

Summary
In Duchenne muscular dystrophy (DMD), the search for new biomarkers to follow the evolution of the disease is of fundamental importance in the light of the evolving gene and pharmacological therapies. In addition to the lack of dystrophin, secondary events including changes in calcium levels, inflammation and fibrosis greatly contribute to DMD progression and the molecules involved in these events may represent potential biomarkers. In this study, we performed a comparative evaluation of the progression of dystrophy within muscles that are differently affected by dystrophy (diaphragm; DIA and quadriceps; QDR) or spared (intrinsic laryngeal muscles) using the mdx mice model of DMD. We assessed muscle levels of calsequestrin (calcium‐related protein), tumour necrosis factor (TNF‐α; pro‐inflammatory cytokine), tumour growth factor (TGF‐β; pro‐fibrotic factor) and MyoD (muscle proliferation) vs. histopathology at early (1 and 4 months of age) and late (9 months of age) stages of dystrophy. Fibrosis was the primary feature in the DIA of mdx mice (9 months: 32% fibrosis), which was greater than in the QDR (9 months: 0.6% fibrosis). Muscle regeneration was the primary feature in the QDR (9 months: 90% of centrally nucleated fibres areas vs. 33% in the DIA). The QDR expressed higher levels of calsequestrin than the DIA. Laryngeal muscles showed normal levels of TNF‐α, TGF‐β and MyoD. A positive correlation between histopathology and cytokine levels was observed only in the diaphragm, suggesting that TNF‐α and TGF‐β serve as markers of dystrophy primarily for the diaphragm.
Keywords: calsequestrin, dystrophy, mdx, tumour growth factor‐β, tumour necrosis factor‐α
Duchenne muscular dystrophy (DMD) is the most common genetic muscle disease in childhood, affecting one in 3500–4000 males born (Engel et al. 1994; Mendell et al. 2012). DMD is characterized by a progressive wasting of skeletal and cardiac muscles, cardiorespiratory failure and death around the third decade of life (Engel et al. 1994; Rall & Grimm 2012). Although there has been extensive research on gene‐, cell‐ and drug‐mediated therapies for various muscular dystrophies (Konieczny et al. 2013), there is still no effective treatment for DMD. The use of experimental models, such as mdx mice (Bulfield et al. 1984), greatly improves the knowledge of DMD pathophysiology and therapy.
Duchenne muscular dystrophy and mdx mice are characterized by the lack of dystrophin due to a mutation in the dystrophin gene present in the X chromosome (Hoffman et al. 1987). The absence of dystrophin results in sarcolemma instability, an increased influx of calcium, the activation of proteases and myonecrosis (Whitehead et al. 2006). During the early stages of dystrophy, muscle regeneration compensates for the degeneration, but with the progression of disease, muscle degeneration and fibrosis eventually predominate (Engel et al. 1994; Pastoret & Sebille 1995). Although the lack of dystrophin is the primary event underlying the pathogenesis of DMD, downstream secondary pathways such as inflammation (Porter et al. 2002; Tidball 2005) and fibrosis (Bernasconi et al. 1995) also contribute to the disease progression.
Gene expression profile studies have revealed that most of the secondary pathways of DMD involve inflammatory responses (Tian et al. 2014). Inflammation responses in dystrophic muscles contain several types of immune cells that release pro‐inflammatory cytokines such as tumour necrosis factor‐alpha (Porter et al. 2002; Tidball 2005; Hodgetts et al. 2006; Villalta et al. 2009). These pro‐inflammatory cytokines activate metalloproteinases, which breakdown the extracellular matrix and promote the production of pro‐fibrotic factors, including tumour growth factor‐beta, that will further contribute to myonecrosis and fibrotic deposition (Gosselin et al. 2004; Li et al. 2009). In addition, calcium homoeostasis pathways, which are differently regulated in presymptomatic and symptomatic DMD patients (Tian et al. 2014) and have a role in the pathogenesis of mdx mice (Whitehead et al. 2006), have been suggested to be potential targets for further molecular diagnostic tests (Tian et al. 2014).
One interesting aspect of dystrophy is the observation that the pathology of the disease differs depending on the muscle, despite the fact that dystrophin is absent in all muscles. For instance, respiratory muscles, such as the diaphragm, are more affected at later stages than are limb muscles (Stedman et al. 1991; Zhou et al. 2006), but the extraocular and intrinsic laryngeal (ILM) muscles are spared and show no signs of muscle degeneration, possibly related to improved calcium homoeostasis (Khurana et al. 1995; Marques et al. 2007; Ferretti et al. 2009; Zeiger et al. 2010).
In the present study, we hypothesized that the pathways related to inflammation, fibrosis and calcium regulation might be differently affected in muscles that show different pathological intensities over the course of the disease, such as the respiratory (diaphragm), limb (quadriceps) and spared (laryngeal) muscles. We performed a comparative evaluation of the progression of dystrophy in the diaphragm, quadriceps and ILM of mdx mice and assessed whether their expression of tumour necrosis factor‐alpha (TNF‐α; inflammation), tumour growth factor‐beta (TGF‐β; fibrosis), calsequestrin (CSQ; calcium regulation) and MyoD (muscle cell proliferation) correlated with their histopathology at very early (1 month), early (4 months) and later (9 months) stages of the disease.
Material and methods
Animals
Male and female mdx mice (C57BL/10‐Dmdmdx/PasUnib) at 1, 4 and 9 months of age were used in these experiments. Male and female C57BL/10 mice (C57BL/10ScCr/PasUnib) at the same ages (1, 4 and 9 months) were used as controls. They were obtained from a breeding colony, maintained by our institutional animal care facility, and housed according to institutional guidelines with free access to food and water. No differences were seen between males and females regarding the parameters studied, and they were mixed equally at the different ages.
Histopathology
Quadriceps femoris (QDR), diaphragm (DIA) and intrinsic laryngeal (IL) muscles were studied. These muscles were chosen because they represent appendicular (QDR) and respiratory (DIA) muscles that are affected differently in DMD, with respiratory muscles being more severely impaired (Stedman et al. 1991). The intrinsic laryngeal muscles (ILM) are spared from dystrophy, showing no signs of muscle degeneration and regeneration (Marques et al. 2007; Ferretti et al. 2009).
Cryostat transverse sections (7 μm) of QDR and DIA from C57BL/10 (n = 15) and mdx (n = 15) mice at the ages of 1 (n = 5), 4 (n = 5) and 9 (n = 5) months were stained with haematoxylin–eosin. Slides were placed in a Nikon Eclipse E 400 microscope and viewed with a video camera (Nikon Express Series; Tokyo, Japan). Non‐overlapping images were tiled together using the ImagePro Express software (Media Cybernetic, Silver Spring, MD, USA). The following areas within each transverse section were characterized as previously described (Machado et al. 2011) and measured using the imagepro express software Media Cybernetics, Silver Spring, Maryland, USA: (i) peripheral nucleated areas, which are the areas containing fibres with peripheral cell nuclei (Figure 1a,b) indicative of fibres that have not undergone cycles of muscle degeneration–regeneration; (ii) inflammation (Figure 1c) and regeneration (Figure 1d) areas, which are the areas containing primarily inflammatory cell infiltration (inflammation) or the areas containing myotubes–myofibres with a small diameter, central cell nuclei and small size (indicative of regeneration); and (iii) central nucleated areas, which are the areas with fully regenerated fibres with central nuclei and normal size (Figure 1e). Myotubes–myofibres appeared in the cross sections as plump, basophilic cells with at least one centrally located nucleus.
Figure 1.
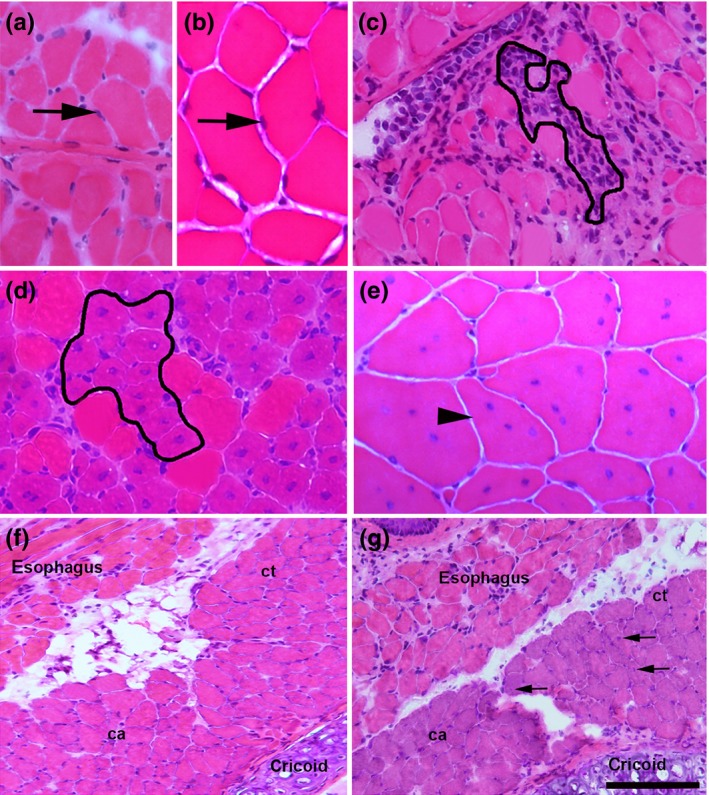
Histological appearance of a 9‐month‐old diaphragm (a) and quadriceps (b) muscle in controls, displaying fibres with peripheral nuclei (arrows). c, histological appearance of a 4‐month‐old mdx diaphragm with an area of inflammation outlined showing inflammatory cells. d, histological appearance of a 1‐month‐old mdx quadriceps with clusters of myofibres under regeneration (outlined), which are observed as plump cells with at least one centrally located nucleus and a small diameter; note the poor inflammatory infiltration. e, histological appearance of a 9‐month‐old mdx quadriceps showing centrally nucleated (arrowhead) polygonal cells with normal size. f and g, images from 18‐month‐old intrinsic laryngeal muscles in control (f) and in mdx (g). The posterior cricoarytenoid (ca) and cricothyroid (ct) muscles are shown. Some central nucleated fibres (arrows in g) in the non‐spared laryngeal muscle. Scale bar (shown only in g), 40 μm (a, b, c, e), 50 μm (d) and 210 μm (f, g). H&E‐stained representative sections.
Cryostat transverse sections of QDR and DIA from C57BL/10 and mdx mice at the ages of 1, 4 and 9 months were stained with Masson's trichrome to observe fibrosis as previously reported (Taniguti et al. 2011). Other sections were labelled for intracellular immunoglobulin G (IgG) using an anti‐mouse IgG (whole molecule) FITC conjugate antibody developed in goat (F‐0257; Sigma) (1:100) to observe degenerating muscle fibres as previously described (Janssen et al. 2014). Areas containing fibres positively labelled were quantified as described above.
The areas of inflammation, regeneration, centrally nucleated, peripherally nucleated, fibrosis and myonecrosis (FITC‐anti‐mouse IgG‐positive) were expressed as a percentage of the total transverse‐sectional area. All counting and measurements were performed by a blinded observer.
Western blot analysis
The levels of tumour necrosis factor‐alpha (TNF‐α), transforming growth factor‐beta (TGF‐β), MyoD and calsequestrin (CSQ) were quantified using Western blots from control C57BL/10 (n = 30) and mdx mice at the ages of 1 (n = 10), 4 (n = 10) and 9 (n = 10) months. For the ILM, the ages studied were 2 (5 mdx and 5 C57BL/10 mice) and 20 (5 mdx and 5 C57BL/10 mice) months. Western blots were performed as previously described (Ferretti et al. 2009). Briefly, muscles were lysed in assay lysis buffer (1% Triton, 10 mM sodium pyrophosphate, 100 mM NaF, 10 g/ml aprotinin, 1 mM PMSF and 0.25 mM Na 3 VO). The samples were centrifuged at 12,581 g for 20 min, and the soluble fraction was resuspended in Laemmli loading buffer (2% SDS, 20% glycerol, 0.04 mg/ml bromophenol blue, 0.12 M Tris‐HCl, pH 6.8 and 0.28 M mercaptoethanol). An aliquot (30 μg) of the total protein homogenate from C57BL/10 and mdx DIA and QDR was loaded onto 12% SDS–polyacrylamide gels. The proteins were transferred to a nitrocellulose membrane (electrotransfer apparatus from Bio‐Rad Laboratories, Hercules, CA, USA). The membranes were blocked with 5% skim milk/Tris‐HCl‐buffered saline–Tween buffer (TBST; 10 mM Tris‐HCl, pH 8, 150 mM NaCl and 0.05% Tween‐20) and incubated with the primary antibodies overnight at 4°C, washed in TBST, incubated with peroxidase‐conjugated secondary antibodies and developed using the SuperSignal West Pico Chemiluminescent Substrate kit (Pierce Biotechnology, Rockford, IL, USA). To control for Western blot transfer and non‐specific changes in protein levels, the blots were stripped and reprobed for glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH). The luminescent signal was captured (G:Box iChemi camera; Syngene, Cambridge, UK), and band intensities were quantified using the analysis software that was provided by the manufacturer (gene tools Version 4.01; Syngene). The following primary antibodies were used for Western blotting: (i) TNF‐α (rabbit anti‐mouse polyclonal; Millipore, CA, USA), (ii) TGF‐β (mouse monoclonal; Sigma‐Aldrich, St Louis, MI, USA), (iii) calsequestrin (VIII12; Affinity BioReagents), (iv) MyoD (rabbit polyclonal M‐318, sc‐760; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and (v) GAPDH (rabbit polyclonal; Santa Cruz Biotechnology). The corresponding secondary antibody that was used for Western blotting was an appropriate peroxidase‐labelled affinity‐purified IgG antibody (H+L) (KPL, Gaithersburg, MD, USA).
Statistical analyses
All data are expressed as the means ± standard deviation (SD). Statistical analyses for direct comparison between the means of two groups were performed using Student's t‐test, and anova was used for multiple statistical comparisons between groups. P < 0.05 was considered statistically significant.
Ethical Approval Statement
The animal experiments and the procedures that are described here have been approved by and were performed in accordance with the guidelines of the Brazilian College for Animal Experimentation (COBEA; protocol #2579‐1) and the guidelines set forth by our institution.
Results
Control muscles showed no signs of histological changes over time, presenting polygonal fibres with normal diameters that were opposed to each other with peripheral cell nuclei (Figure 1). Morphometric analysis also indicated that with ageing, control DIA and QDR do not display areas of fibrosis, myonecrosis (FITC‐anti‐mouse IgG‐positive fibres) or muscle regeneration (as indicated by areas with centrally nucleated fibres), with 99.9% of their cross‐sectional area containing fibres with peripheral nuclei.
The histopathological features of QDR and DIA dystrophic muscles included the presence of central nucleated fibres (indicative of muscle regeneration), fibres with peripheral nuclei (indicative of fibres that did not suffer necrosis and regeneration) and variations in fibre diameter and areas containing inflammatory cells (Figure 1). QDR showed multiple clusters of regenerating myofibres. In agreement with our previous observations (Marques et al. 2007; Ferretti et al. 2009), no signs of myofibre damage were detected in dystrophic ILM (Figure 1 shows posterior cricoarytenoid muscle), except for the cricothyroid muscle that showed some central nucleated fibres (Figure 1). Fibrosis (Figure 2) and myonecrosis (Figure 3) were also observed in dystrophic QDR and DIA muscles. ILM showed no fibrosis, even at a later stage of dystrophy (Figure 2).
Figure 2.
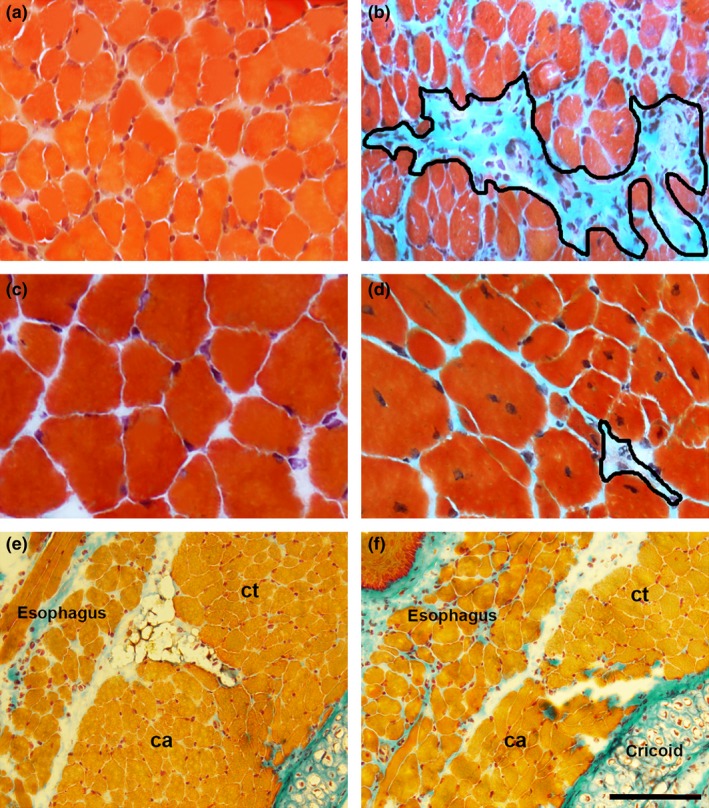
Representative images of diaphragm (a, b) and quadriceps (c, d) muscles from controls (a, c) and 9‐month‐old mdx mice (b, d). e and f, images from 18‐month‐old intrinsic laryngeal muscles in control (e) and mdx (f). Shown are posterior cricoarytenoid (ca) and cricothyroid (ct) muscles. In Masson trichrome‐stained sections, the blue colour denotes fibrosis. Fibrosis areas (outlined in b and d) were prominent in the diaphragm. Note lack of fibrosis in mdx intrinsic laryngeal. Scale bar (shown only in f), 40 μm (a–d) and 210 μm (e, f).
Figure 3.
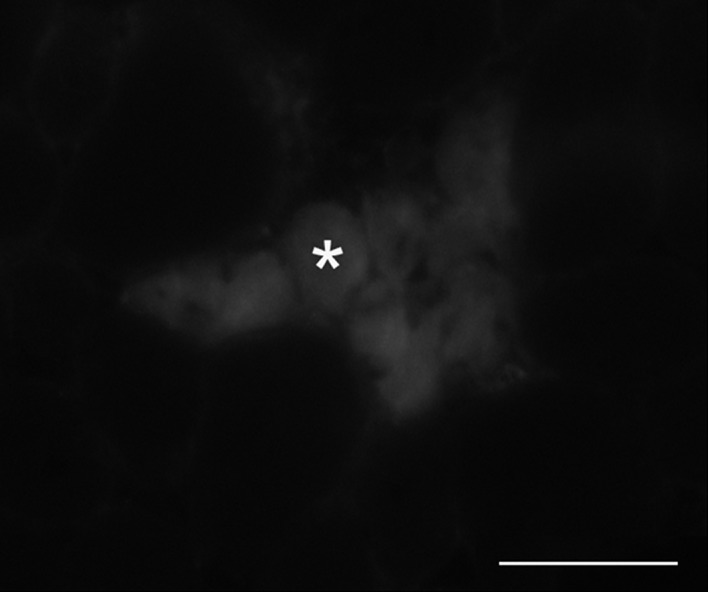
Immunofluorescence labelling of IgG in quadriceps from mdx mice at 9 months of age. Clusters of myofibres positively labelled with IgG (asterisk) indicative of muscle damage. Scale bar, 40 μm.
Morphometric analysis showed that dystrophic DIA presented a greater area of fibrosis over time than did QDR, reaching approximately 32% (32.3 ± 7.4% of fibrosis) by 9 months of age. This is in comparison with QDR at the same period (0.6 ± 0.1% of fibrosis; Table 1). In QDR, the primary feature was a significant increase in areas containing central nucleated muscle fibres, reaching 90% (90.4 ± 1.9% of central nucleated areas) by 9 months of age (Table 1). This occurred concomitantly with a decrease in the areas containing peripheral cell nuclei (Table 1). In DIA, the areas containing central nucleated fibres corresponded to 33% of the total cross‐sectional area at the later stage of the disease (9 months; Table 1). The areas containing fibres positively labelled with FITC‐anti‐mouse IgG (indicative of myonecrosis) were significantly increased in DIA (1.5 ± 0.8% at 4 months) and in QDR (0.7 ± 0.2% at 4 months and 0.6 ± 0.2% at 9 months); all were significantly different from their respective control muscles (no myonecrosis was observed in control muscles). The area of regeneration predominated in QDR at 1 month (22.9 ± 1.1% of the total cross‐sectional area) but decreased by 9 months (3.5 ± 0.9%). In DIA, the area of inflammation was maximal at 4 months (9.5 ± 3.4%), with similar areas (approximately 4.7 ± 1.4%) at 1 and 9 months. In QDR, inflammation was not significant (approximately 1% in all ages).
Table 1.
Morphometric analysis of dystrophy progression in mdx diaphragm (DIA) and quadriceps (QDR) muscles. Indicated are areas containing fibres with peripheral cell nuclei (Peripheral nuclei), areas with centrally nucleated fibres completely regenerated (Central nuclei), areas with clusters of fibres under regeneration (Regeneration) and areas with fibrosis (Fibrosis), at 1, 4 and 9 months of age
| 1 | 4 | 9 | ||||
|---|---|---|---|---|---|---|
| DIA | QDR | DIA | QDR | DIA | QDR | |
| Peripheral nuclei | 88.6 ± 1.4* | 61.3 ± 2.3* | 74.5 ± 2.2 | 7.8 ± 2.9† | 66.2 ± 2.0 | 1.0 ± 0.5 |
| Central nuclei | 1.7 ± 0.09* | 9.9 ± 1.4* | 25.5 ± 2.2 | 79.3 ± 2.5 | 33.8 ± 2.0 | 90.4 ± 1.9 |
| Regeneration | 1.1 ± 0.6* | 22.9 ± 1.1* | 2.2 ± 0.4† | 7.1 ± 1.8 | 4.7 ± 0.3 | 3.5 ± 0.9 |
| Fibrosis | 1.8 ± 0.1* | 0.5 ± 0.1 | 18.2 ± 2.5† | 0.6 ± 0.2 | 32.3 ± 7.4 | 0.6 ± 0.1 |
Values are expressed as the percentage (mean ± SD) of the total cross‐sectional area. *Significantly different from 4 and 9 months of age; †Significantly different from 9 months of age (P < 0.05, anova).
Western blot analysis of TNF‐α showed that mdx DIA and QDR presented elevated levels of TNF‐α compared to their respective controls at 1, 4 and 9 months of age (Figure 4). TGF‐β did not change with ageing in control DIA and QDR (Figure 4). Mdx DIA and QDR presented elevated levels of TGF‐β compared to controls only at 4 (DIA) and 9 months (DIA and QDR; Figure 4). Dystrophic DIA expressed higher levels of TNF‐α (3.0 ± 0.4 arbitrary units in 9‐month‐old mdx DIA) and TGF‐β (1.6 ± 0.8 arbitrary units in 4‐month‐old mdx DIA) than mdx QDR (2.1 ± 0.7 arbitrary units of TNF‐α in 9‐month‐old mdx and 0.8 ± 0.3 arbitrary units of TGF‐β in 4‐month‐old mdx QDR; P < 0.05 compared to mdx DIA at the same age) (Figure 4). CSQ levels were higher in QDR (approximately 2.7 arbitrary units in 9‐month‐old control and mdx) than in DIA (approximately 1.0 arbitrary unit in 9‐month‐old control and mdx mice; P < 0.05 compared to mdx DIA at the same ages; Figure 4). The levels of MyoD in dystrophic DIA and QDR muscles were comparable to those seen in their respective controls, at the ages studied (Figure 5). In the ILM, the levels of TNF‐α, TGF‐β and MyoD were similar to control values at the time points studied (Figure 6).
Figure 4.
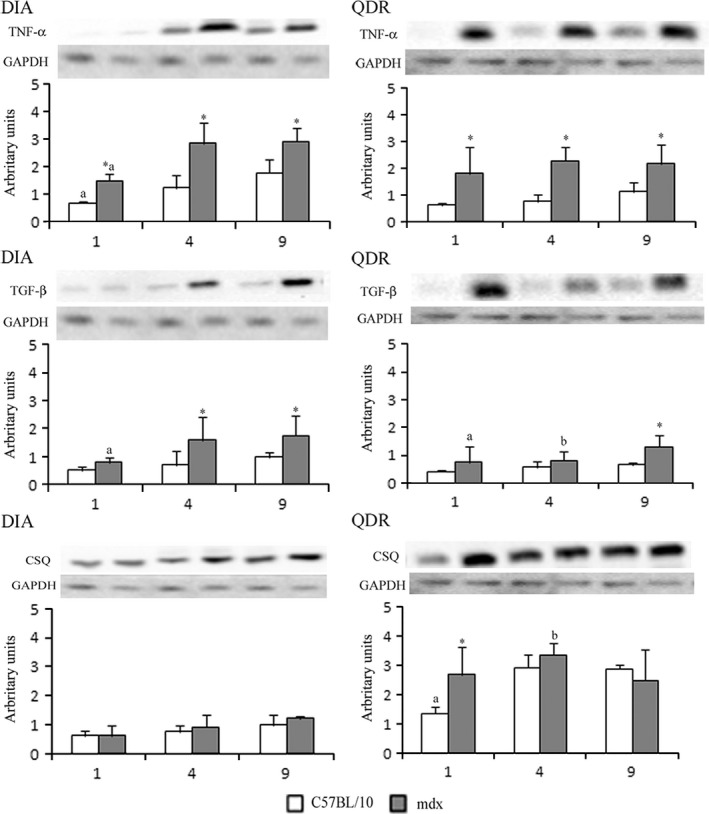
Western blot analysis of tumour necrosis factor‐α (TNF‐α), tumour growth factor‐β (TGF‐β) and calsequestrin (CSQ) in crude extracts of diaphragm (DIA) and quadriceps (QDR) from C57BL/10 and mdx mice at 1, 4 and 9 months of age. Bands indicate Western blot of TNF‐α, TGF‐β and CSQ and the same blot reprobed for GAPDH as a loading control. Graphs represent the level of each indicated protein expressed in arbitrary units normalized to GAPDH levels.*Significantly different from C57BL/10 (P < 0.05, anova). aSignificantly different from 4 and 9 months of age (P < 0.05, anova). bSignificantly different from 9 months of age (P < 0.05, anova).
Figure 5.
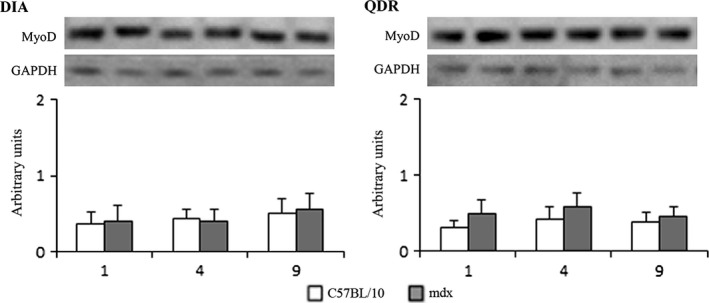
Western blot analysis of MyoD in crude extracts of diaphragm (DIA) and quadriceps (QDR) from C57BL/10 and mdx mice at 1, 4 and 9 months of age. Bands indicate Western blot of MyoD and the same blot reprobed for GAPDH as a loading control. Graphs represent the level of each indicated protein expressed in arbitrary units normalized to GAPDH levels. There were no differences in MyoD levels between control and dystrophic muscles, at the ages studied.
Figure 6.
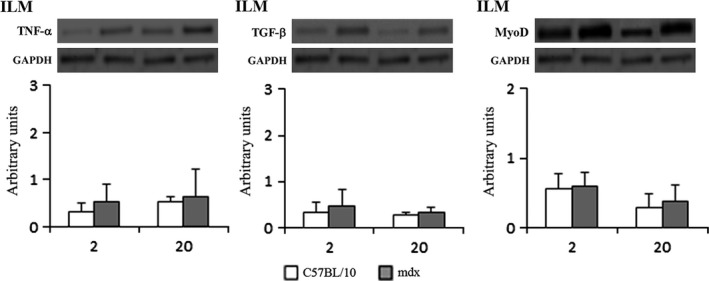
Western blot analysis of tumour necrosis factor‐α (TNF‐α), tumour growth factor‐β (TGF‐β) and MyoD in crude extracts of intrinsic laryngeal muscles (ILM) from C57BL/10 and mdx mice at 2 and 20 months of age. Bands indicate Western blot of TNF‐α, TGF‐β and MyoD and the same blot reprobed for GAPDH as a loading control. Graphs represent the level of each indicated protein expressed in arbitrary units normalized to GAPDH levels. No differences were seen in the levels of the markers between control and dystrophic muscles, at the ages studied.
Discussion
We observed that both of the affected dystrophic muscles studied (diaphragm and quadriceps) expressed increased levels of TNF‐α in comparison with control muscles very early (1 month) in the progression of disease, reaching even higher levels at 4 months of age that were maintained at later stage (9 months) dystrophy. Histopathological analyses revealed that only DIA showed a significant increase in inflammatory areas, which were primarily observed at 4 months of age. The mdx quadriceps muscle showed smaller amounts of inflammatory infiltration than DIA throughout the progression of dystrophy. This difference in the areas of inflammation between mdx DIA and mdx QDR correlates with the higher levels of TNF‐α in DIA vs. QDR. Inflammatory cells are responsible for releasing several pro‐inflammatory cytokines including TNF‐α (Porter et al. 2002; Tidball 2005; Hodgetts et al. 2006; Villalta et al. 2009), and anti‐inflammatory drugs ameliorate the progression of dystrophy by suppressing TNF‐α production (Grounds et al. 2005; Machado et al. 2011). Our finding that dystrophic DIA and QDR share increased levels of TNF‐α from very early disease stages (1 month) is consistent with the finding that inflammatory cascades are induced soon after birth in cases of dystrophy (Chen et al. 2005).
Fibrosis deposition following myofibre degeneration is a major pathological characteristic of DMD (Stedman et al. 1991). TGF‐β is a key cytokine that promotes fibroblast proliferation and fibrosis formation and also has roles in inflammation and immunomodulation (Border & Noble 1994). Previous studies have demonstrated that increased expression of TGF‐β was accompanied by progressive fibrosis in the diaphragm in 6‐ and 12‐week‐old mdx mice (Hartel et al. 2001; Andreetta et al. 2006). Elevated TGF‐β mRNA in the mdx diaphragm has been demonstrated at 6 and 9 but not 12 weeks of age (Gosselin et al. 2004). In the present study, we observed increased levels of TGF‐β earlier in the dystrophic diaphragm (significant at 4 months) than in the quadriceps (a significant increase in the levels of TGF‐β was only observed at 9 months). Concomitantly, the histopathology revealed an increase in the area of fibrosis over time primarily in the mdx DIA, which reached 32% by 9 months of age vs. 0.6% of fibrosis in QDR over the same time. This result demonstrates a strong relation between TGF‐β levels and fibrosis only in respiratory muscle and further supports previous observations regarding differential susceptibility of quadriceps and diaphragm muscles to fibrosis induced by TGF‐β (Zhou et al. 2006). Late‐stage therapy with the anti‐fibrotic drug suramin can ameliorate the diaphragm dysfunction (Taniguti et al. 2012). The present results suggest that early therapy with anti‐fibrotic drugs may protect the mdx diaphragm muscle against fibrosis, which is of relevance to DMD therapy because patients eventually suffer from respiratory failure due to extensive fibrosis.
A striking finding of the present study was the regenerative capacity of the dystrophic quadriceps muscle compared to the diaphragm (and even compared to other limb muscles that we studied previously; Machado et al. 2011); this finding is in agreement with a prior qualitative observation (Zhou et al. 2006). In the mdx quadriceps regeneration occurred in nearly all fibres (99% of central nucleated areas) by the later stage (9 months) of disease. This regenerative profile appears to be expressed very early in the progression of dystrophy within the mdx QDR because we observed regeneration in approximately 23% of the cross‐sectional areas at 1 month of age in QDR (vs. 1.1% in DIA at the same period). This result highlights the lack of correlation between the increased levels of TNF‐α and TGF‐β and histopathology (muscle regeneration) in the dystrophic quadriceps muscle at the time points studied, but QDR degeneration must have occurred at time points not covered by the present investigation.
Some studies have reported that the upregulation of TGF‐β in the mdx QDR improves muscle regeneration (Zhou et al. 2006). Other studies have suggested that TGF‐β may negatively affect muscle regeneration given its inhibitory effects on myoblast differentiation (Heino & Massague 1990; Florini et al. 1991). Treatment with an anti‐TGF‐β antibody did not affect muscle regeneration in the diaphragm of mdx mice (Andreetta et al. 2006). The present morphometric analysis demonstrates a remarkable difference in muscle regeneration between mdx DIA and mdx QDR, both of which express elevated levels of TGF‐β. At least for the QDR muscle, we could postulate that the regenerative capacity of this muscle might be related to elevated levels of TGF‐β or other factors such as TNF receptor‐associated factor 6, which is increased in mdx muscles and is involved in satellite cell proliferation and myofibre regeneration in young mdx mice (Hindi et al. 2013).
Previously, we demonstrated that the mdx DIA shows increased levels of the stretch‐activated calcium channel TRPC1 compared to limb muscles (Matsumura et al. 2011). These receptors are activated in response to muscle contraction, which leads to an increased influx of calcium into the dystrophic fibre (Yeung & Allen 2004). One possibility that could explain the poor muscle regeneration observed in the DIA compared to the QDR could be related to an elevated DIA workload compared to QDR. This would facilitate additional cycles of muscle degeneration (possibly due to increased TRPC1 activation in DIA) and perhaps an earlier exhaustion of satellite cells in the DIA compared to the QDR. This is in line with our previous observations that satellite cells become exhausted and fail to proliferate due to the continual cycles of muscle degeneration–regeneration (Luz et al. 2002) and that treadmill exercise increases TGF‐β and fibrosis in limb muscle (Taniguti et al. 2011). Alternatively, it is possible that the QDR muscle contains a different population of satellite cells that allows them to regenerate better compared to DIA, similar to what is reported for the laryngeal and EOM muscles (Goding et al. 2005; McLoon et al. 2007), but this deserves further elucidation.
With respect to the spared dystrophic ILM, we observed that the levels of TNF‐α and TGF‐β were comparable to normal, even at a very late stage of dystrophy (20 months of age). This is in agreement with our previous and present histopathological analysis, where no signs of muscle degeneration or inflammation were seen in the dystrophic ILM (Marques et al. 2007). Furthermore, the levels of MyoD, a major regulator of the myogenic process (Guttridge et al. 2000; Aziz et al. 2010), were comparable to normal in mdx ILM, possibly due to the lack degeneration in the spared muscle. Levels of MyoD similar to those found in control, non‐dystrophic muscles were also detected in mdx DIA and QDR. Nevertheless, mdx QDR showed better regenerative capacity (as evidenced by the predominance of regenerated fibres already at 4 months of age), compared to mdx DIA. While the reasons for that are unclear, it is possible that in the mdx DIA, MyoD might be negatively affected by TNF‐α, as it has been demonstrated that drugs that inhibit TNF‐α and NF‐kB improved muscle regeneration in the mdx (Messina et al. 2011).
Calsequestrin, the most abundant calcium‐binding protein located in the sarcoplasmic reticulum, maintains low levels of free calcium in the skeletal muscle fibre (Beard et al. 2004). Spared dystrophic muscles, such as the laryngeal and EOM, show higher levels of CSQ, which may explain their protection against myonecrosis, possibly due to a better ability to buffer calcium increases (Khurana et al. 1995; Ferretti et al. 2009; Zeiger et al. 2010). Here, we observed that the levels of CSQ in the mdx DIA were comparable to those in controls throughout the progression of dystrophy. In the mdx QDR, CSQ was increased very early on (1 month), but this was not retained in the aged mdx QDR. In the present study, normal and/or increased levels of CSQ might explain the milder phenotype observed in the mdx QDR during the course of dystrophy. Normal levels of CSQ in the mdx DIA at the early stage of disease have also been reported by a proteomic study and could be related to the mild phenotype observed during this period (Matsumura et al. 2013). Conversely, a subproteomic analysis revealed a decrease in CSQ levels in mdx skeletal muscles at 9 weeks of age (Doran et al. 2004); these differences may be related to the techniques, age and muscles studied.
Overall, we show that spared (ILM) and affected (DIA and QDR) dystrophic muscles show different profiles of the markers of the pathways related to inflammation (increased TNF‐α in affected), fibrosis (increased TGF‐β in affected) and calcium regulation (normal CSQ in affected), in a way compatible to their phenotype. However, while affected DIA and QDR muscles display similar profiles of these markers, they do not share histopathological features during the progression of dystrophy, possibly due to the decreased levels of TNF‐α and/or greater levels of calsequestrin in mdx QDR than in mdx DIA. As observed in DMD symptomatic vs. presymptomatic patients, pro‐inflammatory and pro‐fibrotic cytokines can be elevated independent of the progression of their dystrophic phenotype (Tian et al. 2014). A positive correlation between histopathology and cytokine levels was observed only in the diaphragm, suggesting that TNF‐α and TGF‐β serve as markers of dystrophy primarily for this muscle.
Conflict of interest
The authors have no conflicts of interest to disclose.
Acknowledgements
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, grants 04/15526‐9, 08/58491‐1, 11/51697‐6, 14/04782‐6). H. S. N. and M. J. M. were recipients of fellowships from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, grants 303320/2013‐3, 302831/2013‐4). D. O. M., A. F. M. and S. C. C. were the recipients of Coordenação de Aperfeiçoamento de Pessoal de Ensino Superior (CAPES), CNPq (142282/2012‐0) and FAPESP Fellowships (grants 2012/03498‐7; 2012/13577‐1; 2012/15492‐3). CAPES DINTER Institucional UNICAMP/UPE.
References
- Andreetta F., Bernasconi P., Baggi F. et al (2006) Immunomodulation of TGF‐β1 in mdx mouse inhibits connective tissue proliferation in diaphragm but increases inflammatory response: implications for antifibrotic therapy. J. Neuroimmunol. 175, 77–86. [DOI] [PubMed] [Google Scholar]
- Aziz A., Liu Q.C., Dilworth F.J. (2010) Regulating a master regulator: establishing tissue‐specific gene expression in skeletal muscle. Epigenetics 5, 691–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beard N.A., Laver D.R., Dulhunty A.F. (2004) Calsequestrin and the calcium release channel of skeletal and cardiac muscle. Prog. Biophys. Mol. Biol. 85, 33–69. [DOI] [PubMed] [Google Scholar]
- Bernasconi P., Torchiana E., Confalonieri P. et al (1995) Expression of transforming growth factor‐β1 in dystrophic patients muscles correlates with fibrosis. J. Clin. Invest. 96, 1137–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Border W.A. & Noble N.A. (1994) Transforming growth factor beta in tissue fibrosis. N. Engl. J. Med. 331, 1286–1292. [DOI] [PubMed] [Google Scholar]
- Bulfield G., Siller W.G., Wight P.A.L., Moore K.J. (1984) X chromosome‐liked muscular dystrophy (mdx) in the mouse. Proc. Natl Acad. Sci. USA 81, 1189–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.W., Nagaraju K., Bakay M. et al (2005) Early onset of inflammation and later involvement of TGFbeta in Duchenne muscular dystrophy. Neurology 65, 826–834. [DOI] [PubMed] [Google Scholar]
- Doran P., Dowling P., Lohan J., McDonnell K., Poetsch S., Ohlendieck K. (2004) Subproteomics analysis of Ca+‐binding proteins demonstrates decreased calsequestrin expression in dystrophic mouse skeletal muscle. Eur. J. Biochem. 271, 3943–3952. [DOI] [PubMed] [Google Scholar]
- Engel A.G., Yamamoto M., Fischbeck K.H. (1994) Dystrophinopathies In: Myology, pp. 1133–1187. (eds Engel A.G. & Franzini‐Armstrong C.), New York: McGraw‐Hill. [Google Scholar]
- Ferretti R., Marques M.J., Pertille A., Santo Neto H. (2009) Sarcoplasmic‐endoplasmic‐reticulum Ca2+‐ATPase and calsequestrin are overexpressed in spared intrinsic laryngeal muscles of dystrophin‐deficient mdx mice. Muscle Nerve 39, 609–615. [DOI] [PubMed] [Google Scholar]
- Florini J.R., Ewton D.Z., Magri K.A. (1991) Hormones, growth factors, and myogenic differentiation. Annu. Rev. Physiol. 53, 201–216. [DOI] [PubMed] [Google Scholar]
- Goding G.S. Jr, Al‐Sharif K.I., McLoon L.K. (2005) Myonuclear addition to uninjured laryngeal myofibers in adult rabbits. Ann. Otol. Rhinol. Laryngol. 114, 552–557. [DOI] [PubMed] [Google Scholar]
- Gosselin L.E., Williams J.E., Deering M., Brazeau D., Koury S., Martinez D.A. (2004) Localization and early time course of TGF‐β 1 mRNA expression in dystrophic muscle. Muscle Nerve 30, 645–653. [DOI] [PubMed] [Google Scholar]
- Grounds M.D., Davies M., Torrisi J., Shavlakadze T., White J., Hodgetts S. (2005) Silencing TNF activity by using Remicade or Enbrel blocks inflammation in whole muscle grafts: an in vivo bioassay to assess the efficacy of anti‐cytokine drugs in mice. Cell Tissue Res. 320, 509–515. [DOI] [PubMed] [Google Scholar]
- Guttridge D.C., Mayo M.W., Madrid L.V., Wang C.Y., Baldwin A.S. Jr (2000) NF‐kappaB‐ induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science 29, 2363–2366. [DOI] [PubMed] [Google Scholar]
- Hartel J.V., Granchelli J.A., Hudecki M.S., Pollina C.M., Gosselin L.E. (2001) Impact of prednisone on TGF‐β1 and collagen in diaphragm muscle from mdx mice. Muscle Nerve 24, 428–432. [DOI] [PubMed] [Google Scholar]
- Heino J. & Massague J. (1990) Cell adhesion to collagen and decreased myogenic gene expression implicated in the control of myogenesis by transforming growth factor beta. J. Biol. Chem. 265, 10181–10184. [PubMed] [Google Scholar]
- Hindi S.M., Shin J., Ogura Y., Li H., Kumar A. (2013) Matrix metalloproteinase‐9 inhibition improves proliferation and engraftment of myogenic cells in dystrophic muscle of mdx mice. PLoS ONE 8, e72121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgetts S., Radley H., Davies M., Grounds M.D. (2006) Reduced necrosis of dystrophic muscle by depletion of host neutrophils, or blocking TNF‐alpha function with Etanercept in mdx mice. Neuromuscul. Disord. 16, 591–602. [DOI] [PubMed] [Google Scholar]
- Hoffman E.P., Brown R.H. Jr, Kunkel L.M. (1987) Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51, 919–928. [DOI] [PubMed] [Google Scholar]
- Janssen P.M., Murray J.D., Schill K.E. et al (2014) Prednisolone attenuates improvement of cardiac and skeletal contractile function and histopathology by lisinopril and spironolactone in the mdx mouse model of Duchenne muscular dystrophy. PLoS ONE 2, e88360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana T.S., Prendergast R.A., Alameddine H.S. et al (1995) Absence of extraocular muscle pathology in Duchenne's muscular dystrophy: role for calcium homeostasis in extraocular muscle sparing. J. Exp. Med. 182, 467–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konieczny P., Swiderski K., Chamberlain J.S. (2013) Gene and cell‐mediated therapies for muscular dystrophy. Muscle Nerve 47, 649–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Mittal A., Makonchuk D.Y., Bhatnagar S., Kumar A. (2009) Matrix metalloproteinase‐9 inhibition ameliorates pathogenesis and improves skeletal muscle regeneration in muscular dystrophy. Hum. Mol. Genet. 18, 2584–2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luz M.A., Marques M.J., Santo Neto H. (2002) Impaired regeneration of dystrophin‐deficient muscle fibers is caused by exhaustion of myogenic cells. Braz. J. Med. Biol. Res. 35, 691–695. [DOI] [PubMed] [Google Scholar]
- Machado R.V., Maurício A.F., Taniguti A.P., Ferretti R., Neto H.S., Marques M.J. (2011) Eicosapentaenoic acid decreases TNF‐α and protects dystrophic muscle of mdx mice from degeneration. J. Neuroimmunol. 232, 145–150. [DOI] [PubMed] [Google Scholar]
- Marques M.J., Ferretti R., Vomero V.U., Minatel E., Neto H.S. (2007) Intrinsic laryngeal muscles are spared from myonecrosis in the mdx mouse model of Duchenne Muscular Dystrophy. Muscle Nerve 35, 349–353. [DOI] [PubMed] [Google Scholar]
- Matsumura C.Y., Taniguti A.P., Pertille A., Santo Neto H., Marques M.J. (2011) The stretch‐activated calcium channel protein TRPC1 is correlated with the different degrees of the dystrophic phenotype in mdx mice. Am. J. Physiol. Cell Physiol. 301, C1344–C1350. [DOI] [PubMed] [Google Scholar]
- Matsumura C.Y., Menezes de Oliveira B., Durbeej M., Marques M.J. (2013) Isobaric tagging‐based quantification for proteomic analysis: a comparative study of spared and affected muscles from mdx mice at the early phase of dystrophy. PLoS ONE 6, e65831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLoon L.K., Thorstenson K.M., Solomon A., Lewis M.P. (2007) Myogenic precursor cells in craniofacial muscles. Oral Dis. 13, 134–140. [DOI] [PubMed] [Google Scholar]
- Mendell J.R., Shilling C., Leslie N.D. et al (2012) Evidence‐based path to newborn screening for Duchenne muscular dystrophy. Ann. Neurol. 3, 304–313. [DOI] [PubMed] [Google Scholar]
- Messina S., Bitto A., Aguennouz M. et al (2011) The soy isoflavone genistein blunts nuclear factor kappa‐B, MAPKs and TNF‐a activation and ameliorates muscle function and morphology in mdx mice. Neuromuscul. Disord. 21, 579–589. [DOI] [PubMed] [Google Scholar]
- Pastoret C. & Sebille A. (1995) Mdx mice show weakness and muscle deterioration with age. J. Neurol. Sci. 129, 97–105. [DOI] [PubMed] [Google Scholar]
- Porter J.D., Khanna S., Kaminski H.J. et al (2002) A chronic inflammatory response dominates the skeletal muscle molecular signature in dystrophin‐deficient mdx mice. Hum. Mol. Genet. 11, 263–272. [DOI] [PubMed] [Google Scholar]
- Rall S. & Grimm T. (2012) Survival in Duchenne muscular dystrophy. Acta Myol. 31, 117–120. [PMC free article] [PubMed] [Google Scholar]
- Stedman H.H., Sweeney H.L., Shrager J.B. et al (1991) The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature 352, 536–539. [DOI] [PubMed] [Google Scholar]
- Taniguti A.P., Pertille A., Matsumura C.Y., Santo Neto H., Marques M.J. (2011) Prevention of muscle fibrosis and myonecrosis in mdx mice by suramin, a TGF‐β1 blocker. Muscle Nerve 43, 82–87. [DOI] [PubMed] [Google Scholar]
- Taniguti A.P., Matsumura C.Y., Rodrigues‐Simioni L., Santo Neto H., Marques M.J. (2012) Suramin affects metalloproteinase‐9 activity and increases beta‐dystroglycan levels in the diaphragm of the dystrophin‐deficient mdx mouse. Muscle Nerve 46, 810–813. [DOI] [PubMed] [Google Scholar]
- Tian L.J., Cao J.H., Deng X.Q. et al (2014) Gene expression profiling of Duchenne muscular dystrophy reveals characteristics along disease progression. Genet. Mol. Res. 28, 1402–1411. [DOI] [PubMed] [Google Scholar]
- Tidball J.G. (2005) Inflammatory processes in muscle injury and repair. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288, R345–R353. [DOI] [PubMed] [Google Scholar]
- Villalta S.A., Nguyen H.X., Deng B., Gotoh T., Tidball J.G. (2009) Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum. Mol. Genet. 18, 482–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead N.P., Yeung E.W., Allen D.G. (2006) Muscle damage in mdx (dystrophic) mice: role of calcium and reactive oxygen species. Clin. Exp. Pharmacol. Physiol. 33, 657–662. [DOI] [PubMed] [Google Scholar]
- Yeung E.W. & Allen D.G. (2004) Stretch‐activated channels in stretch‐induced muscle damage: role in muscular dystrophy. Clin. Exp. Pharmacol. Physiol. 31, 551–556. [DOI] [PubMed] [Google Scholar]
- Zeiger U., Mitchell C.H., Khurana T.S. (2010) Superior calcium homeostasis of extraocular muscles. Exp. Eye Res. 91, 613–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L., Porter J.D., Cheng G. et al (2006) Temporal and spatial mRNA expression patterns of TGF‐β 1, 2, 3 and TbRI, II, III in skeletal muscles of mdx mice. Neuromuscul. Disord. 16, 32–38. [DOI] [PubMed] [Google Scholar]