

Abstract
Aims
AZD0837 is a novel oral anticoagulant investigated in clinical studies for stroke prevention in patients with atrial fibrillation (AF). It is bioconverted to its active form, AR‐H067637, a potent, specific and reversible thrombin inhibitor. The effects on coagulation biomarkers were correlated with the pharmacokinetic (PK) exposure of AR‐H067637 to guide selection of the effective dose regimen for a confirmatory efficacy study in AF patients.
Methods
Blood samples were obtained from 601 AF patients randomized to one of four doses of AZD0837 (blinded treatment) or dose‐adjusted vitamin K antagonists (VKA, open treatment) for 3–9 months. A pharmacodynamic model was developed to describe the time course of the AR‐H067637 exposure dependent effects and the effect of VKA on fibrin D‐dimer. The thrombin generation measured ex vivo in venous plasma was also investigated.
Results
The PK exposure of AR‐H067637 was stable with an interindividual variability of 33% and no or minor influence of patient demographics or comedications. For AZD0837, D‐dimer levels decreased with more rapid onset than for VKA. The decrease in D‐dimer levels correlated with steady‐state plasma concentrations (C ss) of AR‐H067637, with a maximum decrease of baseline D‐dimer levels estimated to approximately 60% for both AZD0837 and VKA therapy. The effect on thrombin generation correlated closely with the plasma concentration of AR‐H067637.
Conclusions
The effects on thrombin generation and fibrin D‐dimer levels correlated with the plasma concentration of its active form and provided comparable effects to well‐controlled VKA therapy at an exposure at least corresponding to the 300 mg once daily dose of AZD0837.
Keywords: anticoagulants, atrial fibrillation, exposure–response, pharmacodynamics, pharmacokinetic, thrombin
What is Already Known About This Subject
AZD0837 is a novel oral anticoagulant that after bioconversion to its active form is a potent and reversible thrombin inhibitor.
Effects of AZD0837 treatment on thrombin generation and fibrin D‐dimer levels were investigated and compared with the effect of vitamin K antagonist (VKA) therapy (INR 2.0–3.0, in a phase II dose‐guiding study.
Thrombin generation and fibrin D‐dimer are biomarkers of target activation and thrombogenicity, respectively
What This Study Adds
The PK of the active form of AZD0837 was stable with no or minor influence of patient demographic factors and comedications.
Following oral therapy with AZD0837, inhibitory effects on thrombin generation and fibrin D‐dimer levels were correlated with the plasma concentration of its active form.
At an exposure at least corresponding to the 300 mg once daily dose, AZD0837 provided comparable effects to well‐controlled VKA therapy.
Introduction
AZD0837 is a novel oral anticoagulant that has been investigated in phase II studies for prevention of stroke and systemic embolic events in patients with atrial fibrillation (AF) 1, 2, 3. It is rapidly metabolized via the metabolite AR‐H069927 to AR‐H067637, a selective and reversible direct thrombin inhibitor 4, 5, 6. The oral bioavailability of AZD0837 was 22–52% in healthy subjects given single ascending doses (range 15 to 750 mg) and the half‐life of AR‐H067637 was 9–14 h 5. Peak plasma concentrations and area under the curve for AR‐H067637 showed a low to moderate inter‐individual variability. A slight decrease in bioavailability of the active form was observed with increasing dose, which is presumably because of a dose‐dependent decrease in the formation. Elimination of AZD0837 is mainly by cytochrome P450 metabolism to the intermediate metabolite AR‐H069927, which is subsequently metabolized by N‐hydroxylamine reductase to the active form that is eliminated via both hepatic and renal routes.
In a dose‐guiding trial, AZD0837 demonstrated a good anticoagulant effect and a potentially lower bleeding risk compared with a dose‐adjusted vitamin K antagonist (VKA) 2, 3. Dose‐finding phase II trials based on the primary efficacy variable (i.e. stroke and systemic embolism) require large patient groups and long treatment time because of the low frequency of stroke and systemic embolic events. To overcome this, the use of surrogate indices of thrombogenesis, which would be correlated with pharmacokinetic plasma exposure of the drug, could be supportive information to guide selection of the effective dose range for a confirmatory clinical study of efficacy on stroke and systemic embolism.
In the phase II dose‐guiding study (NCT00684307), primarily aiming to evaluate the safety and tolerability of AZD0837 therapy, the effects on thrombin generation (TG) and fibrin D‐dimer levels were investigated and compared with the effect of VKA therapy (INR 2.0–3.0) 2, 3. According to the classification of biomarkers described by Danhof et al. 7, thrombin generation is a biomarker of target activation or function (type 3 biomarker) and is useful to demonstrate the effect of a thrombin inhibitor 8. Endogenous thrombin generation is measured ex vivo after initiation of the coagulation in plasma collected from the patient. Thrombin accelerates its own formation by positive feedback activation of other coagulation factors and thrombin inhibition results in a decrease of thrombin generation. Fibrin D‐dimer is a fibrin degradation product that has been used as a biomarker of thrombogenicity 9 and may be classified as a pathophysiological response (type 5 biomarker) 7. Plasma fibrin D‐dimer is an index of the degree of hypercoagulability and has been related to adverse thrombotic outcomes 10, 11. Changes in fibrin D‐dimer levels with therapy have also been used to assess new antithrombotic regimes 12, 13, as well as the effects of new oral anticoagulants 14, 15. To our knowledge, the exposure–response relationship between plasma concentrations of a thrombin inhibitor and the effect on fibrin D‐dimer levels has not previously been demonstrated.
The objective of the present analysis of the exposure–response relationships for the biomarkers of thrombin activity and thrombogenesis measured in the phase II study was to gain knowledge of the antithrombotic properties of AZD0837 compared with VKA therapy, and characterize the therapeutic plasma concentration range to guide selection of an effective dose regimen. The pharmacokinetics (PK) of the active form of AZD0837 (AR‐H067637) were evaluated with special regard to patient demographics and concomitant medications on the inter‐patient variability in systemic plasma exposure. A pharmacodynamic (PD) model was developed to describe the exposure–response of AR‐H067637 with regard to fibrin D‐dimer levels. Furthermore, the concentration–effect relationship for thrombin generation measured in venous plasma was assessed.
Methods
Pharmacokinetic and pharmacodynamic data were obtained in a phase II randomized, controlled, parallel, dose‐guiding study to evaluate the safety and tolerability of AZD0837 extended release vs. dose‐adjusted VKA (INR 2.0–3.0) in AF patients with more than one risk factor for stroke 3. In brief, 955 AF patients were randomized to receive one of four doses of AZD0837 (150, 300 or 450 mg once daily or 200 mg twice daily, blinded treatment; n = 631) or VKA (INR 2.0–3.0, target 2.5, open treatment) for 3–9 months. Approximately 30% were naïve to VKA treatment. The primary outcomes were safety and tolerability, whilst PK and PD variables were measured as secondary variables.
Blood sampling
For the PK and PD variables, blood samples were taken at randomization, 2, 4, 8 and 12 weeks and then every 8th week until the end of treatment. The 2, 12 and 36 week samples were taken pre‐dose, and at the 2 week visit also at 2 and 4 h post‐dose. Otherwise, samples were taken at any time. Plasma samples for evaluation of fibrin D‐dimer and ex vivo thrombin generation (TG) were obtained at the same times as PK samples. In addition, fibrin D‐dimer level and TG were determined at enrolment (baseline value), i.e. without anticoagulation for VKA‐naïve patients.
Bioanalysis
Plasma concentrations of AR‐H067637 were determined with a liquid chromatography tandem mass spectrometry method at Eurofins Medinet B.V., the Netherlands 5. This method also determined concentrations of AZD0837 (prodrug) and AR‐H069927 (intermediate metabolite) that were not used in the present investigation of exposure–response relationships. The lower limit of quantification (LLOQ) was 10 nmol l−1 and the range was linear to 4000 nmol l−1 for the three analytes. The method has excellent accuracy and precision, which has been reported previously 5.
Pharmacodynamic analyses
Fibrin D‐dimer plasma analyses (Trinity Biotech, Umeå, Sweden) were performed by a central laboratory (Covance, Switzerland). The LLOQ of the method was 75 ng ml−1. The upper limit of normal (ULN) for the method was 130 ng ml−1. TG was measured ex vivo in plasma with the calibrated automated thrombogram method as previously described by Hemker et al. 16 with 5 pm tissue factor activator and using commercially available reagents (Thrombinoscope BV, Maastricht, the Netherlands). The instructions from the manufacturer were followed. The peak height of the thrombogram (the maximal free thrombin activity) was measured with the calibrated automated method as described previously. Peak thrombin activity was expressed in percent relative to normal measured in pooled human platelet poor plasma from untreated healthy volunteers.
Population pharmacokinetic and pharmacodynamic models
Population models were developed for the PK of AR‐H067637 and its pharmacodynamic effect on fibrin D‐dimer levels. The non‐linear mixed‐effect modelling approach was applied using the software nonmem version VI (Globomax, Hanover, MD, USA). The first order conditional estimation method with interaction (FOCE INTER) was used during the model building. Model comparisons were based on the objective function value (OFV), goodness‐of‐fit plots, precision in parameter estimates and scientific plausibility. A difference in OFV between two nested models is approximately χ2‐distributed. A difference in OFV greater than 3.84 and 6.63 (one degree of freedom) is thus significant at the 5% and 1% level, respectively. In the covariate modelling, the 5% level was used in the inclusion step and 1% level at the exclusion step. Visual predictive check (VPC) plots were done to evaluate the predictive performance of the key models in the analyses. Data were simulated 500 times using the doses and covariate data from the patients who were used in the analysis data set with the same study design. The observed and simulated dependent variable (DV) vs. time profiles were compared graphically with percentiles of the observed data and 95% confidence intervals for the corresponding percentiles based on the model simulations.
Population PK model
In total, 601 patients and 4165 observations of AR‐H067637 plasma concentrations contributed to the population PK analysis. Different compartmental PK models and absorption models were evaluated. Effects of time and dose were evaluated. The relationship of bioavailability with respect to dose was evaluated with linear and non‐linear models. The potential influence of time on the PK was evaluated by estimating different CL/F or F during the day and night (clock‐times). The effect of food was investigated on CL/F, F and k a. A potential difference in F between once daily and twice daily dosing was investigated where a separate fixed effect was estimated for twice daily compared with once daily. Inter‐individual variability (IIV) was assumed to be log‐normally distributed. Different residual error models, an additive, a proportional and a combined additive and proportional error model on the natural logarithm‐transformed data were evaluated.
The influence of patient covariates, genotype of P‐gp transporter protein and concomitant medications were evaluated. The following patient covariates were evaluated: gender, age, body weight, renal function (eGFR, estimated as glomerular filtration rate from serum creatinine using the MDRD algorithm 17). Creatinine clearance (CLcr) was calculated using the Cockroft–Gault equation 18. In addition to statistical significance, covariates were also assessed with respect to clinical significance. Inclusion of a covariate was judged from the estimated magnitude of the effect and was included if the mean covariate effect of categorical covariates was outside the −0.1 to 0.11 (+10%) range in the forward inclusion step and outside the −0.2 to 0.25 (+20%) range for the backwards deletion step.
The influence of concomitant medications and ABCB1 (MDR1) 3435C > T genotype on AR‐H067637XX CL/F was evaluated based on the final covariate model. A stepwise, forward inclusion and backward deletion procedure was employed for evaluation of co‐medications and genotype covariates, one by one, as described for demographic covariates. Inclusion was also based on if the magnitude of the mean covariate effect of categorical covariates was outside the −0.1 to 0.11 (±10%) range, it was further evaluated. Backward deletion was done for covariate effects not outside the −0.2 to 0.25 (±20%) range.
The comedication drugs investigated (see Table S1, Supplementary information for details) included alprenolol, amiodarone, amlodipine, atenolol, enalapril, furosemide, losartan, metformin, metoprolol, simvastatin, sotalol and verapamil. In addition, evaluations were done for the following drug classes and groups: ACE inhibitors, angiotensin II antagonists, calcium antagonists, SSRIs, statins, CYP3A4 inhibitors/substrates and P‐gp inhibitors/substrates. Start of treatment and end of treatment were mapped to the closest visit in order to generate a time‐varying covariate.
The MDR1genotypes C/C, C/T or T/T were determined in 514 of the patients. For the remaining patients with missing genotype data, the frequency from all patients in the study (available genotype data, n = 774 patients) was used. The following genotype frequencies were used C/C = 0.21, C/T = 0.52 and T/T = 0.27. Due to a relatively large proportion of the data being missing, two approaches were undertaken to account for the missing data, as follows: (i) A mixture model with the $MIXTURE subroutine within nonmem with fixed frequencies was used and (ii) All patients with missing genotype information were deleted and only individuals with genotype information were used in the model.
Population PK/PD model
Fibrin D‐dimer data from 265 patients and in total 2411 observations including data below LOQ (75 µg l−1) were included with 171 patients (1445 observations) on AZD0837 treatment and 629 observations in 94 patients on VKA treatment. The number of samples below LLOQ was 1102 (53%). The actual values below LLOQ were used in the modelling where the higher uncertainty for these observations was accounted for by defining separate residual error models above and below LLOQ. This method was judged to be superior to not including these or imputing values by other methods.
Non‐linear mixed effects modeling was used to evaluate the relationship between the exposure of AR‐H067637, using the individual estimates of average concentration at steady‐state (C ss) obtained from the PK model, and the effect on fibrin D‐dimer plasma concentrations over time in VKA‐naïve patients. The effect of VKA treatment in VKA‐naïve patients was also described by the model. Figure 1 visualizes the indirect response PK/PD model that was applied to describe the time course of response.
Figure 1.
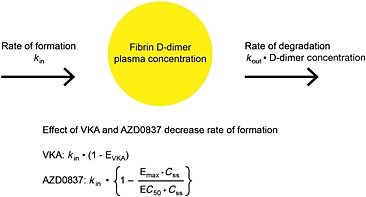
Schematic description of the PK/PD model for the effect of VKA and AZD0837 on the plasma concentration of fibrin D‐dimer
The change in plasma D‐dimer levels (D) over time (t) and effect of the drugs was modelled as:
where k in and k out are the production and elimination rate of plasma D‐dimer, respectively. Emax and C ss are the maximal effect and average steady‐state concentration of AR‐H067637 and EC50 is the concentration at half of the Emax. EVKA is the effect of VKA treatment and was modelled using logit transformation in order to restrict the effect to be between 0–100% as:
where η1 is the individual between patient variability in EVKA and ρ is
where TVEVKA is the VKA effect in a typical patient (mean effect).
During early exploratory analysis it was revealed that the D‐dimer baseline was skewed. Therefore, the D‐dimer baseline (BASE) was modelled using a Cox–Box distribution where
where ρ2 is
SHP is the shape parameter of the Box–Cox distribution and ρ1 is
Influence of patient demographics and dose regimen were evaluated. Age and gender were evaluated as covariates on baseline and EC 50. The potential difference between once daily and twice daily in EC 50 was evaluated. Inclusion of covariates in the model was based on the same procedure as describe above for the population PK modelling.
Effect on thrombin generation
The concentration–effect relationship for the effect on thrombin generation vs. plasma concentrations of AR‐H067637 was evaluated graphically and by linear regression analysis of log‐transformed peak thrombin activity.
Results
Patient population
A summary of the demographic characteristics for the patient population included in the population PK analysis is shown in Table 1. The PK population included 601 of the 631 patients in the study patient population that were randomized and treated with AZD0837. The patient demographics were balanced in the four dose groups and comparable with the 318 patients randomized to VKA treatment 3. About 30% of the patients were VKA naïve and 265 of these patients were included in the population PK/PD model analysis. The patient demographics in this subpopulation were comparable with the overall study population.
Table 1.
Summary of patient demographic characteristics (PK population, n = 601)
| Covariate | |
|---|---|
| Gender, women [n (%)] | 183 (30) |
| Age (years) | 71 [34–93] |
| Body weight (kg) | 85 [47–167] |
| CLcr (ml min−1) | 76 [28–187] |
| eGFR (ml min−1) | 70 [20–135] |
Results are presented as absolute numbers (%) or as the median [range]. The patients included in the PK/PD modelling of fibrin D‐dimer effect (n = 265), had similar patient characteristics. CLcr, creatinine clearance calculated using the Cockroft–Gault equation 18; eGFR, estimated glomerular filtration rate calculated using the MDRD equation 17.
Population pharmacokinetics
Pharmacokinetics of AR‐H067637 were adequately described by a one compartmental model, where the formation from orally absorbed AZD0837 via the intermediary metabolite was described with a zero order input rate constant that subsequently was followed by a first order process. Figure 2 shows good consistency between the model‐predicted plasma concentrations of AR‐H067637 and corresponding observations for the four dose groups (see Figure S1 and Figure S2, Supplementary information for additional goodness‐of‐fit plots). Parameters for the final population PK model are presented in Table 2. A correlation between oral clearance (CL/F) and oral volume of distribution (V/F) was significant. The best residual error model was a combined additive plus proportional model.
Figure 2.
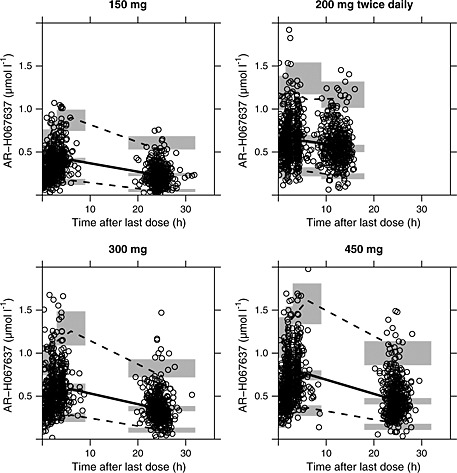
AR‐H067637 plasma concentrations over time after last dose (circles) including model predicted means (solid line) and 5th and 95th percentiles (dashed lines). The shaded grey areas show the 95% CI limits for the mean and percentiles
Table 2.
Population estimates of pharmacokinetic parameters for AR‐H067637
| Parameter | Estimate (% RSE)* | Inter‐individual variability % (% RSE) |
|---|---|---|
| CL/F (l h−1) ‡ | 39.2 (2.6) | 31 (7.8)† |
| V/F (l) ‡ | 77.8 (10) | 26 (52)† |
| ka (1 h−1) ‡ | 0.0308 (6.3) | 52 (17) |
| Duration (h) ‡ | 1.4 (4.7) | 52 (20) |
| Covariate effects | ||
| Dose on Frel § | −0.42 (7.3) | ‐ |
| Dose 200 mg twice daily on Frel (%) § | −27.9 (9.1) | ‐ |
| eGFR on CL/F (% per ml min−1) ¶ | 0.45 (21) | ‐ |
| Age on CL/F (% per year) ¶ | −0.94 (20) | ‐ |
| Residual error | ||
| Additive (µmol l−1) | 0.0777 (6.8) | ‐ |
| Proportional (%) | 17.2 (6.3) | ‐ |
CL/F, oral clearance; Duration, duration of zero order formation process; eGFR, estimated glomerular filtration rate calculated using the MDRD equation [17]; k a, first order rate constant of the formation process; V/F, oral volume of distribution.
RSE = relative standard error;
Covariance between CL/F and V/F was 26 (RSE 52);
Shrinkage of η was 6.8% for CL/F, 8.6% for V/F, 30% for k a, and 66% for duration;
Relative bioavailability compared with 150 mg AZD0837 once daily, F rel = (dose/150)‐0.42 excluding 200 mg twice daily. F rel 200 mg was 28% lower compared with 150 mg;
Covariate effects of age and renal function on CL/F, relative to the median eGFR of 70 ml min−1 and the median age of 71 years: CL/F = θCL/F × (1 + θeGFR × (eGFR – 65)) × (1 + θAge × (Age – 71)).
The bioavailability of AR‐H067637 decreased with increasing dose. The relative bioavailability compared with 150 mg once daily was 75% for 300 mg once daily, 72% for 200 mg twice daily and 63% for 450 mg once daily.
The inter‐individual variability in CL/F, which determines the average steady‐state plasma concentration (C ss) of AR‐H067637, was moderate (33%). The C ss of AR‐H067637 was stable over the study period. Dosing with food or fasting did not have a significant influence on the pharmacokinetics.
Age and renal function were statistically significant predictors of between patient variability in CL/F and included in the final PK model. However, inclusion of these two covariates decreased the estimated inter‐individual variability in CL/F by only 2%, from 33% to 31%. In the ranges of age and renal function accounting for the majority of patients (80%), the changes in exposure were less than 10% relative to the average patient with a median age (71 years) and renal function (70 ml min−1). After inclusion of age and renal function as covariates in the model, gender and body weight did not have a significant influence on the pharmacokinetics of AR‐H067637. Figure 3 shows simulations of the median steady‐state plasma concentration profiles with the 10th and 90th percentiles to illustrate the reference range for a typical AF patient receiving 300 mg once daily and 200 mg twice daily doses.
Figure 3.
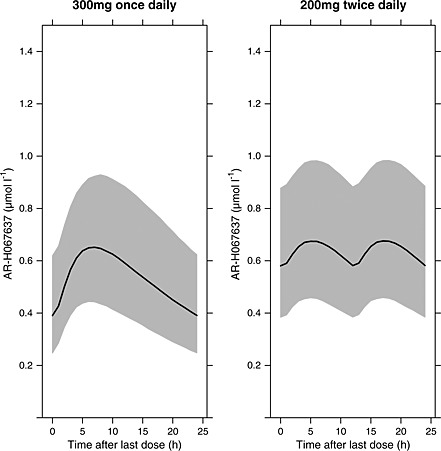
Simulated median steady‐state plasma concentration profiles (solid lines) with the 10th and 90th percentiles (shaded area) to illustrate the reference range for a typical AF patient receiving 300 mg once daily and 200 mg twice daily doses
The only co‐medication found to have significant influence on CL/F of AR‐H067637XX was verapamil, which decreased CL/F by 25% (90% CI 11%, 38%), corresponding to a 33% (90% CI 12%, 62%) increase in C ss of AR‐H067637XX. Pharmacogenetic analysis of P‐glycoprotein gene polymorphism in population PK modelling did not support any influence of ABCB1(C3435T) genotype on oral clearance (CL/F) of AR‐H067637.
Based on the population PK model and the individual observed plasma concentrations, predictions of C ss of AR‐H067637 were obtained for each individual patient. The covariate effect of verapamil was not included in the model used to predict individual exposures, as the estimated change in CL/F did not exceed 25% and the percentage of patients treated with verapamil was low (6%). Medians of the individual C ss estimates were approximately 0.3, 0.5, 0.6 and 0.6 µmol l−1 for patients receiving AZD0837 150 mg once daily, 300 mg once daily, 450 mg once daily and 200 mg twice daily, respectively.
Population PK/PD model for fibrin D‐dimer
In VKA‐naïve patients, fibrin D‐dimer levels decreased after start of treatment in all treatment groups. A rapid onset of the D‐dimer reductive effect was seen within 2 to 4 weeks in all treatment groups, with a faster onset for AZD0837 compared with VKA treated patients. The different onset was accounted for by a slower rate constant for the elimination of the fibrin D‐dimer response variable for patients treated with VKA. The half‐life of onset was about 5 and 11 days for treatment with AZD0837 and VKA, respectively. Consequently, steady‐state for the response was estimated to occur around 15–20 days after start of treatment with AZD0837. For VKA steady‐state was estimated to be achieved after 30–40 days of treatment. Maximum effect on D‐dimer was an approximately 60% decrease of baseline levels for both AZD0837 and VKA therapy.
Figure 4 shows the model‐predicted relationship between D‐dimer and C ss of AR‐H067637 or VKA respectively, including observations collected after at least 4 weeks of AZD0837 treatment and at least 8 weeks of VKA treatment when steady‐state effects are estimated to be achieved (see goodness‐of‐fit plot Figure S3 included in Supplementary information).
Figure 4.
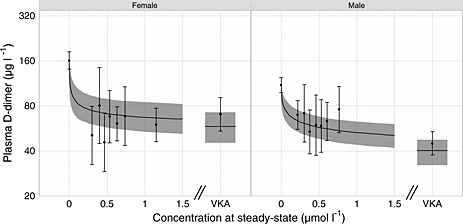
Steady‐state relationship between fibrin D‐dimer levels vs. AZD0837 and VKA treatment groups showing male and female patients in separate graphs. Horizontal bars are 90% CI of the observations. The solid line and shaded areas represent mean model predictions and 90% CI, respectively
The population parameter estimates for the PK/PD model are presented in Table 3. The mean baseline plasma D‐dimer level was estimated to 94 µg l−1 in a typical male patient and 42% higher (133 µg l−1) in a typical female patient. Age was also found to influence D‐dimer levels significantly, but the data did not support inclusion of both age and gender as predictive covariates. Gender was included in the model as the effect was estimated to be larger than for age.
Table 3.
Parameter estimates for PK/PD model for fibrin D‐dimer
| Parameter | Estimate (% RSE) * | Inter‐individual variability % (% RSE) * |
|---|---|---|
| BASEmale † (µg l−1) | 94 (92) | 65 (6.2) |
| kout, AZD (day−1) | 0.142 (11) | ‐ |
| kout, VKA (day−1) | 0.0622 (14) | ‐ |
| Emax, AZD | 0.60 (5.5) | ‐ |
| EC50 males ‡, AZD | 0.13 (14) | 238 (23) |
| Effect VKA | 0.58 (5.4) | 65 (32.6) |
| SHP | 0.37 (39) | ‐ |
| Covariate effects | ||
| BASE‐GENDER † | 0.42 (32) | ‐ |
| EC50 females (%) ‡ | −0.79 (21) | |
| Residual error | ||
| Error above LOQ | 0.52 (8.2) | 45 (8.2) |
| Error below LOQ | 0.31 (6.6) | ‐ |
AZD, AZD0837 treatment; BASE, D‐dimer baseline; EC 50, the concentration at which half of the maximum effect is achieved; Effect VKA, D‐dimer response for VKA treatment; Emax, maximal effect on D‐dimer; k out, D‐dimer elimination rate constant; SHP, shape parameter of Cox–Box distribution; VKA, VKA treatment.
Relative standard error (%);
Baseline was 42% higher in females patients: BASE = θBASE × (1 + θGENDER=FEMALE);
Females had on average a lower EC 50 compared with males: EC 50 = θEC50 × (1 + θGENDER=FEMALE).
A gender difference for the EC 50 was found, where female patients appeared on average to be more sensitive to AZD0837 treatment compared with males (0.03 vs. 0.13 µmol l−1). The net effect of the gender difference in both baseline and EC 50 resulted in similar level of D‐dimer levels on treatment as seen in Figure 4. The variability in plasma D‐dimer was in general high, also between patients which can be seen in the estimate of inter‐individual variability in EC 50 of more than 200%. Inclusion of D‐dimer values below LLOQ (75 ng ml−1) was valuable to allow characterization of the full concentration–effect relationship. No gender difference in the effect of VKA treatment was found and both males and females had the same relative reduction in D‐dimer.
Thrombin generation
Thrombin generation, expressed as maximal thrombin activity (peak of the thrombin generation curve) in percent of the maximal thrombin activity in normal plasma, was decreased by AZD0837 and VKA treatment. For all patients, treated with VKA or AZD0837, the onset of effects on peak TG were observed after 2 weeks (first sampling time) and remained relatively stable during the treatment periods of up to 36 weeks (data not shown). In VKA naïve patients, model‐based estimates of the absolute decrease in peak TG compared with baseline before start of treatment (90% CI, n = number of observations) were 11% (3, 19, n = 20), 26% (19, 34, n = 24), 40% (33, 47, n = 23) and 33% (26, 41, n = 28) when measured at trough (just before next dose) after 12 weeks of treatment with AZD0837 150 mg oce daily, 300 mg once daily, 450 mg once daily and 200 mg twice daily, respectively. For VKA naïve patients receiving VKA therapy for 12 weeks, the estimated decrease in TG after 12 weeks of treatment was 50% (45, 55, n = 56).
The effects on peak TG correlated with the plasma concentrations of AR‐H067637. Figure 5 shows the concentration–effect relationship for peak TG data in all patients receiving AZD0837 and with available PK observations (including both VKA naïve and VKA‐treated before inclusion). The natural logarithm of TG was linearly related to the plasma concentration of AR‐H067637 by the linear regression analysis (r 2 value of 0.45). The equation for the regression line is shown in Figure 5. The intercept (TG at zero plasma concentration) was 100% and a 50% inhibition of TG was estimated at a plasma concentration of 0.57 µmol l−1, which is in the range of median C ss for the 300 mg once daily and higher daily doses.
Figure 5.
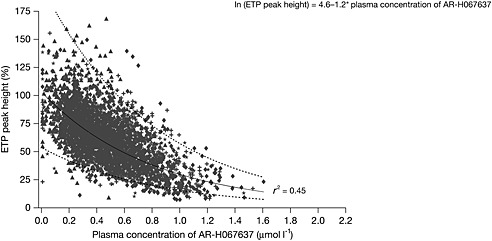
Thrombin generation, measured as peak height and expressed as percentage relative to normal plasma, vs. plasma concentrations of AR‐H067637.  , AZD0837 150 mg once daily;
, AZD0837 150 mg once daily;  , AZD0837 300 mg once daily;
, AZD0837 300 mg once daily;  , AZD0837 450 mg once daily;
, AZD0837 450 mg once daily;  , AZD0837 200 mg twice daily;
, AZD0837 200 mg twice daily;  , predicted values;
, predicted values;  , 95% prediction interval, upper and lower limit
, 95% prediction interval, upper and lower limit
Discussion
In the present ancillary analysis of a phase II trial, the PK profile of the active form of AZD0837 was stable with no or minor influence of patient demographic factors and comedications. Fibrin D‐dimer levels decreased following oral therapy with AZD0837, whereby the inhibitory effects of doses of 300 or 450 mg once daily or 200 mg twice daily on D‐dimer levels were comparable with the effect of VKA with INR 2–3 and correlated with the PK exposure of the active form. Analyses of the exposure–response relationship for the D‐dimer indicate that AZD0837 treatment at an exposure at least corresponding to the 300 mg once daily dose provides similar suppression of thrombogenesis and thrombin generation as VKA treatment aiming for INR 2.0–3.0. For daily doses of 300 to 450 mg, maximum effects were achieved while about 40% decrease was observed for the lowest dose group receiving 150 mg 3.
The population pharmacokinetic model of AR‐H067637 showed that the inter‐individual variability in CL/F, which determines C ss and overall exposure of AR‐H067637, was moderate with a CV of 33%. Food had no substantial influence on the pharmacokinetics. The predictable PK profile, with moderate variability and slight dose dependent decrease in bioavailabilty, is consistent with that found in healthy young volunteers given single escalating doses of AZD0837 in solution 5. Of the investigated patient demographic factors, only age and renal function were included in the PK model. However these covariates were weak predictors of inter‐individual variability for CL/F and were estimated to result in relatively small changes in exposure (<10%). As the active form is eliminated via both hepatic and renal routes 5, some influence of age and renal function is expected but renal clearance is not a major route of elimination, which is consistent with the relatively small changes estimated for renal function and age. The variability in exposure estimated for the active form of AZD0837 shows about a two‐fold range for the 10th to 90th percentiles in the typical AF patient in the present study, which was lower than the three‐fold range reported for the oral thrombin inhibitor dabigatran 19. Furthermore, the influence of renal function was greater for dabigatran with 1.8 fold higher exposure in patients with moderate renal impairment, compared with a 20% increase estimated for AZD0837.
Verapamil caused an increase in C ss of AR‐H067637 while other comedications showed no significant influence on the exposure. The influence of verapamil may be explained by the hepatobiliary disposition of AZD0837. Inhibition of biliary excretion, shown to occur mainly for the active form and mediated by transporter proteins, was suggested to explain a pharmacokinetic interaction with ketoconazole that increased plasma exposure of the active form by approximately two‐fold 20. The active form of AZD0837 was found to be a substrate of both P‐glycoprotein (P‐gp) and the multidrug and toxin extrusion 1 (MATE1) transporter proteins, which were inhibited by ketoconazole. Verapamil is known to have weaker inhibitory effects on transporters. Similar to the verapamil effect on AZD0837 (33% increase), increases in exposure to the active form of dabigatran have also been observed in AF patients receiving verapamil (23%) and amiodarone (12%), which both are inhibitors of the Pgp transport protein 20.
TG and fibrin D‐dimer were used as biomarkers to investigate the anticoagulant effect of AZD0837 and VKA treatments. The TG assay, a biomarker of the functional status of blood coagulation, measures thrombin activity ex vivo in plasma using tissue factor to initiate the coagulation. The assay is used to demonstrate an anticoagulant effect and also suggested to identify risk of thrombosis and bleeding 21, 22, 23, 24. In the presence of a thrombin inhibitor the thrombin generation assay may underestimate the effect because of an interaction with α2‐macroglobulin 25. Compared with the measurement of the area under the TG curve, the peak thrombin activity is less influenced by the measurement error and was the variable evaluated in the present study. The effect on peak thrombin activity correlated with the plasma concentration of AR‐H067637, with 50% inhibition at about 0.6 µmol l−1. This is consistent with the concentration–effect relationship for TG measured ex vivo in plasma from healthy young subjects given single doses of AZD0837 5 and in vitro in platelet poor plasma 6. At daily doses of 300 mg and higher, the median C ss estimates were close to the concentration needed for 50% inhibition and comparable with the decrease observed for VKA naïve patients treated with VKA.
There is extensive literature on fibrin D‐dimer as an index of thrombogenesis, which support that it is a useful biomarker to guide dose selection for new antithrombotic regimes or new oral anticoagulants 12, 15. Indeed, the effect on fibrin D‐dimer in the present study suggests an antithrombotic efficacy comparable with warfarin for daily doses of AZD0837 of ≥300 mg. When compared with warfarin the onset of effect was more rapid for AZD0837, which is expected based on its direct and reversible inhibition of thrombin. The onset is reflected by the turnover of fibrin D‐dimer while for warfarin the onset is dependent on its indirect effect on the activity of coagulation factors. Steady‐state for the fibrin D‐dimer level was estimated to be achieved after about 2–3 weeks of treatment with AZD0837 and about 5–6 weeks with VKA treatment.
The fibrin D‐dimer assay is used mainly to identify patients with increased levels and not developed to be sensitive for measurement of low concentrations. The LLOQ of the assay (75 ng ml−1) was about 60% of the ULN of the assay (130 ng ml−1), and a large fraction (about 50%) of the observations were below LLOQ. The actual values were used and the uncertainty for these observations was accounted for by separate residual error models above and below the LLOQ. Assuming a graded response on D‐dimer this was considered the best option. The estimated residual error was actually lower (31 vs. 52%) for D‐dimer observations below LOQ, presumably because of the anticoagulant effects of AZD0837 and VKA treated patients were close to the maximum reduction in D‐dimer levels.
Fibrin D‐dimer is generated from degradation of fibrin by plasmin and may be elevated as a consequence of either increased fibrin formation (thrombogenisis) or enhanced fibrinolysis 9. The effect of AZD0837 and other direct thrombin inhibitors on fibrin formation may be underestimated from investigating D‐dimer levels due to their properties to facilitate also fibrinolysis mediated by thrombin activable fibrinolysis inhibitor (TAFI). For the groups receiving daily doses of 300 mg or higher, the effects on fibrin D‐dimer levels were comparable with the effect in patients treated with VKA.
The baseline plasma D‐dimer level was estimated to be 94 µg l−1 in a typical male patient and 42% higher in a typical female patient. Indeed, D‐dimer concentrations are higher in women and increase with age. Age was found to influence D‐dimer levels significantly, but was not included in the model as the data did not support inclusion of both age and gender as predictive covariates. Gender was included in the model as the effect was estimated to be larger than for age. A gender difference in EC 50 was also found, where female patients appeared to be more sensitive to AZD0837 treatment compared with males (0.03 vs. 0.13 µmol l−1). The net effect of the gender difference in both baseline and EC 50 resulted in similar level of D‐dimer levels on treatment. For patients treated with VKA, no difference in sensitivity was found between males and females, respectively. In addition, it should be noted that the inter‐individual variability in EC 50 was large (CV > 200%). During treatment many patients had similar values of D‐dimer close to the maximum achievable reduction and similar to the D‐dimer levels in patients receiving VKA. Therefore it is possible that the large variability in EC 50 is at least in part a result of the very wide range of D‐dimer levels at baseline with 10% of patients having measured D‐dimer greater than 400 µg dl−1 (with the maximum observation being 1870 µg dl−1). As similar D‐dimer levels were achieved for most treated patients, the model will estimate much lower EC 50s for patients with higher baseline values.
The model assumes that the relative treatment effect is independent of the baseline value, which showed large inter‐individual variability. The baseline values were similar for the VKA naïve patients in the different treatment groups, with the highest median value for the 150 mg dose group for AZD0837 (139.3 µg dl−1) and the lowest for the patients receiving VKA (104.6 µg dl−1). The D‐dimer assay used was not optimized to determine high concentrations accurately, making the higher values more uncertain. Furthermore, the collection of the plasma samples may cause ex vivo formation of D‐dimer that may result in erroneously high D‐dimer levels in the samples collected at baseline when no anticoagulant is present.
According to the PK/PD model, the maximum response was similar for VKA and AZD0837 treatments suggesting that at high exposure of AR‐H06763 the effect on fibrin D‐dimer approaches the VKA response. In the C ss range of 0.5 to 0.6 µmol l−1, which covers the range of median C ss concentrations for the three highest AZD0837 doses studied (300 mg and 450 mg once daily and 200 mg twice daily), the response for AZD0837 is estimated to be approximately 80 to 95% of the maximum response in male and female patients, respectively. Thus, higher AZD0837 doses than investigated in the present study are predicted to give only a minor additional effect on fibrin D‐dimer levels, while the lowest dose group of 150 mg once daily showed a lower anticoagulant effect. For the 150 mg dose group, the median C ss concentration was 0.3 µmol l−1 which is about two times higher than the EC 50 estimate for male patients (0.13 µmol l−1) and predicted to give about 75% of maximum response. Although the PK/PD modelling was done for data obtained in VKA naïve patients to observe the onset of effect, the dose‐dependent anticoagulant effect on D‐dimer was consistent with that observed in VKA pre‐treated patients 2, 3.
In conclusion, following oral therapy with AZD0837, the inhibitory effects on thrombin generation and fibrin D‐dimer levels were correlated with the plasma concentration of its active form and provided comparable effects to well‐controlled VKA therapy at an exposure at least corresponding to the 300 mg once daily dose AZD0837. A confirmatory study would be needed to demonstrate efficacy at this dose regimen.
Competing Interests
All authors have completed the Unified Competing Interest form at www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare that the study was supported by AstraZeneca R&D, Mölndal, Sweden. GYHL acted as the Chair of the Executive Steering Committee, and has received funding for consultancy, meetings and symposia from Astra Zeneca. LHR was a member of the Executive Steering Committee and has received funding for consultancy from AstraZeneca. SBO was a member of the Executive Steering Committee, and is a shareholder in AstraZeneca and has received financial support for clinical research from the company. EJ, BH, UGE and KW are employees of AstraZeneca, the sponsor of the clinical study presented, with stock ownership in the company.
We would like to acknowledge Ulrika Simonsson and Margareta Elg for valuable scientific contributions to the modelling and interpretation of the results.
Contributors
GYHL, LHR, SBO – study steering committee, drafting and revision of manuscript. GYHL was Chairman of the committee, and wrote the first draft of the paper
EJ – study physician, drafting and revision of manuscript.
BH, UGE – pharmacokinetic modelling, drafting and revision of manuscript.
KW – project oversight, drafting and revision of manuscript.
Supporting information
Supporting info item
Lip, G. Y. H. , Rasmussen, L. H. , Olsson, S. B. , Jensen, E. , Hamrén, B. , Eriksson, U. G. , and Wåhlander, K. (2015) Exposure–response for biomarkers of anticoagulant effects by the oral direct thrombin inhibitor AZD0837 in patients with atrial fibrillation. Br J Clin Pharmacol, 80: 1362–1373. doi: 10.1111/bcp.12719.
References
- 1. Olsson SB, Rasmussen LH, Tveit A, Jensen E, Wessman P, Panfilov S, Wahlander K. Safety and tolerability of an immediate‐release formulation of theoral direct thrombin inhibitor AZD0837 in the prevention of stroke and systemic embolism in patients with atrial fibrillation. Thromb Haemost 2010; 103: 604–12. [DOI] [PubMed] [Google Scholar]
- 2. Lip GY, Rasmussen LH, Olsson SB, Zetterstrand S, Stahre C, Bylock A, Aunes‐Jansson M, Eriksson U, Wahlander K, Steering C. Oral direct thrombin inhibitor AZD0837 for the prevention of stroke and systemic embolism in patients with non‐valvular atrial fibrillation: a Phase II study of AZD0837 in patients who are appropriate for but unable or unwilling to take vitamin K antagonist therapy. Thromb Res 2011; 127: 91–9. [DOI] [PubMed] [Google Scholar]
- 3. Lip GY, Rasmussen LH, Olsson SB, Jensen EC, Persson AL, Eriksson U, Wahlander KF. Oral direct thrombin inhibitor AZD0837 for the prevention of stroke and systemic embolism in patients with non‐valvular atrial fibrillation: a randomized dose‐guiding, safety, and tolerability study of four doses of AZD0837 vs. vitamin K antagonists. Eur Heart J 2009; 30: 2897–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pehrsson S, Johansson K, Kjaer M, Elg M. Evaluation of AR‐H067637, the active metabolite of the new direct thrombin inhibitor AZD0837, in models of venous and arterial thrombosis and bleeding in anaesthetised rats. Thromb Haemost 2010; 104: 1242–9. [DOI] [PubMed] [Google Scholar]
- 5. Johansson S, Cullberg M, Eriksson UG, Elg M, Duner K, Jensen E, Wollbratt M, Wahlander K. Single‐dose pharmacokinetics, pharmacodynamics and safety of AZD0837, a novel oral direct thrombin inhibitor, in young healthy male subjects. Int J Clin Pharmacol Ther 2011; 49: 258–67. [DOI] [PubMed] [Google Scholar]
- 6. Deinum J, Mattsson C, Inghardt T, Elg M. Biochemical and pharmacological effects of the direct thrombin inhibitor AR‐H067637. Thromb Haemost 2009; 101: 1051–9. [PubMed] [Google Scholar]
- 7. Danhof M, Alvan G, Dahl SG, Kuhlmann J, Paintaud G. Mechanism‐based pharmacokinetic‐pharmacodynamic modeling‐a new classification of biomarkers. Pharm Res 2005; 22: 1432–7. [DOI] [PubMed] [Google Scholar]
- 8. Wolzt M, Bostrom SL, Svensson M, Wahlander K, Grind M, Sarich TC. Effects of the oral direct thrombin inhibitor ximelagatran on p‐selectin expression and thrombin generation in atrial fibrillation. Pathophysiol Haemost Thromb 2003; 33: 68–74. 73849. [DOI] [PubMed] [Google Scholar]
- 9. Lip GY, Lowe GD. Fibrin D‐dimer: a useful clinical marker of thrombogenesis? Clin Sci 1995; 89: 205–14. [DOI] [PubMed] [Google Scholar]
- 10. Vene N, Mavri A, Kosmelj K, Stegnar M. High D‐dimer levels predict cardiovascular events in patients with chronic atrial fibrillation during oral anticoagulant therapy. Thromb Haemost 2003; 90: 1163–72. [DOI] [PubMed] [Google Scholar]
- 11. Mahe I, Drouet L, Simoneau G, Minh‐Muzeaux S, Caulin C, Bergmann JF. D‐dimer can predict survival in patients with chronic atrial fibrillation. Blood Coagul Fibrinolysis 2004; 15: 413–7. [DOI] [PubMed] [Google Scholar]
- 12. Kamath S, Blann AD, Chin BS, Lip GY. A prospective randomized trial of aspirin‐clopidogrel combination therapy and dose‐adjusted warfarin on indices of thrombogenesis and platelet activation in atrial fibrillation. J Am Coll Cardiol 2002; 40: 484–90. [DOI] [PubMed] [Google Scholar]
- 13. Lip GY, Lip PL, Zarifis J, Watson RD, Bareford D, Lowe GD, Beevers DG. Fibrin D‐dimer and beta‐thromboglobulin as markers of thrombogenesis and platelet activation in atrial fibrillation. Effects of introducing ultra‐low‐dose warfarin and aspirin. Circulation 1996; 94: 425–31. [DOI] [PubMed] [Google Scholar]
- 14. Ezekowitz MD, Reilly PA, Nehmiz G, Simmers TA, Nagarakanti R, Parcham‐Azad K, Pedersen KE, Lionetti DA, Stangier J, Wallentin L. Dabigatran with or without concomitant aspirin compared with warfarin alone in patients with nonvalvular atrial fibrillation (PETRO Study). Am J Cardiol 2007; 100: 1419–26. [DOI] [PubMed] [Google Scholar]
- 15. Weitz JI, Connolly SJ, Patel I, Salazar D, Rohatagi S, Mendell J, Kastrissios H, Jin J, Kunitada S. Randomised, parallel‐group, multicentre, multinational phase 2 study comparing edoxaban, an oral factor Xa inhibitor, with warfarin for stroke prevention in patients with atrial fibrillation. Thromb Haemost 2010; 104: 633–41. [DOI] [PubMed] [Google Scholar]
- 16. Hemker HC, Giesen P, Al Dieri R, Regnault V, de Smedt E, Wagenvoord R, Lecompte T, Beguin S. Calibrated automated thrombin generation measurement in clotting plasma. Pathophysiol Haemost Thromb 2003; 33: 4–15. 71636. [DOI] [PubMed] [Google Scholar]
- 17. Levey AS, Bosch JP, Lewis JB, Greene T, Rogers N, Roth D. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of Diet in Renal Disease Study Group. Ann Intern Med 1999; 130: 461–70. [DOI] [PubMed] [Google Scholar]
- 18. Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron 1976; 16: 31–41. [DOI] [PubMed] [Google Scholar]
- 19. Liesenfeld KH, Lehr T, Dansirikul C, Reilly PA, Connolly SJ, Ezekowitz MD, Yusuf S, Wallentin L, Haertter S, Staab A. Population pharmacokinetic analysis of the oral thrombin inhibitor dabigatran etexilate in patients with non‐valvular atrial fibrillation from the RE‐LY trial. J Thromb Haemost 2011; 9: 2168–75. [DOI] [PubMed] [Google Scholar]
- 20. Matsson EM, Eriksson UG, Palm JE, Artursson P, Karlgren M, Lazorova L, Brännström M, Ekdahl A, Dunér K, Knutson L, Johansson S, Schützer K‐M, Lennernäs H. Combined in vitro‐in vivo approach to assess the hepatobiliary disposition of a novel oral thrombin inhibitor. Mol Pharmacol 2013; 10: 4252–62. [DOI] [PubMed] [Google Scholar]
- 21. Hron G, Kollars M, Binder BR, Eichinger S, Kyrle PA. Identification of patients at low risk for recurrent venous thromboembolism by measuring thrombin generation. JAMA 2006; 296: 397–402. [DOI] [PubMed] [Google Scholar]
- 22. van Hylckama VA, Christiansen SC, Luddington R, Cannegieter SC, Rosendaal FR, Baglin TP. Elevated endogenous thrombin potential is associated with an increased risk of a first deep venous thrombosis but not with the risk of recurrence. Br J Haematol 2007; 138: 769–74. [DOI] [PubMed] [Google Scholar]
- 23. Dargaud Y, Lienhart A, Meunier S, Hequet O, Chavanne H, Chamouard V, Marin S, Negrier C. Major surgery in a severe haemophilia A patient with high titre inhibitor: use of the thrombin generation test in the therapeutic decision. Haemophilia 2005; 11: 552–8. [DOI] [PubMed] [Google Scholar]
- 24. Dargaud Y, Beguin S, Lienhart A, Al Dieri R, Trzeciak C, Bordet JC, Hemker HC, Negrier C. Evaluation of thrombin generating capacity in plasma from patients with haemophilia A and B. Thromb Haemost 2005; 93: 475–80. [DOI] [PubMed] [Google Scholar]
- 25. Wagenvoord RJ, Deinum J, Elg M, Hemker HC. The paradoxical stimulation by a reversible thrombin inhibitor of thrombin generation in plasma measured with thrombinography is caused by alpha‐macroglobulin‐thrombin. J Thromb Haemost 2010; 8: 1281–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item