to foggy memories. J. Immunol. 193, 2622–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odgren PR, Pratt CH, Mackay CA, Mason‐Savas A, Curtain M, Shopland L, Ichicki T, Sundberg JP, Donahue LR (2010) Disheveled hair and ear (Dhe), a spontaneous mouse Lmna mutation modeling human laminopathies. PLoS One 5, e9959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Po JL, Gardner EM, Anaraki F, Katsikis PD, Murasko DM (2002) Age‐associated decrease in virus‐specific CD8 + T lymphocytes during primary influenza infection. Mech. Ageing Dev. 123, 1167–1181. [DOI] [PubMed] [Google Scholar]
- Purnama C, Ng SL, Tetlak P, Setiagani YA, Kandasamy M, Baalasubramanian S, Karjalainen K, Ruedl C (2014) Transient ablation of alveolar macrophages leads to massive pathology of influenza infection without affecting cellular adaptive immunity. Eur. J. Immunol. 44, 2003–2012. [DOI] [PubMed] [Google Scholar]
- Schneider C, Nobs SP, Heer AK, Kurrer M, Klinke G, van Rooijen N, Vogel J, Kopf M (2014) Alveolar macrophages are essential for protection from respiratory failure and associated morbidity following influenza virus infection. PLoS Pathog. 10, e1004053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson WW, Shay DK, Weintraub E, Brammer L, Cox N, Anderson LJ, Fukuda K (2003) Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 289, 179–186. [DOI] [PubMed] [Google Scholar]
- Varela I, Cadinanos J, Pendas AM, Gutierrez‐Fernandez A, Folgueras AR, Sanchez LM, Zhou Z, Rodriguez FJ, Stewart CL, Vega JA, Tryggvason K, Freije JM, Lopez‐Otin C (2005) Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature 437, 564–568. [DOI] [PubMed] [Google Scholar]
- Vignali DA, Collison LW, Workman CJ (2008) How regulatory T cells work. Nat. Rev. Immunol. 8, 523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yager EJ, Ahmed M, Lanzer K, Randall TD, Woodland DL, Blackman MA (2008) Age‐associated decline in T cell repertoire diversity leads to holes in the repertoire and impaired immunity to influenza virus. J. Exp. Med. 205, 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Similar numbers of interstitial macrophage and dendritic cells in the lungs of naturally aged and accelerated aging Lmna Dhe mice.
Fig. S2 Expression of regulatory T cell‐intrinsic phenotypic markers in naturally aged and Lmna Dhe mice.
Fig. S3 Cell‐extrinsic factors drive Foxp3+ T cell shifts in Lmna Dhe mice.
Fig. S4 Discordant accumulation and phenotypical shifts for CD8+ T cells between naturally aged and Lmna Dhe mice.
Fig. S5 Selectively increased expansion of influenza PA224–233 compared with NP366–374‐specific CD8+ T cell from naturally aged and accelerated aging Lmna Dhe mice.