

Abstract
Ten years ago, there was an emerging view that the molecular basis for adult mitochondrial disorders was largely known and that the clinical phenotypes had been well described. Nothing could have been further from the truth. The establishment of large cohorts of patients has revealed new aspects of the clinical presentation that were not previously appreciated. Over time, this approach is starting to provide an accurate understanding of the natural history of mitochondrial disease in adults. Advances in molecular diagnostics, underpinned by next generation sequencing technology, have identified novel molecular mechanisms. Recently described mitochondrial disease phenotypes have disparate causes, and yet share common mechanistic themes. In particular, disorders of mtDNA maintenance have emerged as a major cause of mitochondrial disease in adults. Progressive mtDNA depletion and the accumulation of mtDNA mutations explain some of the clinical features, but the genetic and cellular processes responsible for the mtDNA abnormalities are not entirely clear in each instance. Unfortunately, apart from a few specific examples, treatments for adult mitochondrial disease have not been forthcoming. However, the establishment of international consortia, and the first multinational randomised controlled trial, have paved the way for major progress in the near future, underpinned by growing interest from the pharmaceutical industry. Adult mitochondrial medicine is, therefore, in its infancy, and the challenge is to harness the new understanding of its molecular and cellular basis to develop treatments of real benefit to patients.
Keywords: mitochondrial disease, mitochondrial DNA, mitochondrial encephalomyopathy, myopathy, neurometabolic
Subject Categories: Genetics, Gene Therapy & Genetic Disease; Metabolism
Glossary
- 31P‐MRS
Phosphorus magnetic resonance spectroscopy is a functional MRI technique that can be used to indirectly measure mitochondrial ATP synthesis in skeletal and cardiac muscle in vivo
- Heteroplasmy (and the threshold effect)
A mixture of wild‐type and mutated mtDNA. At a cellular level, the percentage level of heteroplasmy determines whether there is a biochemical defect affecting oxidative phosphorylation and ATP synthesis
- Homoplasmy
When all mtDNA molecules are identical
- Kearns‐Sayre syndrome
A clinical syndrome involving progressive external ophthalmoplegia, ptosis (drooping eyelids), pigmentary retinopathy, cardiac conduction abnormalities, ataxia, an elevated cerebrospinal fluid protein, diabetes mellitus, sensorineural hearing loss and myopathy
- LHON
Leber hereditary optic neuropathy. A mtDNA disorder causing maternally inherited blindness that typically develops in mid‐adult life and preferentially affects men
- MELAS
Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke‐like episodes. A classical mitochondrial disorder usually due to the m.3243A>G mtDNA mutation
- MNGIE
Mitochondrial neurogastrointestinal encephalopathy. A rare autosomal recessive disorder caused by mutations in TP, which codes for thymidine phosphorylase
- MRI
Magnetic resonance imaging
- mtDNA depletion disorders
Autosomal recessive diseases causing a reduction in the amount of mtDNA
- mtDNA maintenance disorders
An emerging and complex group of nuclear genetic mitochondrial disorders which cause secondary point mutations or deletions of mtDNA. The secondary mtDNA abnormalities contribute to the clinical features of the disease
- mtDNA
Human mitochondrial DNA is a 16.5‐Kb molecule of double‐stranded DNA. MtDNA codes for 13 respiratory chain proteins and both tRNA and RNAs required by the mitochondrion for intra‐mitochondrial protein synthesis. Multiple copies of mtDNA are present in each cell. The number is tightly regulated and cell specific
- Oxidative phosphorylation
Metabolic pathway on the inner mitochondrial membrane which generates ATP through the oxidation of cofactors generated by intermediary metabolism
- PEO
Progressive external ophthalmoplegia. A clinical syndrome with weakness of the external eye muscles, often accompanied by ptosis (drooping eyelids)
- POLG
Nuclear gene encoding the mitochondrial DNA polymerase γ
Introduction
The year 1988 saw the first description of patients with pathogenic mutations of mitochondrial DNA (mtDNA) (Holt et al, 1988; Wallace et al, 1988), opening the floodgates, and paving the way for the new discipline of clinical mitochondrial medicine. The following decades saw major advances in our understanding of the molecular basis of mitochondrial disorders, initially focused on the smaller, more tractable mitochondrial genome. In later years, this was accompanied by the identification of novel nuclear gene defects (Koopman et al, 2012), which have emerged as a major cause of mitochondrial disorders (Gorman et al, 2015c). Review articles published a over decade ago described a molecular dichotomy between adults and children (Leonard & Schapira, 2000a,b), with mtDNA defects typically presenting in adult life, and children typically having autosomal recessive disorders due to presumed defects of the nuclear DNA. Over the last 10 years, it has become clear that mtDNA mutations can present throughout life and that autosomal recessive, dominant and X‐linked nuclear genetic disorders frequently present in adult life. This is but one of many examples where our initial impression of a clinical genotype–phenotype relationship has become blurred as our knowledge base has increased. Broad, overlapping phenotypes are caused by a myriad of molecular lesions affecting both genomes that present throughout the human life course (Fig 1). One could argue that separate review articles on childhood and adult mitochondrial diseases simply perpetuate the old dogma. However, despite this complexity, certain patterns do still exist, linking curious facets of the clinical presentation with associated biochemical abnormalities and their underlying molecular defects. All of this is, however, work in progress—and it remains to be seen whether the strict separation of childhood and paediatric mitochondrial disease will stand the test of time.
Figure 1. Mitochondrial biogenesis and the clinical features of mitochondrial disease in adults.
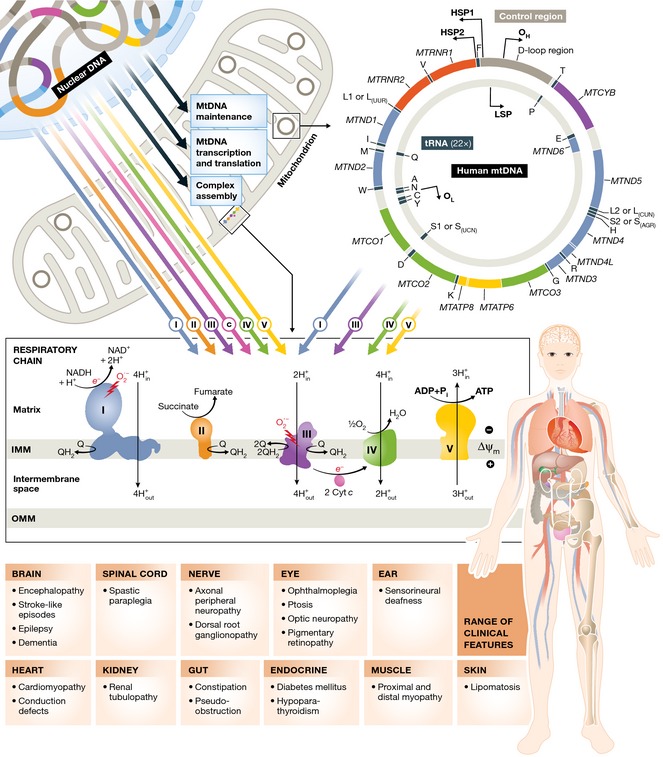
Upper panel: Adenosine triphosphate (ATP) is generated by the process of oxidative phosphorylation. This is achieved by the concerted action of ~90 proteins arranged into five respiratory chain complexes on the inner mitochondrial membrane. Thirteen of these proteins are encoded by the mitochondrial genome (mtDNA, right), which is present in high copy number in the mitochondrial matrix (100s to 1,000s per cell, depending on the cell type). The remaining mitochondrial proteins are synthesised in the cytoplasm from nuclear gene transcripts (left) and include the remaining structural subunits; complex assembly factors; proteins involved in the replication, maintenance and expression of mtDNA; and functional and structural components of the mitochondrial membrane. Mutations in genes encoding all of these proteins can cause mitochondrial diseases. Lower panel: the range of clinical features varies from patient to patient. Some have only one or a few of the features listed, whereas other patients have many in a multi‐system disease. Although some genetic defects cause specific phenotypes (e.g. the mtDNA mutations causing Leber hereditary optic neuropathy, which principally affect a single cell type in the vast majority of patients), other genetic defects cause an overlapping spectrum of phenotypes that can be caused by mtDNA and nuclear DNA mutations. The reasons for the tissue selectivity are not well understood.
This review focuses specifically on recent insight gained in mitochondrial disorders presenting predominantly in adult life. The huge growth of literature means that it cannot be comprehensive, but the examples illustrate some emerging principles.
Epidemiology
Until 1999, it was generally thought that mitochondrial disorders were extremely rare, perhaps affecting a few patients in every million of the population. However, a number of epidemiological studies performed in Europe and the antipodes challenged this view (Majamaa et al, 1998; Chinnery et al, 2000; Skladal et al, 2000; Darin et al, 2001). Initially focusing on the presence of mtDNA‐related disease, the most up to date figures for 2015 incorporate both mitochondrial DNA and nuclear DNA mutations, which are found in approximately 1:4,300 of the population (Gorman et al, 2015c). In the north of England, 23% of adults with mitochondrial disease had known or presumed nuclear genetic causes for their mitochondrial disease, with the most prevalent being mutations in SPG7 (Pfeffer et al, 2015), closely followed by PEO1 and OPA1 (Yu‐Wai‐Man et al, 2010a; Gorman et al, 2015c). For mtDNA, point mutations causing Leber's hereditary optic neuropathy (LHON, m.11778G>A, m.3460G>A and m.14484T>C) were found in ~40% of patients affected by mitochondrial disease (Man et al, 2003; Gorman et al, 2015c). Overall, these recent findings support the original view that mtDNA mutations are the major cause of mitochondrial disease in adults, but demonstrate clearly the importance of a diverse array of nuclear gene defects that can present to the clinic in adult life. The implication of these findings is profound. Different mutations are associated with different disease features and progress at different rates, and defining the genetic basis of the mitochondrial disorder has important implications for genetic counselling and prenatal diagnosis.
New phenotypes–new genotypes
Refined phenotypes
With the widespread availability of molecular diagnostic tests, increasing numbers of patients have been diagnosed worldwide. A number of major centres have built large cohorts of patients and have described the clinical features affecting these individuals. To a large extent, these endeavours have reinforced what was already known about the major characteristics of mitochondrial disease in adults, but a number of features had been under‐emphasised in the past. In part, this reflects the phenotypic overlap between mitochondrial disorders and common symptoms, which initially obscured the clear relationship with mitochondrial dysfunction.
Gastrointestinal (GI) features have emerged as a major element of many mitochondrial diseases (Kornblum et al, 2001). Both dysphagia and recurrent vomiting are now well‐recognised signs and can help to distinguish different molecular groups. For example, patients with chronic progressive external ophthalmoplegia and early dysphagia are more likely to have RR2BM mutations rather than a large single deletion of mtDNA as a cause of their mitochondrial disease (Pitceathly et al, 2012). Lower GI dysmotility is also a more common feature than was previously anticipated, with many patients describing constipation that can be very difficult to treat medically. This can lead to gastrointestinal pseudo‐obstruction and require surgical resection on occasions. Originally thought to be restricted to patients with mitochondrial neurogastrointestinal encephalopathy (MNGIE) caused by TP mutations (Nishino et al, 1999), gastrointestinal signs are now well recognised to be a frequent finding in patients with m.3243A>G (Kaufmann et al, 2009; Nesbitt et al, 2013), which, like MNGIE, may present with an acute abdomen (Garcia‐Velasco et al, 2003; Dindyal et al, 2014). Despite being called the “MELAS” mutation, m.3243A>G rarely causes mitochondrial encephalomyopathy with lactic acidosis and stroke‐like episodes (< 20% of affected mutation carriers), and much more commonly causes maternally inherited diabetes and deafness (Gorman et al, 2015c).
Cardiac anomalies were also under recognised in the past, and several studies have shown subclinical structural and functional abnormalities of the heart in patients with common mtDNA mutations. This has been observed in LHON (Nikoskelainen et al, 1994; Nemes et al, 2008), and m.3243A>G mutation carriers (Lodi et al, 2004; Vydt et al, 2007; Nemes et al, 2009; Bates et al, 2013). Whilst there is no doubt that some patients with mitochondrial disease develop heart failure which can be fatal if not treated effectively, the natural history of the subclinical cardiomyopathy is not well described, and it remains to be determined whether this is a major contributor to morbidity and mortality. There have also been an increased number of reports of sudden unexplained death in patients with mitochondrial disorders (Ng et al, 2015). Patients with Kearns‐Sayre syndrome are particularly vulnerable to heart block (Kabunga et al, 2015), which can cause syncope and sudden death without cardiac pacing, so it is now routine practice to monitor cardiac function in patients with mitochondrial disease with regular electrocardiography and echocardiography.
New genotypes
The number of different genetic abnormalities found in patients with mitochondrial disease has expanded exponentially in the last decade. The widespread availability of complete mtDNA sequencing has revealed rare, but recurrent, mtDNA mutations across the globe, and a greater number of families are now known to have extremely rare or private mtDNA disorders. In one recent epidemiological study, approximately one‐third of the mtDNA mutations detected were only found in single cases (Gorman et al, 2015c). Although many of these patients have classical features of mitochondrial disease, some have unique or unusual phenotypes, and in some, the phenotype is organ specific.
With the exception of LHON (Wallace et al, 1988), and maternally inherited deafness due to the m.1555A>G mtDNA mutation (Taylor et al, 2003), pathogenic homoplasmic mutations were thought to be a rare cause of mitochondrial disease and not to cause multi‐system disorders. However, it is now clear that this is not the case, with several well‐documented cases of multi‐system mtDNA disease due to homoplasmic mutations (Tiranti et al, 2000; McFarland et al, 2002; Limongelli et al, 2004). This has been an important finding, because the presence of heteroplasmy was used as a discriminating feature in diagnostic algorithms when investigating patients with multi‐system mitochondrial disorders, and homoplasmic mutations may have been inappropriately rejected as benign polymorphisms in the past. Finally, it is now clear that mtDNA mutations are much more common in the general population than was previously thought, and are found in between 0.5 and 1% of the population at > 1% heteroplasmy levels (Elliott et al, 2008). In some instances, the carriers harbour low levels of mtDNA heteroplasmy, but this is not always the case, pointing towards extraneous factors—perhaps in the nuclear genome or perhaps in the environment—that contribute to the pathogenesis of mtDNA disorders in the population.
With the advent of exome and whole‐genome sequencing, novel nuclear mitochondrial disease genes have been identified at an unanticipated rate, with one or two new genes being described every month over the last 2 years. Many of these disorders present in adult life. Again, although some patients have classical mitochondrial clinical syndromes, many have discriminating clinical features or a totally distinct phenotype. Perhaps the best example is a group of patients with biochemical defects affecting multiple components of the mitochondrial respiratory chain due to a disorder of intra‐mitochondrial protein translation (Rotig, 2010; Kemp et al, 2011; Hallberg & Larsson, 2014; Taylor et al, 2014). Although some of these patients present in early childhood, this is not always the case, and a detailed consideration illustrates the emerging principles. From first principles, one would expect these patients to have a very similar phenotype because the underlying mechanism and final common pathways are identical. However, patients with EARS2 mutations demonstrate new unique features on MRI imaging that discriminate them from other tRNA synthase disorders (Steenweg et al, 2012). Cardiomyopathy is particularly common in patients with AARS2 and MTO1 mutations (Gotz et al, 2011; Baruffini et al, 2013), which also cause a defect of intra‐mitochondrial protein synthesis. Mutations in RMND1 cause deafness (Janer et al, 2012), and mutations in YARS2 and PUS1 cause sideroblastic anaemia (Bykhovskaya et al, 2004; Riley et al, 2010), which is unusual in mitochondrial disease. It is not clear why these differences exist, given that all of these gene defects affect intra‐mitochondrial protein synthesis. However, the dual function of some of these enzymes may provide an explanation, with phenotypic modulation mediated through cytoplasmic effects.
Old genes new diseases
A further emerging field stems from the identification of mutations in well‐known nuclear disease genes in patients who also have a mitochondrial disorder based on conventional diagnostic criteria. OPA1 provides one example. For over a decade, mutations in OPA1 were known only to cause autosomal dominant optic atrophy, a very specific disorder that only causes blindness through the death of a specific cell type: the retinal ganglion cell (Alexander et al, 2000). However, parallel studies in Italy and the United Kingdom identified OPA1 mutations in several families with a multi‐system mitochondrial disease (Amati‐Bonneau et al, 2008; Hudson et al, 2008). These patients not only had optic atrophy, but also developed deafness, ptosis, ophthalmoplegia, ataxia and a peripheral neuropathy in later life (Yu‐Wai‐Man et al, 2010b). Routine mitochondrial investigations identified multiple deletions of mtDNA in the skeletal muscle biopsies, explaining the observed mosaic cytochrome c oxidase defect (Yu‐Wai‐Man et al, 2010c). Subsequent studies in animal models strongly suggest that different components of the phenotype are caused by different mechanisms. Disrupted fusion and fission of mitochondria, linked to apoptosis, probably explains the optic neuropathy (Alavi et al, 2009). On the other hand, the chronic progressive external ophthalmoplegia is most likely to be due to the secondary accumulation of multiple deletions of mtDNA (Yu‐Wai‐Man et al, 2010b). The secondary mtDNA defects are probably an indirect consequence of the fission–fission abnormality, adding OPA1 to the growing list of genes known to cause defects of mtDNA maintenance (discussed further below). Mutations in OPA1 have also recently been described in families with autosomal dominant Parkinsonism (Carelli et al, 2015), reminiscent of the Parkinsonism described in Scandinavian families with autosomal dominant POLG mutations (Luoma et al, 2004). It is clearly important to dissect out these mechanisms if we are to develop treatments effective that tackle the problems considered to be most important by patients.
A second example is SPG7, known for over a decade to cause autosomal recessive hereditary spastic paraplegia (Casari et al, 1998). Whole‐exome sequencing in a cohort of patients with unexplained ptosis and external ophthalmoplegia, and multiple deletions of mtDNA in skeletal muscle, led to the identification of SPG7 as a further disorder of mtDNA maintenance (Pfeffer et al, 2014). Intriguingly, SPG7 appears to be the most common nuclear genetic cause of mitochondrial disease in adults to date (Gorman et al, 2015c; Pfeffer et al, 2015).
What phenotypes are important for patients?
Although the expanding genotypic and phenotypic spectrum reinforces the long held view that mitochondrial disorders cause a complex overlapping myriad of disorders, a central question still remains: which elements are the most important for patients? As will be discussed later, this is critical for the development of new treatments for mitochondrial disease. The establishment of large cohorts of patients in Europe (http://mitocohort.ncl.ac.uk/; http://mitonet.org/) and North America (http://www.rarediseasesnetwork.org/namdc/index.htm) has provided the impetus for studies to tackle this problem. Perhaps surprisingly, muscle fatigue and weakness are at the top of the list, having the greatest impact on quality of life (Gorman et al, 2015a). These symptoms rarely form the focus of attention in a busy clinic (nor do they fascinate most clinicians and scientists), but ignoring the mechanisms behind these symptoms will distort priorities when it comes to the development of new treatments.
Disease mechanisms
The identification of novel genetic causes for mitochondrial disorders has opened a Pandora's box of novel mechanisms. Initial success identifying mutations affecting the structural subunits of the mitochondrial respiratory chain (Koopman et al, 2012) was accompanied by the molecular dissection of the assembly appara‐tus for complexes IV (Ghezzi & Zeviani, 2012), then I (Dieteren et al, 2012), III (Fernandez‐Vizarra & Zeviani, 2015) and finally V (Fujikawa et al, 2015). Several assembly proteins also appear to have dual functions and multiple functions and remarkably, multiple functions have also been observed for the structural peptide components of complex I (Elguindy & Nakamaru‐Ogiso, 2015). What has been perhaps the most interesting is the identification of increasingly more sophisticated disease mechanisms only indirectly connected to oxidative phosphorylation and ATP synthesis. In patients presenting in adult life, disorders affecting the maintenance of mtDNA have emerged as the single most important group.
For over 25 years, it has been known that some patients with chronic progressive external ophthalmoplegia have multiple deletions of mtDNA in skeletal muscle and that the disorder can be inherited as autosomal dominant or autosomal recessive trait. In the late 1990s, reverse genetic and candidate gene studies identified mutations in PEO1/Twinkle (Spelbrink et al, 2001), encoding the mtDNA helicase; and POLG (Van Goethem et al, 2001), the mtDNA polymerase. In other families, mutations were found in the adenine nucleotide translocase gene SLC25A4/ANT1 (Kaukonen et al, 2000). This raised the possibility that an imbalance of cytoplasmic nucleotides could cause a secondary defect of mtDNA. This mechanism was reaffirmed by the subsequent identification of autosomal recessive mutations in TK2 (Saada et al, 2001), encoding thymidine kinase; and DGUOK (Mandel et al, 2001), encoding deoxyguanosine kinase, in children with mtDNA depletion. The emerging pattern at this stage was of recessive mutations causing the loss of mtDNA (mtDNA depletion) and dominant mutations causing secondary mtDNA deletions and point mutations—a conclusion supported by the recent finding of single heterozygous mutations in TK2 and RRM2B causing late‐onset PEO with multiple deletions in muscle (Pitceathly et al, 2012), and on the other hand, recessive mutations in PEO1 causing infantile onset spinocerebellar ataxia (Hakonen et al, 2008). However, several examples have recently challenged this distinction. For example, patients with MNGIE typically present in early adult life with a progressive disorder mediated by a combination of both mtDNA depletion and mtDNA mutations (Mancuso et al, 2002; Gardner et al, 2015). Likewise, autosomal recessive mutations in POLG mediate their effect through both mtDNA depletion and secondary mtDNA mutation, as shown at the single‐cell level (Tzoulis et al, 2014). The balance between the depletion and secondary mutations appears to be cell and tissue specific, adding further complexity to the situation, but providing a potential explanation for the heterogeneous phenotypes.
Secondary mtDNA damage also appears to be intimately linked to mitochondrial structure, as illustrated by the multi‐system disease seen in ~20% of patients with autosomal dominant mutations in the mitochondrial fusion gene OPA1 (Yu‐Wai‐Man et al, 2010b), patients with autosomal recessive mutations in SPG7 which encodes the AAA‐protein paraplegin (Pfeffer et al, 2014), and dominant mutations in AFG3L2 (Gorman et al, 2015b), the paraplegin binding partner. It is not clear how the secondary mtDNA defects arise in these disorders, although this appears to be intimately linked to the defective fusion and fission of the organelles themselves. Finally, recent exome studies have identified new, unrelated disease mechanisms for mtDNA maintenance disorders, including autosomal dominant mutations in DNA2 (Ronchi et al, 2013), which codes for the DNA replication ATP‐dependent helicase/nuclease DNA2; and autosomal recessive mutations in MGME1 which cause secondary mtDNA deletions by disrupting the cleavage of single‐stranded mtDNA and the processing of DNA flap substrates (Kornblum et al, 2013).
When considered together, these interesting findings raise several important issues for adults with mtDNA diseases. Different elements of the clinical phenotype may be caused by different mechanisms: although some may be linked to the primary function of the mutated protein, others may be due to mtDNA depletion, or the accumulation of secondary mtDNA deletions and point mutations. This raises the possibility that therapies aimed at preventing or repairing the mtDNA damage may be applicable across several disease states. The mtDNA deletions and point mutations usually require time to accumulate, explaining clinical progression and also providing a window of therapeutic opportunity. Finally, given recent reports that similar mtDNA mutations accumulate with ageing and may contribute to the ageing process (Payne & Chinnery, 2015), these disorders may be useful models to understand and slowdown the human ageing process itself.
Natural history and biomarkers
Until recently, our understanding of the natural history of mitochondrial disease was largely based on anecdote, but longitudinal studies of large patient cohorts are now documenting clinical progression in mitochondrial disorders caused by both mtDNA and nuclear gene mutations (e.g. Grady et al, 2014). Although these studies are in their early stages, emerging evidence is reaffirming the view that some adult mitochondrial disorders have a fluctuating encephalopathic course, before settling into a more progressive neurodegenerative phenotype, whereas others present very much like a multi‐system neurodegenerative disorder. These data sets are providing a clearer view of the life course of a patient with mitochondrial disease. This can be helpful from a diagnostic perspective (e.g. the emergence of symptoms in a particular order may suggest one diagnosis more over another; for example, patients with ophthalmoplegia and hearing loss could have mutations in RRM2B or OPA1, but by the time the hearing loss emerges, all OPA1 patients have an optic neuropathy). The findings will also optimise the long‐term follow‐up of patients, allowing clinicians to look for specific complications at particular stages in their disease course. The interplay between genotype and phenotype is also more complex than expected, with evidence of phenotypic modifiers for both nuclear and mtDNA disorders, and the suggestion that clinical penetrance may even decrease with subsequent generations (reverse anticipation) (Howell & Mackey, 1998). This may be in response to improving environmental conditions, although this has yet to be substantiated.
Natural history studies are being supplemented by biomarker studies. Standard diagnostic biomarkers (creatine kinase, blood and cerebrospinal fluid lactate levels) are insensitive both diagnostically, and for measuring disease progression in adults (Jackson et al, 1995). Serum biomarkers such as FGF21 may be useful in diagnosing childhood mitochondrial myopathy (Suomalainen et al, 2011), but the levels in plasma do not change over short periods of time in adults (Koene et al, 2014). Non‐invasive 31P‐MRS shows great potential in patients with a demonstrable bioenergetic defect in skeletal and cardiac muscle (Hollingsworth et al, 2012), but at present, exercise physiology and muscle biopsy are the gold standard for measuring disease progression. Unfortunately, both techniques are logistically challenging and require considerable commitment from individual patients. Hopefully, novel metabolomic profiling methods will identify the “holy grail”—a sensitive, reliable non‐invasive biomarker linked to the known disease mechanism that can be used as a surrogate in early phase experimental medicine studies testing new treatments.
Current treatment options
The increased diagnostic yield through advanced genomics means that the majority of patients with suspected mitochondrial disease now have a molecular diagnosis. This is critically important for prognostic and genetic counselling and disease prevention, which has recently been reviewed elsewhere (Poulton et al, 2010). In certain circumstances, specific treatment approaches have been shown to be beneficial. These include the use of co‐enzyme Q10 in disorders of ubiquinone biosynthesis (Quinzii et al, 2007), allogenic bone marrow transplantation in MNGIE (Halter et al, 2015) and riboflavin supplementation in adults with riboflavin transporter disorders (Foley et al, 2014). However, despite over 1,000 articles describing possible treatments for mitochondrial disease published over five decades, there is very little objective evidence that any of the vitamins, co‐factors and dietary supplements are of any benefit in mitochondrial disorders (Pfeffer et al, 2012, 2013). Although this may suggest that more rigorous studies are needed, it can also be argued that the lack of any known treatment effect is evidence that these treatments are likely to only have subtle benefits at best, at least when applied across the whole patient group. It thus remains to be seen whether specific vitamins or cocktails of vitamins are helpful for defined molecular categories. One exception is idebenone, which has recently been recommended for approval by the European Medicines Agency Committee for Medicinal Products for Human Use (CHMP) for visual failure in LHON (http://www.ema.europa.eu/docs/en_GB/document_library/Summary_of_opinion_Initial_authorisation/human/003834/WC500188673.pdf). This is based on a randomised, placebo‐controlled, double‐blind, trial (900 mg/day) which failed its primary endpoint which was defined as the best recovery in visual acuity (Klopstock et al, 2011). However, data generated through an open‐labelled named‐patient programme convinced the reviewing committee that the treatment should be used in LHON. In most countries, there are still several hurdles to vault before idebenone can be prescribed for LHON, including obtaining approval by the health services in the various European Union member states. The development of idebenone as a treatment for LHON has taken over a decade, highlighting how complex, expensive and time‐consuming it is to take a medicine from early clinical use into clinical practice, despite the existence of “rare” or “orphan” disease legislation. Nevertheless, the published idebenone study demonstrated that multicentre, multinational randomised controlled trials are possible in mitochondrial disease, laying the foundation for the five trials ongoing in the United States studying a range of different compounds in different mitochondrial disorders (ClinicalTrials.gov) (Table 1).
Table 1.
Registered treatment studies for mitochondrial diseases (ClinicalTrials.gov 2015)
| Study title | Phase | Design | Medicine | Primary outcome(s) | Sites |
|---|---|---|---|---|---|
| Safety, Tolerability, Efficacy, PK and PD of RP103 in Children With Inherited Mitochondrial Disease (RP103‐MITO‐001) | II/III | Open Label | RP103 | Change in NPMDS | USA |
| EPI‐743 for Mitochondrial Respiratory Chain Diseases | II | Open Label | EPI‐743 | Change in Neuromuscular examination; AEs; Change in NPMDS | USA |
| Safety and Efficacy Study of EPI‐743 in Children With Leigh Syndrome | II | R, PC, DB | EPI‐743 | Change in NPMDS | USA |
| A Study Investigating the Safety, Tolerability, and Efficacy of MTP‐131 for the Treatment of Mitochondrial Myopathy | I/II | R, PC, DB | MTP‐131 | AEs; Change in vital signs; Changes in clinical laboratory evaluations | USA |
| RTA 408 Capsules in Patients With Mitochondrial Myopathy ‐ MOTOR | II | R, PC, DB | RTA408 | Change in peak workload (watts/kg) |
USA Denmark |
| EPI‐743 for Metabolism or Mitochondrial Disorders | II | R, PC, DB cross‐over | EPI‐743 | Change in NPMDS | USA |
| Phase 2 Study of EPI‐743 in Children With Pearson Syndrome | II | Open Label | EPI‐743 | Occurrence of episodes of sepsis, metabolic crisis or hepatic failure | USA |
| Safety Evaluation of Gene Therapy in Leber Hereditary Optic Neuropathy (LHON) Patients | I/II | Open Label | GS010 (AAV‐ND4) | Incidence of local and general adverse events and Serious Adverse Events | France |
| Safety and Efficacy Study of rAAV2‐ND4 Treatment of Leber Hereditary Optic Neuropathy (LHON) | I/II | Open Label | RAAV2‐ND4 | Visual acuity | China |
| Trial of Cyclosporine in the Acute Phase of Leber Hereditary Optic Neuropathy (CICLO‐NOHL) | II | Open Label | Cyclosporine | Visual acuity | France |
| Safety Study of an Adeno‐associated Virus Vector for Gene Therapy of Leber's Hereditary Optic Neuropathy (LHON) Caused by the G11778A Mutation (LHON GTT) | I | Open Label | scAAV2‐P1ND4v2 | AEs & SAEs | USA |
| MNGIE Allogeneic Hematopoietic Stem Cell Transplant Safety Study (MASS) | I | Open Label | Hematopoietic allogeneic stem cells | Neutrophil count (cells/l) | USA |
AE, adverse event; DB, double blind; NPMDS, Newcastle Paediatric Mitochondrial Disease Scale; PC, placebo controlled; PD, pharmacodynamics; PK, pharmacokinetics; R, randomised.
Conclusions
Reflecting back over the last decade, advances in molecular diagnostics have proceeded at an extraordinary pace, providing new diagnoses for singleton cases and small families. These findings are dissecting out the complex molecular mechanisms responsible for defects of oxidative phosphorylation. Large cohorts have now been assembled internationally, with over 3,000 patients being followed prospectively (see above for the web links). This has refined our understanding of phenotypic subgroups and the relationship to genotype, providing rich natural history data sets and underpinning first interventional clinical trials. Adult mitochondrial medicine has matured. We can now explain more to our patients about their disease than ever before, for some groups there are new treatment options, and there is a clear pathway towards a more comprehensive treatment in the near future.
Pending issues.
Comprehensive molecular diagnosis: With the advent of whole‐genome sequencing, a comprehensive molecular diagnosis for all patients with mitochondrial disease is within our grasp. International efforts to share data will facilitate this effort, enabling different groups to confirm or refute putative novel pathogenic mutations as early as possible.
Tissue selectivity: Ultimately all mitochondrial diseases are thought to be due to a bioenergetic defect, but different mitochondrial disorders have strikingly different phenotypes. The reasons for this are not known, but are the key to understanding pathogenesis and developing new treatments.
Biomarker development: Laboratory and preclinical animal studies have identified several new approaches to treat mitochondrial diseases. The major bottleneck is evaluating the potential clinical impact of these treatments in early phase human studies. Sensitive and clinically meaningful biomarkers linked to the underlying pathophysiology will accelerate this, allowing the early identification of effective and ineffective treatments.
Natural history studies: There is currently a limited understanding of the natural history of mitochondrial diseases. Detailed studies of genetically homogeneous clinical cohorts are required to identify which features change over a 6‐ to 12‐month period and could be used as clinical trial endpoints. Natural history studies should be linked to biomarker development.
Conflict of interest
The author was Chief Investigator for the Santhera Pharmaceuticals sponsored trial Rescue of Hereditary Optic Disease Outpatient Study (NCT00747487), but has remained an independent academic investigator throughout. He currently has a collaborative research project with GlaxoSmithKline on the natural history of mitochondrial disease. He has no conflict of interest with the work described in this article.
Acknowledgements
PFC is a Wellcome Trust Senior Fellow in Clinical Science (101876/Z/13/Z), and a UK NIHR Senior Investigator, who receives support from the Medical Research Council (UK) Mitochondrial Biology Unit (MC_UP_1501/2). He receives additional support from the Wellcome Trust Centre for Mitochondrial Research (096919Z/11/Z), the Medical Research Council (UK) Centre for Translational Muscle Disease Research (G0601943), EU FP7 TIRCON and the National Institute for Health Research (NIHR) Newcastle Biomedical Research Centre based at Cambridge University Hospitals NHS Foundation Trust and the University of Cambridge. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
EMBO Mol Med (2015) 7: 1503–1512
See the Glossary for abbreviations used in this article.
References
- Alavi MV, Fuhrmann N, Nguyen HP, Yu‐Wai‐Man P, Heiduschka P, Chinnery PF, Wissinger B (2009) Subtle neurological and metabolic abnormalities in an Opa1 mouse model of autosomal dominant optic atrophy. Exp Neurol 220: 404–409 [DOI] [PubMed] [Google Scholar]
- Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, Rodriguez M, Kellner U, Leo‐Kottler B, Auburger G et al (2000) OPA1, encoding a dynamin‐related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet 26: 211–215 [DOI] [PubMed] [Google Scholar]
- Amati‐Bonneau P, Valentino ML, Reynier P, Gallardo ME, Bornstein B, Boissiere A, Campos Y, Rivera H, de la Aleja JG, Carroccia R et al (2008) OPA1 mutations induce mitochondrial DNA instability and optic atrophy “plus” phenotypes. Brain 131: 338–351 [DOI] [PubMed] [Google Scholar]
- Baruffini E, Dallabona C, Invernizzi F, Yarham JW, Melchionda L, Blakely EL, Lamantea E, Donnini C, Santra S, Vijayaraghavan S et al (2013) MTO1 mutations are associated with hypertrophic cardiomyopathy and lactic acidosis and cause respiratory chain deficiency in humans and yeast. Hum Mutat 34: 1501–1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates MG, Hollingsworth KG, Newman JH, Jakovljevic DG, Blamire AM, Macgowan GA, Keavney BD, Chinnery PF, Turnbull DM, Taylor RW et al (2013) Concentric hypertrophic remodelling and subendocardial dysfunction in mitochondrial DNA point mutation carriers. Eur Heart J Cardiovasc Imaging 14: 650–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bykhovskaya Y, Casas K, Mengesha E, Inbal A, Fischel‐Ghodsian N (2004) Missense mutation in pseudouridine synthase 1 (PUS1) causes mitochondrial myopathy and sideroblastic anemia (MLASA). Am J Hum Genet 74: 1303–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carelli V, Musumeci O, Caporali L, Zanna C, La Morgia C, Del Dotto V, Porcelli AM, Rugolo M, Valentino ML, Iommarini L et al (2015) Syndromic parkinsonism and dementia associated with OPA1 missense mutations. Ann Neurol 78: 21–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casari G, De Fusco M, Ciarmatori S, Zeviani M, Mora M, Fernandez P, De Michele G, Filla A, Cocozza S, Marconi R et al (1998) Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear‐encoded mitochondrial metalloprotease. Cell 93: 973–983 [DOI] [PubMed] [Google Scholar]
- Chinnery PF, Johnson MA, Wardell TM, Singh‐Kler R, Hayes C, Brown DT, Taylor RW, Bindoff LA, Turnbull DM (2000) Epidemiology of pathogenic mitochondrial DNA mutations. Ann Neurol 48: 188–193 [PubMed] [Google Scholar]
- Darin N, Oldfors A, Moslemi AR, Holme E, Tulinius M (2001) The incidence of mitochondrial encephalomyopathies in childhood: clinical features and morphological, biochemical, and DNA anbormalities. Ann Neurol 49: 377–383 [PubMed] [Google Scholar]
- Dieteren CE, Koopman WJ, Swarts HG, Peters JG, Maczuga P, van Gemst JJ, Masereeuw R, Smeitink JA, Nijtmans LG, Willems PH (2012) Subunit‐specific incorporation efficiency and kinetics in mitochondrial complex I homeostasis. J Biol Chem 287: 41851–41860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dindyal S, Mistry K, Angamuthu N, Smith G, Hilton D, Arumugam P, Mathew J (2014) MELAS syndrome presenting as an acute surgical abdomen. Ann R Coll Surg Engl 96: 101E–103E [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elguindy MM, Nakamaru‐Ogiso E (2015) Apoptosis‐inducing factor (AIF) and its family member protein, AMID, are rotenone‐sensitive NADH: ubiquinone oxidoreductases (NDH‐2). J Biol Chem 290: 20815–20826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott HR, Samuels DC, Eden JA, Relton CL, Chinnery PF (2008) Pathogenic mitochondrial DNA mutations are common in the general population. Am J Hum Genet 83: 254–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez‐Vizarra E, Zeviani M (2015) Nuclear gene mutations as the cause of mitochondrial complex III deficiency. Front Genet 6: 134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley AR, Menezes MP, Pandraud A, Gonzalez MA, Al‐Odaib A, Abrams AJ, Sugano K, Yonezawa A, Manzur AY, Burns J et al (2014) Treatable childhood neuronopathy caused by mutations in riboflavin transporter RFVT2. Brain 137: 44–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujikawa M, Sugawara K, Tanabe T, Yoshida M (2015) Assembly of human mitochondrial ATP synthase through two separate intermediates, F‐c‐ring and b‐e‐g complex. FEBS Lett 589: 2707–2712 [DOI] [PubMed] [Google Scholar]
- Garcia‐Velasco A, Gomez‐Escalonilla C, Guerra‐Vales JM, Cabello A, Campos Y, Arenas J (2003) Intestinal pseudo‐obstruction and urinary retention: cardinal features of a mitochondrial DNA‐related disease. J Intern Med 253: 381–385 [DOI] [PubMed] [Google Scholar]
- Gardner K, Payne BA, Horvath R, Chinnery PF (2015) Use of stereotypical mutational motifs to define resolution limits for the ultra‐deep resequencing of mitochondrial DNA. Eur J Hum Genet 23: 413–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghezzi D, Zeviani M (2012) Assembly factors of human mitochondrial respiratory chain complexes: physiology and pathophysiology. Adv Exp Med Biol 748: 65–106 [DOI] [PubMed] [Google Scholar]
- Gorman GS, Elson JL, Newman J, Payne B, McFarland R, Newton JL, Turnbull DM (2015a) Perceived fatigue is highly prevalent and debilitating in patients with mitochondrial disease. Neuromuscul Disord 25: 563–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman GS, Pfeffer G, Griffin H, Blakely EL, Kurzawa‐Akanbi M, Gabriel J, Sitarz K, Roberts M, Schoser B, Pyle A et al (2015b) Clonal expansion of secondary mitochondrial DNA deletions associated with spinocerebellar ataxia type 28. JAMA Neurol 72: 106–111 [DOI] [PubMed] [Google Scholar]
- Gorman GS, Schaefer AM, Ng Y, Gomez N, Blakely EL, Alston CL, Feeney C, Horvath R, Yu‐Wai‐Man P, Chinnery PF et al (2015c) Prevalence of nuclear and mtDNA mutations related to adult mitochondrial disease. Ann Neurol 77: 753–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotz A, Tyynismaa H, Euro L, Ellonen P, Hyotylainen T, Ojala T, Hamalainen RH, Tommiska J, Raivio T, Oresic M et al (2011) Exome sequencing identifies mitochondrial alanyl‐tRNA synthetase mutations in infantile mitochondrial cardiomyopathy. Am J Hum Genet 88: 635–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady JP, Campbell G, Ratnaike T, Blakely EL, Falkous G, Nesbitt V, Schaefer AM, McNally RJ, Gorman GS, Taylor RW et al (2014) Disease progression in patients with single, large‐scale mitochondrial DNA deletions. Brain 137: 323–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakonen AH, Goffart S, Marjavaara S, Paetau A, Cooper H, Mattila K, Lampinen M, Sajantila A, Lonnqvist T, Spelbrink JN et al (2008) Infantile‐onset spinocerebellar ataxia and mitochondrial recessive ataxia syndrome are associated with neuronal complex I defect and mtDNA depletion. Hum Mol Genet 17: 3822–3835 [DOI] [PubMed] [Google Scholar]
- Hallberg BM, Larsson NG (2014) Making proteins in the powerhouse. Cell Metab 20: 226–240 [DOI] [PubMed] [Google Scholar]
- Halter JP, Michael W, Schupbach M, Mandel H, Casali C, Orchard K, Collin M, Valcarcel D, Rovelli A, Filosto M et al (2015) Allogeneic haematopoietic stem cell transplantation for mitochondrial neurogastrointestinal encephalomyopathy. Brain 138: 2847–2858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth KG, Gorman GS, Trenell MI, McFarland R, Taylor RW, Turnbull DM, MacGowan GA, Blamire AM, Chinnery PF (2012) Cardiomyopathy is common in patients with the mitochondrial DNA m.3243A>G mutation and correlates with mutation load. Neuromuscul Disord 22: 592–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt I, Harding AE, Morgan‐Hughes JA (1988) Deletion of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 331: 717–719 [DOI] [PubMed] [Google Scholar]
- Howell N, Mackey DA (1998) Low‐penetrance branches in matrilineal pedigrees with Leber Hereditary Optic Neuropathy. Am J Hum Genet 63: 1220–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson G, Amati‐Bonneau P, Blakely EL, Stewart JD, He L, Schaefer AM, Griffiths PG, Ahlqvist K, Suomalainen A, Reynier P et al (2008) Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: a novel disorder of mtDNA maintenance. Brain 131: 329–337 [DOI] [PubMed] [Google Scholar]
- Jackson MJ, Schaefer JA, Johnson MA, Morris AAM, Turnbull DM, Bindoff LA (1995) Presentation and clinical investigation of mitochondrial respiratory chain disease. Brain 118: 339–357 [DOI] [PubMed] [Google Scholar]
- Janer A, Antonicka H, Lalonde E, Nishimura T, Sasarman F, Brown GK, Brown RM, Majewski J, Shoubridge EA (2012) An RMND1 Mutation causes encephalopathy associated with multiple oxidative phosphorylation complex deficiencies and a mitochondrial translation defect. Am J Hum Genet 91: 737–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabunga P, Lau AK, Phan K, Puranik R, Liang C, Davis RL, Sue CM, Sy RW (2015) Systematic review of cardiac electrical disease in Kearns‐Sayre syndrome and mitochondrial cytopathy. Int J Cardiol 181: 303–310 [DOI] [PubMed] [Google Scholar]
- Kaufmann P, Engelstad K, Wei Y, Kulikova R, Oskoui M, Battista V, Koenigsberger DY, Pascual JM, Sano M, Hirano M et al (2009) Protean phenotypic features of the A3243G mitochondrial DNA mutation. Arch Neurol 66: 85–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaukonen J, Juselius JK, Tiranti V, Kyttala A, Zeviani M, Comi GP, Keranen S, Peltonen L, Suomalainen A (2000) Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science 289: 782–785 [DOI] [PubMed] [Google Scholar]
- Kemp JP, Smith PM, Pyle A, Neeve VC, Tuppen HA, Schara U, Talim B, Topaloglu H, Holinski‐Feder E, Abicht A et al (2011) Nuclear factors involved in mitochondrial translation cause a subgroup of combined respiratory chain deficiency. Brain 134: 183–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klopstock T, Yu‐Wai‐Man P, Dimitriadis K, Rouleau J, Heck S, Bailie M, Atawan A, Chattopadhyay S, Schubert M, Garip A et al (2011) A randomized placebo‐controlled trial of idebenone in Leber's hereditary optic neuropathy. Brain 134: 2677–2686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koene S, de Laat P, van Tienoven DH, Vriens D, Brandt AM, Sweep FC, Rodenburg RJ, Donders AR, Janssen MC, Smeitink JA (2014) Serum FGF21 levels in adult m.3243A>G carriers: clinical implications. Neurology 83: 125–133 [DOI] [PubMed] [Google Scholar]
- Koopman WJ, Willems PH, Smeitink JA (2012) Monogenic mitochondrial disorders. N Engl J Med 366: 1132–1141 [DOI] [PubMed] [Google Scholar]
- Kornblum C, Broicher R, Walther E, Seibel P, Reichmann H, Klockgether T, Herberhold C, Schroder R (2001) Cricopharyngeal achalasia is a common cause of dysphagia in patients with mtDNA deletions. Neurology 56: 1409–1412 [DOI] [PubMed] [Google Scholar]
- Kornblum C, Nicholls TJ, Haack TB, Scholer S, Peeva V, Danhauser K, Hallmann K, Zsurka G, Rorbach J, Iuso A et al (2013) Loss‐of‐function mutations in MGME1 impair mtDNA replication and cause multisystemic mitochondrial disease. Nat Genet 45: 214–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard JV, Schapira AH (2000a) Mitochondrial respiratory chain disorders I: mitochondrial DNA defects. Lancet 355: 299–304 [DOI] [PubMed] [Google Scholar]
- Leonard JV, Schapira AVH (2000b) Mitochondrial respiratory chain disorders II: neurodegenerative disorders and nuclear gene defects. Lancet 355: 389–394 [DOI] [PubMed] [Google Scholar]
- Limongelli A, Schaefer J, Jackson S, Invernizzi F, Kirino Y, Suzuki T, Reichmann H, Zeviani M (2004) Variable penetrance of a familial progressive necrotising encephalopathy due to a novel tRNA(Ile) homoplasmic mutation in the mitochondrial genome. J Med Genet 41: 342–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodi R, Rajagopalan B, Blamire AM, Crilley JG, Styles P, Chinnery PF (2004) Abnormal cardiac energetics in patients carrying the A3243G mtDNA mutation measured in vivo using phosphorus MR spectroscopy. Biochim Biophys Acta 1657: 146–150 [DOI] [PubMed] [Google Scholar]
- Luoma P, Melberg A, Rinne JO, Kaukonen JA, Nupponen NN, Chalmers RM, Oldfors A, Rautakorpi I, Peltonen L, Majamaa K et al (2004) Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: clinical and molecular genetic study. Lancet 364: 875–882 [DOI] [PubMed] [Google Scholar]
- Majamaa K, Moilanen JS, Uimonen S, Remes AM, Salmela PI, Karppa M, Majamaa‐Volti KAM, Rusanen H, Sorri M, Peuhkurinen KJ et al (1998) Epidemiology of A3243G, the mutation for mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes: prevalence of the mutation in an adult population. Am J Hum Genet 63: 447–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man PY, Griffiths PG, Brown DT, Howell N, Turnbull DM, Chinnery PF (2003) The epidemiology of leber hereditary optic neuropathy in the north East of England. Am J Hum Genet 72: 333–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso M, Salviati L, Sacconi S, Otaegui D, Camano P, Marina A, Bacman S, Moraes CT, Carlo JR, Garcia M et al (2002) Mitochondrial DNA depletion: mutations in thymidine kinase gene with myopathy and SMA. Neurology 59: 1197–1202 [DOI] [PubMed] [Google Scholar]
- Mandel H, Szargel R, Labay V, Elpeleg O, Saada A, Shalata A, Anbinder Y, Berkowitz D, Hartman C, Barak M et al (2001) The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat Genet 29: 337–341 [DOI] [PubMed] [Google Scholar]
- McFarland R, Clark KM, Morris AA, Taylor RW, Macphail S, Lightowlers RN, Turnbull DM (2002) Multiple neonatal deaths due to a homoplasmic mitochondrial DNA mutation. Nat Genet 30: 145–146 [DOI] [PubMed] [Google Scholar]
- Nemes A, De Coo IF, Spruijt L, Smeets HJ, Chinnery PF, Soliman OI, Geleijnse ML, Ten Cate FJ (2008) Is there alteration in aortic stiffness in Leber hereditary optic neuropathy? Eur J Ophthalmol 18: 309–312 [DOI] [PubMed] [Google Scholar]
- Nemes A, Geleijnse ML, Sluiter W, Vydt TC, Soliman OI, van Dalen BM, Vletter WB, ten Cate FJ, Smeets HJ, de Coo RF (2009) Aortic distensibility alterations in adults with m.3243A>G MELAS gene mutation. Swiss Med Wkly 139: 117–120 [DOI] [PubMed] [Google Scholar]
- Nesbitt V, Pitceathly RD, Turnbull DM, Taylor RW, Sweeney MG, Mudanohwo EE, Rahman S, Hanna MG, McFarland R (2013) The UK MRC Mitochondrial Disease Patient Cohort Study: clinical phenotypes associated with the m.3243A>G mutation–implications for diagnosis and management. J Neurol Neurosurg Psychiatry 84: 936–938 [DOI] [PubMed] [Google Scholar]
- Ng YS, Grady JP, Lax NZ, Bourke JP, Alston CL, Hardy SA, Falkous G, Schaefer AG, Radunovic A, Mohiddin SA et al (2015) Sudden adult death syndrome in m.3243A>G‐related mitochondrial disease: an unrecognized clinical entity in young, asymptomatic adults. Eur Heart J doi:10.1093/eurheartj/ehv306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikoskelainen EK, Savontaus ML, Huoponen K, Antila K, Hartiala J (1994) Pre‐excitation syndrome in Leber's hereditary optic neuropathy. Lancet 344: 857–858 [DOI] [PubMed] [Google Scholar]
- Nishino I, Spinazzola A, Hirano M (1999) Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science 283: 689–692 [DOI] [PubMed] [Google Scholar]
- Payne BA, Chinnery PF (2015) Mitochondrial dysfunction in aging: much progress but many unresolved questions. Biochim Biophys Acta 1847: 1347–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer G, Majamaa K, Turnbull DM, Thorburn D, Chinnery PF (2012) Treatment for mitochondrial disorders. Cochrane Database Syst Rev 4: CD004426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer G, Horvath R, Klopstock T, Mootha VK, Suomalainen A, Koene S, Hirano M, Zeviani M, Bindoff LA, Yu‐Wai‐Man P et al (2013) New treatments for mitochondrial disease‐no time to drop our standards. Nat Rev Neurol 9: 474–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer G, Gorman GS, Griffin H, Kurzawa‐Akanbi M, Blakely EL, Wilson I, Sitarz K, Moore D, Murphy JL, Alston CL et al (2014) Mutations in the SPG7 gene cause chronic progressive external ophthalmoplegia through disordered mitochondrial DNA maintenance. Brain 137: 1323–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer G, Pyle A, Griffin H, Miller J, Wilson V, Turnbull L, Fawcett K, Sims D, Eglon G, Hadjivassiliou M et al (2015) SPG7 mutations are a common cause of undiagnosed ataxia. Neurology 84: 1174–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitceathly RD, Smith C, Fratter C, Alston CL, He L, Craig K, Blakely EL, Evans JC, Taylor J, Shabbir Z et al (2012) Adults with RRM2B‐related mitochondrial disease have distinct clinical and molecular characteristics. Brain 135: 3392–3403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulton J, Chiaratti MR, Meirelles FV, Kennedy S, Wells D, Holt IJ (2010) Transmission of mitochondrial DNA diseases and ways to prevent them. PLoS Genet 6: e1001066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinzii CM, DiMauro S, Hirano M (2007) Human coenzyme Q10 deficiency. Neurochem Res 32: 723–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley LG, Cooper S, Hickey P, Rudinger‐Thirion J, McKenzie M, Compton A, Lim SC, Thorburn D, Ryan MT, Giege R et al (2010) Mutation of the mitochondrial tyrosyl‐tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia–MLASA syndrome. Am J Hum Genet 87: 52–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronchi D, Di Fonzo A, Lin W, Bordoni A, Liu C, Fassone E, Pagliarani S, Rizzuti M, Zheng L, Filosto M et al (2013) Mutations in DNA2 link progressive myopathy to mitochondrial DNA instability. Am J Hum Genet 92: 293–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotig A (2010) Genetic bases of mitochondrial respiratory chain disorders. Diabetes Metab 36: 97–107 [DOI] [PubMed] [Google Scholar]
- Saada A, Shaag A, Mandel H, Nevo Y, Eriksson S, Elpeleg O (2001) Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nat Genet 29: 342–344 [DOI] [PubMed] [Google Scholar]
- Skladal D, Bernier FP, Halliday JL, Thorburn DR (2000) Birth prevalence of mitochondrial respiratory chain defects in children. J Inher Metab Dis 23: 138 [Google Scholar]
- Spelbrink JN, Li FY, Tiranti V, Nikali K, Yuan QP, Tariq M, Wanrooij S, Garrido N, Comi G, Morandi L et al (2001) Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4‐like protein localized in mitochondria. Nat Genet 28: 223–231 [DOI] [PubMed] [Google Scholar]
- Steenweg ME, Ghezzi D, Haack T, Abbink TE, Martinelli D, van Berkel CG, Bley A, Diogo L, Grillo E, Te Water Naude J et al (2012) Leukoencephalopathy with thalamus and brainstem involvement and high lactate “LTBL” caused by EARS2 mutations. Brain 135: 1387–1394 [DOI] [PubMed] [Google Scholar]
- Suomalainen A, Elo JM, Pietilainen KH, Hakonen AH, Sevastianova K, Korpela M, Isohanni P, Marjavaara SK, Tyni T, Kiuru‐Enari S et al (2011) FGF‐21 as a biomarker for muscle‐manifesting mitochondrial respiratory chain deficiencies: a diagnostic study. Lancet Neurol 10: 806–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RW, Giordano C, Davidson MM, d'Amati G, Bain H, Hayes CM, Leonard H, Barron MJ, Casali C, Santorelli FM et al (2003) A homoplasmic mitochondrial transfer ribonucleic acid mutation as a cause of maternally inherited hypertrophic cardiomyopathy. J Am Coll Cardiol 41: 1786–1796 [DOI] [PubMed] [Google Scholar]
- Taylor RW, Pyle A, Griffin H, Blakely EL, Duff J, He L, Smertenko T, Alston CL, Neeve VC, Best A et al (2014) Use of whole‐exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. JAMA 312: 68–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiranti V, Corona P, Greco M, Taanman JW, Carrara F, Lamantea E, Nijtmans L, Uziel G, Zeviani M (2000) A novel frameshift mutation of the mtDNA COIII gene leads to impaired assembly of cytochrome c oxidase in a patient affected by Leigh‐like syndrome. Hum Mol Genet 9: 2733–2742 [DOI] [PubMed] [Google Scholar]
- Tzoulis C, Tran GT, Coxhead J, Bertelsen B, Lilleng PK, Balafkan N, Payne B, Miletic H, Chinnery PF, Bindoff LA (2014) Molecular pathogenesis of polymerase gamma‐related neurodegeneration. Ann Neurol 76: 66–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Goethem G, Dermaut B, Lofgren A, Martin J‐J, Van Broeckhoven C (2001) Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat Genet 28: 211–212 [DOI] [PubMed] [Google Scholar]
- Vydt TC, de Coo RF, Soliman OI, Ten Cate FJ, van Geuns RJ, Vletter WB, Schoonderwoerd K, van den Bosch BJ, Smeets HJ, Geleijnse ML (2007) Cardiac involvement in adults with m.3243A>G MELAS gene mutation. Am J Cardiol 99: 264–269 [DOI] [PubMed] [Google Scholar]
- Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, Elsas LJD, Nikoskelainen EK (1988) Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science 242: 1427–1430 [DOI] [PubMed] [Google Scholar]
- Yu‐Wai‐Man P, Griffiths PG, Burke A, Sellar PW, Clarke MP, Gnanaraj L, Ah‐Kine D, Hudson G, Czermin B, Taylor RW et al (2010a) The prevalence and natural history of dominant optic atrophy due to OPA1 mutations. Ophthalmology 117: 1538–1546, 1546 e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu‐Wai‐Man P, Griffiths PG, Gorman GS, Lourenco CM, Wright AF, Auer‐Grumbach M, Toscano A, Musumeci O, Valentino ML, Caporali L et al (2010b) Multi‐system neurological disease is common in patients with OPA1 mutations. Brain 133: 771–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu‐Wai‐Man P, Sitarz KS, Samuels DC, Griffiths PG, Reeve AK, Bindoff LA, Horvath R, Chinnery PF (2010c) OPA1 mutations cause cytochrome c oxidase deficiency due to loss of wild‐type mtDNA molecules. Hum Mol Genet 19: 3043–3052 [DOI] [PMC free article] [PubMed] [Google Scholar]