

Abstract
Objective
Oxidative stress in the brain is highly prevalent in many neurodegenerative disorders including lysosomal storage disorders, in which neurodegeneration is a devastating manifestation. Despite intense studies, a precise mechanism linking oxidative stress to neuropathology in specific neurodegenerative diseases remains largely unclear.
Methods
Infantile neuronal ceroid lipofuscinosis (INCL) is a devastating neurodegenerative lysosomal storage disease caused by mutations in the ceroid lipofuscinosis neuronal‐1 (CLN1) gene encoding palmitoyl‐protein thioesterase‐1. Previously, we reported that in the brain of Cln1 −/− mice, which mimic INCL, and in postmortem brain tissues from INCL patients, increased oxidative stress is readily detectable. We used molecular, biochemical, immunohistological, and electrophysiological analyses of brain tissues of Cln1 −/− mice to study the role(s) of oxidative stress in mediating neuropathology.
Results
Our results show that in Cln1 −/− mice oxidative stress in the brain via upregulation of the transcription factor, CCAAT/enhancer‐binding protein‐δ, stimulated expression of serpina1, which is an inhibitor of a serine protease, neurotrypsin. Moreover, in the Cln1 −/− mice, suppression of neurotrypsin activity by serpina1 inhibited the cleavage of agrin (a large proteoglycan), which substantially reduced the production of agrin‐22, essential for synaptic homeostasis. Direct whole‐cell recordings at the nerve terminals of Cln1 −/− mice showed inhibition of Ca2+ currents attesting to synaptic dysfunction. Treatment of these mice with a thioesterase‐mimetic small molecule, N‐tert (Butyl) hydroxylamine (NtBuHA), increased agrin‐22 levels.
Interpretation
Our findings provide insight into a novel pathway linking oxidative stress with synaptic pathology in Cln1 −/− mice and suggest that NtBuHA, which increased agrin‐22 levels, may ameliorate synaptic dysfunction in this devastating neurodegenerative disease.
Introduction
Oxygen is essential for life, but paradoxically, as a by‐product of metabolism it generates reactive oxygen species, which is highly toxic to the cells,1 especially in the central nervous system. Indeed, elevated levels of oxidative stress have been reported in many neurodegenerative diseases.2, 3 Although a link between oxidative stress and neurodegeneration has long been suggested, the mechanism(s) by which it contributes to pathogenesis in specific neurodegenerative diseases has not been clearly elucidated. Neuronal ceroid lipofuscinosis (NCLs), commonly known as Batten disease,4 represent a group of the most prevalent (1 in 12,500 births)5 neurodegenerative lysosomal storage disorders (LSDs).6 Mutations in at least 13 different genes (called CLNs) underlie pathogenesis of various types of NCLs.7 The infantile NCL (INCL)8 is caused by mutations in the CLN1 (ceroid lipofuscinosis neuronal‐1) gene,9 which encodes palmitoyl‐protein thioesterase 1 (PPT1).10
Synaptic dysfunction, manifested by myoclonus and seizures, is one of the earliest signs of pathogenesis in patients with neurodegenerative disorders including INCL.4, 11 Despite intense studies, a precise molecular mechanism(s) of synaptic dysfunction in INCL remains largely unclear. Emerging evidence indicates that activity‐regulated proteolytic cleavage at the synapse plays important roles in regulating synaptic structure, number, and function.12 Moreover, proteases with distinct function and localization at the neuronal synapses have been implicated to regulate homeostasis at the nerve terminals.12 Neurotrypsin is a serine protease predominantly expressed in neurons of the cerebral cortex, hippocampus, and amygdala.13, 14 The accumulation of neurotrypsin has been localized to presynaptic boutons especially in the region of or around the synaptic cleft.15, 16 Neurotrypsin has also been reported to catalyze the cleavage of agrin (a large proteoglycan) at or around neuronal synapses.15 Furthermore, neurotrypsin cleaves its only known substrate, agrin, generating a 90 kDa fragment (agrin‐90) and a 22‐kDa fragment (agrin‐22).17, 18 Agrin‐22 has been reported to play critical roles in synaptic homeostasis.12 Notably, truncating mutations in the neurotrypsin gene underlie an autosomal recessive nonsyndromic mental retardation.16
Neurotrypsin activity is regulated by serine protease inhibitors called serpins.19 Previously, using total RNA from brain tissues of Cln1 −/− mice,20 which mimic INCL,21 and those of their wild‐type (WT) littermates, we conducted a cDNA microarray analysis. The results showed that the levels of serine protease inhibitor clade A (commonly known as serpina1) were several fold higher in the brain of Cln1 −/− mice compared to those of their WT littermates. In this study, we sought to determine whether oxidative stress in the brain of Cln1 −/− mice22, 23 mediated serpina1 expression and inhibiting neurotrypsin activity and suppressed neurotrypsin‐mediated agrin‐22 production in the nerve terminals leading to synaptic dysfunction.
Materials and Methods
Animals and treatments
Cln1 −/− mice (a generous gift from Dr. Sandra L. Hofmann, University of Texas Southwestern Medical Center at Dallas, Dallas, TX) were generated by gene targeting in Embryonic Stem cells as previously described.20 These mice were backcrossed for eight generations in Dr. Mark Sand's laboratory to obtain syngeneic C57 background. All mice were maintained and housed in a pathogen‐free facility, and animal procedures were carried out in accordance with institutional guidelines after approval of a study protocol (#10‐012) by the National Institute of Child Health and Human Development (NICHD) Animal Care and Use Committee. Cln1 −/− mice at 3 months of age were treated for 3 months with N‐tert (Butyl) hydroxylamine (NtBuHA) (final concentration: 1 mmol/L NtBuHA) or resveratrol (RSV) (final concentration: 600 mg/day per kg body weight). NtBuHA was given in the drinking water,24, 25 while RSV was mixed with the diet as previously reported.23 Animals of both sexes were used in all of our experiments. Moreover, the mice were age‐ and sex‐matched for each experiment.
Golgi‐Cox staining and analysis of dendritic filopodia and spine
Golgi‐Cox analysis was referred to a modified Golgi staining protocol.26 Briefly, mouse fresh whole brains are immersed in Golgi stain (GS, EZ Golgi kit Solution I) in a capped 50 mL black corning tube for 7–10 days at room temperature in the dark. Following 7–10 days of impregnation in GS, brains were transferred to 30% sucrose solution in double distilled water, and incubated for a minimum of 3 days at 4°C, with the solutions being changed every 12 h after the initial 12 h of incubation. For sagittal section whole brains were then embedded in a 3% agarose solution (low melting point agarose, Sigma, St. Louis, MO) on ice and the tissue blocks were prepared carefully. The block is attached on the sample plate and cut at 150 μm thick sections. Sections are collected in 30% sucrose, and peeled off the surrounding agarose. Serial sections are immediately mounted with 0.3% gelatin (G9391; Sigma‐Aldrich, St. Louis, MO) on glass slides. To avoid complete drying, the brain sections were brushed with 50% sucrose solution, and allowed to air dry for at least 48 h in a dark place. Sucrose was removed using double distilled water and then developed in developing solution (National Diagnostics Atlanta, GA, USA) according to the instructions of the supplier for visualizing the staining. Cells were examined under upright microscope (Nikon Instruments Inc.) under high‐resolution magnification (1200× Melville, NY, USA). Dendritic filopodia were counted from only visible flanking spines along 30‐μm terminal tip segments of five total segments in each cell. Neurons from each brain were randomly selected and a total of five cells from the hippocampus area were used in this analysis.
Electrophysiology using the calyx of Held synapse
Standard whole‐cell patch clamp was used to measure the voltage‐dependent calcium current (ICa) and membrane capacitance of the calyx as previously described.27 Briefly, parasagittal brainstem slices (200 μm thick) containing the medial nucleus of the trapezoid body were prepared from 7 to 10 days old male or female WT mice, or their Cln1 −/− littermates using a vibratome. Capacitance measurements were made with the EPC‐9 amplifier together with the software lock‐in amplifier (PULSE; HEKA, Lambrecht, Germany) that implements Lindau–Neher's technique. The frequency of the sinusoidal stimulus was 1000 Hz and the peak‐to‐peak voltage of the sine wave was 60 mV. To isolate presynaptic Ca2+ current, the bath solution contained: 105 mmol/L NaCl, 20 TEA‐Cl, 2.5 mmol/L KCl, 1 mmol/L MgCl2, 2 mmol/L CaCl2, 25 mmol/L NaHCO3, 1.25 mmol/L NaH2PO4, 25 mmol/L glucose, 0.4 mmol/L ascorbic acid, 3 mmol/L myo‐inositol, 2 mmol/L sodium pyruvate, 0.001 mmol/L tetrodotoxin, and 0.1 mmol/L 3,4‐diaminopyridine. This solution was 300–310 mOsm, and pH 7.4, when bubbled with 95% O2 and 5% CO2. The presynaptic pipette contained: 125 mmol/L Cs‐gluconate, 20 mmol/L CsCl, 4 mmol/L MgATP, 10 mmol/L Na2‐phosphocreatine, 0.3 mmol/L Guanosine‐5'‐triphosphate (GPT), 10 mmol/L 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid (HEPES), 0.05 mmol/L 1,2‐bis(o‐aminophenoxy)ethane‐N,N,N',N'‐tetraacetic acid (BAPTA), 310–320 mOsm, pH 7.2, adjusted with CsOH. Recordings were done at 22–24°C.
Neurosphere culture and astroglia differentiation in vitro
Neurospheres were isolated from the brains of 12‐day‐old embryos from Cln1 −/− mice and those of their WT littermates as previously described.28 Briefly, cortical tissues were mechanically dispersed and the cells were cultured in NeuroCult NSC Basal Medium (Stem‐Cell Technologies, Vancouver, BC, Canada) containing NeuroCult NSC proliferation supplements and human epidermal growth factor (final concentration of 20 ng/mL). To achieve glial differentiation, the proliferating neurospheres were cultured in Dulbecco's Modified Eagle's Medium (DMEM) Medium containing 10% fetal bovine serum. The cultures were incubated at 37°C under an atmosphere of 5% CO2 and 95% air.
Real‐time RT‐PCR
Total RNA from the cerebral cortices of Cln1 −/− mice and those of their WT littermates was isolated using TriZol reagent (Invitrogen, Grand Island, NY) and further purified using QIAGEN RNeasy Mini Kit and treated with DNAse (DNAse I, 30 U/μg total RNA) (QIAGEN Valencia, CA, USA). The RNA was reverse‐transcribed using SuperScript III First‐Strand Synthesis System (Invitrogen). Expression of mRNA was quantitated using SYBR Green PCR Master Mix, performed with ABI Prism 7000 Sequence Detection System (Applied Biosystems, Grand Island, NY, USA) with cDNA equivalent to 10 ng of total RNA. The primers used for the real‐time RT‐PCR are shown in Table 1. The results were analyzed using ABI Prism Software version 1.01 (Applied Biosystems). The relative amounts of mRNA were calculated from the cycle threshold (Ct) values using Glyceraldehyde‐3‐Phosphate Dehydrogenase (GAPDH) or TBP for normalization and presented as fold change in the brain of Cln1 −/− mice compared with those of their WT littermates. All experiments were repeated at least three times.
Table 1.
List of the primers used for real‐time polymerase chain reaction
| Serpina1 | |
| Forward | 5′‐GGG TGC TGC TGA TGG ATT AT‐3′ |
| Reverse | 5′‐ATG GAC AGT CTG GGG AAG TG‐3′ |
| Neurotrypsin | |
| Forward | 5′‐CTG GGG CAC TGT CAC CAG CC‐3′ |
| Reverse | 5′‐TGC CAG CAC AGA CCA TGC CC‐3′ |
| Agrin | |
| Forward | 5′‐CGT AGA GGA GGC TGG TTT TG‐3′ |
| Reverse | 5′‐TCT TCA GCT GGC ATT CAT TG‐3′ |
| C/EBP‐δ | |
| Forward | 5′‐CAG GCA GGG TGG ACA AGC‐3′ |
| Reverse | 5′‐GTC GTA CAT GGC AGG AGT CG‐3′ |
| β‐actin | |
| Forward | 5′‐GAT CTG GCA CCA CAC CTT CT‐3′ |
| Reverse | 5′‐GGG GTG TTG AAG GTC TCA AA‐3′ |
| GAPDH | |
| Forward | 5′‐TGG CCT CCA AGG AGT AAG AA‐3′ |
| Reverse | 5′‐TGT GAG GGA GAT GCT CAG TG‐3′ |
C/EBP‐δ, CCAAT enhancer‐binding protein.
Western blot analysis
The cortical tissues were homogenized in RIPA buffer (Invitrogen) containing protease‐inhibitor cocktails (Sigma‐Aldrich, St. Louis, MO). For the preparation of total lysates from cultured astroglia and neurons from WT or Cln1 −/− mice, cells were homogenized in Phosphorate extraction reagent (EMD Biosciences, Billerica, MA). Protein concentrations were determined by BCA kits (Pierce Biotechnology, Thermo Scientific, Rockford, IL). Total proteins (20 μg) from each sample were resolved by electrophoresis using 4–12% gradient sodiumdodecyl sulfate (SDS)‐polyacrylamide gels (Invitrogen, Life Technologies, Grand Island, NY) under denaturing and reducing conditions. Proteins were then electrotransferred to nitrocellulose membrane. The membranes were blocked with 5% nonfat dry milk (Bio‐Rad, Hercules, CA) and then subjected to immunoblot analysis using standard methods. The primary antibodies used in this study were rabbit anti‐serpina1 (dilution 1:1000, raised in the rabbit; goat anti‐agrin and its active fragment (dilution 1:1500, a generous gift from Dr. P. Sonderegger, Department of Biochemistry, University of Zurich, Winterthurerstrasse 190, CH‐8057 Zurich, Switzerland), rabbit anti‐neurotrypsin (dilution 1:1000, ab59452; Abcam, Cambridge, MA), and mouse anti–β‐actin (dilution 1:2000, A0760‐40; US Biological, Pittsburgh, PA). The following secondary antibodies were from Santa Cruz Biotechnology Inc., Dallas, TX. These antibodies were: donkey anti‐goat (1:2000, sc‐2020), goat anti‐rabbit IgG (1:1500, sc‐2054), and rabbit anti‐mouse IgG (1:3000, sc‐358914). Chemiluminescence was detected using SuperSignal west pico luminol/enhancer solution (Cat#34077, Pierce Biotechnology; Thermo Scientific, Rockford, IL) according to the manufacturer's instructions. Densitometric analysis was performed by Quantity One software (170‐9601; Bio‐Rad, Hercules, CA). Densitometric measurements of the protein bands were obtained from three independent gels and the results were statistically analyzed using the Student's t‐test. Error bars in the figures indicate standard deviation (n = 3).
Cortical neuron culture
Primary cortical neurons were cultured as previously described,29 with minor modifications. Briefly, cerebral cortices were isolated from 15‐ to 17‐day‐old Cln1 −/− and WT embryos, and dissociated by trituration using 0.25% trypsin. The dissociated cells were cultured in either polylysine‐coated slide chambers (Nunc, Rochester, NY, USA) or in Petri dishes using neurobasal medium (Invitrogen) supplemented with 10% heat‐inactivated fetal bovine serum, 300 μmol/L glutamine, 2% B27, and antibiotics. The medium was changed 24 h after plating the cells. On the 4th day after plating, 50% of the medium was replaced with fresh medium. Subsequently, medium was changed every 3–4 days.
Transfection of cells with cDNA constructs
Cultured WT astroglia were transfected with serpina1 and vector control cDNA constructs (SC323039 and pCMV6‐XL4; Origene, Gaithersberg, MD). The Cln1 −/− astrocytes were either transfected with serpina1‐shRNA or scrambled‐shRNA (RMM4532 NM_009243 and RHS4349; Thermo Fisher Scientific, AL, Waltham, MA, USA) using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's protocol. The plasmids used for transfection were prepared using a Plasmid Mini Kit (QIAGEN). The consistency between plasmid preparations was monitored by determining the concentration of plasmids by both spectrophotometry and agarose gel electrophoresis.
Enzymatic activity assay
Neurotrypsin‐like activity was assayed using a fluorometric assay kit (SensoLyte® Red Protease Assay Kit; ANASPEC Inc. Fremont, CA), according to the manufacturer's specifications.
Confocal and fluorescent microscopic imaging
Primary cultures of astroglia and/or neurons from Cln1 −/− mice and those of their WT littermates were plated on slide chambers (Nunc™ Lab‐Tek™; Thermo Scientific, Rockford, IL) and incubated at 37°C in an atmosphere of 5% of CO2 and 95% air. The cells were washed three times with Phosphate‐buffered saline (PBS) (pH 7.6), and fixed using 4% paraformaldehyde solution for 15 min at room temperature. The primary antibodies used were: anti‐serpina1 (1:200; a generous gift from a generous gift from Dr. P. Sonderegger), anti‐neurotrypsin (1:300, ab59452; Abcam, Cambridge, MA), anti‐synaptophysin (1:300, ab23754; Abcam, Cambridge, MA), anti‐agrin (1:300, generous gift from Dr. Sonderegger), anti‐GFAP (1:1000, SAB4501162; Sigma‐Aldrich, St. Louis, MO), and anti‐β‐lll‐tubulin (1:1000,T5076, Sigma, St. Louis, MO). The secondary antibodies bought from Invitrogen (Invitrogen, Life Technologies, Grand Island, NY) were: Alexa Fluor 488–conjugated anti‐goat (1:300, A‐11029), Alexa Fluor 555–conjugated anti‐rabbit (1:300, A‐21206). Nuclei were stained with 4',6‐diamidino‐2‐phenylindole (DAPI) in mounting solution (SKU‐F6057, Sigma‐Aldrich, St. Louis, MO). Fluorescence was visualized with the Zeiss LSM 510 Inverted Meta confocal microscope or Zeiss Axio plus florescence microscope (Carl Zeiss, Thornwood, NY, USA), and the image was processed with the LSM image software (Carl Zeiss, NICHD Microscope Core Facility) or SPOT advanced plus image software (Diagnostic Instruments Inc., Sterling Heights, MI). Astroglia obtained from Cln1 −/− mice and their WT littermates were differentiated from cultured neurospheres. In each experiment, images were acquired using identical settings and the same threshold was used for all groups. All experiments were performed at six different locations and repeated three times with the same effect.
Synaptosome preparation
Isolation of synaptosomes from WT and Cln1 −/− mouse brains was performed according to a previously reported protocol30 with minor modifications. Briefly, cortical tissues (100 mg) from the mouse brain were removed and placed on sucrose homogenization buffer (0.32 mol/L sucrose, 1 mmol/L Ethylenediaminetetraacetic acid (EDTA), 5 mmol/L HEPES) in a glass homogenizer, gently striking 10 times up and down. Homogenate was centrifuged at 1000g 4°C for 10 min, and the supernatant was collected and transferred onto a discontinuous Percoll gradient (3%, 10%, 15% and 23%) for 5 min and centrifuged at 32,500g 4°C. The white cloudy synaptosomes from the 15% and 23% interface were collected and washed with 30 mL wash buffer (122 mmol/L NaCl, 5 mmol/L KCl, 1.15 mmol/L NaH2PO4, 1 mmol/L glucose, 5 mmol/L HEPES, 1× protease inhibitor cocktail) and centrifuged at 15,000g for 15 min. The pellets were resuspended in a 500 μL chilled wash buffer and stored at −20°C for later use.
Immunoprecipitation of serpina1 and neurotrypsin
Total synaptosome preparations (1.5 mg protein) from WT mice or from their Cln1 −/− littermates were precleared and incubated with serpina1 (1: 500 generous gift from a generous gift from Dr. P. Sonderegger) and/or anti‐neurotrypsin (1:1000 Abcam; ab‐59452) antibodies for 1.5 h at 4°C with gentle rotation. Protein A/G (25 μL, Dallas, TX, USA) agarose beads (Santa‐Cruz Biotechnology, Cat# sc‐2003) were added and the mixtures were incubated with rotation overnight at 4°C. The next day, beads were washed six times with 1x PBS and resuspended in 40 μL of 2x electrophoresis sample buffer (Santa‐Cruz Biotechnology, Cat# sc‐24945).
Detection of interneuron loss in Cln1 −/− mouse brain
WT mice and their Cln1 −/− littermates at 1.5–6 months old were perfused using 4% paraformaldehyde, pH 7.2. Brains were removed and postfixed in 4% paraformaldehyde for 4–16 h at 4°C. After washing in 1x PBS, brains were transferred into 25% sucrose until they sunk. 50 μm horizontal sections were cut on a frozen sliding microtome (HM430; MICROM International, GmbH, Walldorf, Germany). Brain slices were thoroughly washed in 1x PBS. Endogenous peroxidase was blocked by incubation in 3% hydrogen peroxide in 1x PBS at room temperature for 30 min. Slices were then blocked and permeablized in 1x PBS containing 1% Bovine serum albumin (BSA), 10% goat serum, and 0.5% TritonX‐100 at room temperature for 2 h. Primary antibodies were diluted in 1x PBS containing 1% BSA, 1% goat serum, and 0.5% TritonX‐100. Brain slices were incubated in primary antibody solution at 4°C for 16 h. For parvalbumin (PV) antibody (rabbit anti‐PV 1:2500; Swant, Switzerland), brain slices at all ages, and for somatostatin (SOM) antibody (rabbit anti‐SOM 1:1000; DAKO, Carpinteria, CA, USA) and COUP transcription factor 2 (COUP TFII) antibody (mouse anti‐ COUP TFII 1:1000; R&D Systems, Minneapolis, MN, USA), brain slices at 1.5‐month only, fluorophore‐conjugated secondary antibodies were used. Alexa‐Fluor 555‐conjugated goat secondary antibodies (Molecular Probes) were diluted in 1x PBS containing 1% BSA and 1% goat serum at 1:1000 and incubated with brain slices at room temperature for 1 h. After staining, slices were mounted on gelatinized slides and layered over with ProLong Gold hard set with DAPI (Invitrogen). For cholecystokinin (CCK) antibody (mouse anti‐CCK 1:5000; CURE UCLA, Los Angeles, CA, USA), brain slices at all ages, and for SOM and COUP TFII antibodies, brain slices older than 1.5‐month, staining was detected by DAB staining using Elite ABC Kit (Vector Laboratory, Burlingame, CA, USA) following the manufacturer's protocol. After mounting on gelatinized slides, stained slices were dehydrated and covered using Permount (Fisher Scientific, Pittsburgh, PA, USA). Stained brain slices were photographed using an Olympus Provis AX70 microscope (Olympus America, Center Valley, PA) and images were captured using QCapture Suite (QImaging, Surrey, BC). Immunopositive interneurons were manually counted using the cell counter plugin of ImageJ. The surface area of the region of interest was measured using ImageJ software (National Institutes of Health, Bethesda, MD) and multiplied by the thickness of the slice. The cell density in the area of interest was calculated as cells/mm3.
Statistical analysis
To estimate the kinetics of endocytosis (Rate_rapid and Rate_slow), we measured the initial rate of the capacitance decay in a time window of 2 or 4 sec after the stimulation with a single 20 msec pulse or a train of 10 pulses, respectively. The data were statistically analyzed by t‐test. All other data are statistically evaluated by one‐way analysis of variance (ANOVA) and post hoc tukey t‐test. P < 0.05 was considered significant. All experiments were repeated at least three times and the results are expressed as the mean ± SEM.
Results
Dendritic spine abnormalities in Cln1 −/− mice
Recent reports indicate that the synaptic dysfunction is preceded by a reduction in the number of dendritic filopodia, and the preponderance of long and thin dendritic spines.31 Thus, we first analyzed the dendritic filopodia in 1‐, 3‐, and 6‐month‐old Cln1 −/− mice20 that mimic INCL21 and in their WT littermates. We found that while the total dendritic spine counts in the hippocampal neurons of 1‐ and 3‐month‐old Cln1 −/− mice were reduced by only ~9% of those in WT littermates (Fig. 1A–D), at 6 months of age, these mice showed a significant decline (35% of WT; P < 0.01) in dendritic spine levels (Fig. 1E and F). Although the majority of the spine‐types affected in Cln1 −/− mouse hippocampus were the long and thin variety, (Fig. 1A, C, and E, right panels) those in their WT littermates contained the short and thick spines on dendrites (Fig. 1A, C and E, left panels). These results suggest that the abnormal dendritic spine morphology in Cln1 −/− mice is appreciable in these mice around 6 months of age when they manifest signs of synaptic dysfunction attested by myoclonus and seizures.
Figure 1.
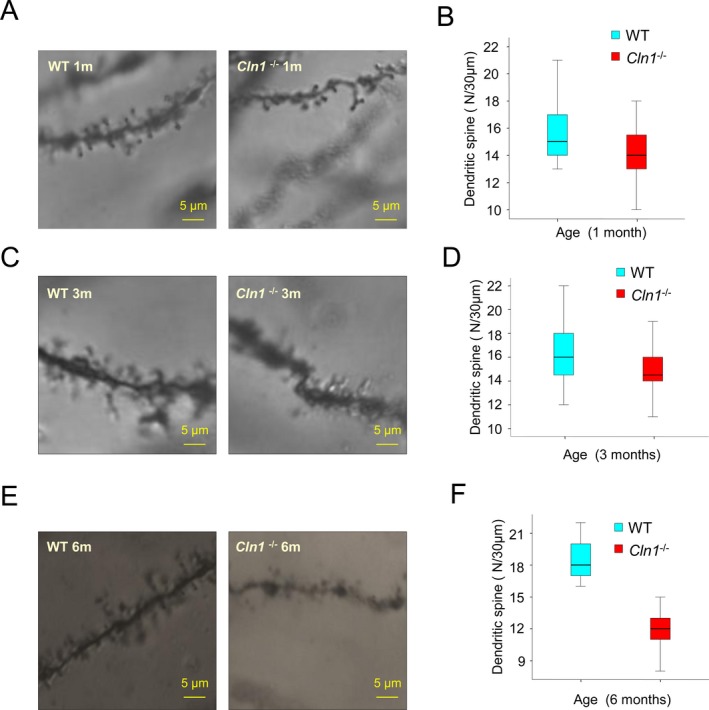
Dendritic filopodia abnormality in the hippocampus of Cln1 −/− mice. Representative Golgi‐Cox‐stained neuron images of dendrites from WT mouse hippocampus (left panels) and their Cln1 −/− littermates (right panels) at 1 month (A), 3 months (C) and 6 months (E) of age. The majority of the spine‐type affected was stubby spines (A, C, and E). The reductions of total spine in the hippocampi of Cln1 −/− mice occur in a time‐dependent manner. The numbers of total spines are 9% less in 1‐month old Cln1 −/− mouse hippocampus compared to those of their WT littermates (B). The total spine levels decline further in 3‐month‐old Cln1 −/− mice (D). There is a significant reduction in dendritic filopodia levels in the hippocampi of 6‐month‐old Cln1 −/− mice (35% of WT; P < 0.01, one way ANOVA) (F). The results are presented as the mean spine counts from five randomly selected hippocampal neurons in each mouse brain. A 30 μm long dendrite was used as a counting standard, and five dendrites were counted separately in each neuron. WT, wild‐type; ANOVA, analysis of variance.
Reduced SV exocytosis in Cln1 −/− mice
To evaluate the synaptic function, we conducted electrophysiological studies using the calyx of Held preparations32, 33 from Cln1 −/− mice and those from their WT littermates. The calyx of Held, a very large glutamatergic axonal terminal in the auditory brain stem,32 which is ideal for patch‐clamp studies to evaluate synaptic function at high resolution.26 Accordingly, we first measured the synaptic vesicle (SV) exocytosis by monitoring the membrane capacitance (C m) in calyx of Held preparations from Cln1 −/− mice and in those of their WT littermates. Notably, in the WT calyx, stimulation with a train of 10 depolarization pulses (from −80 mV to +10 mV for 20 msec, with an interval of 80 msec) triggered a total increase in C m (ΔC m_train) to 1333 ± 109 fF (n = 5) (Fig. 2A and C, left panel), which reflected the sum of vesicle membranes that fused with the plasma membrane during exocytosis. However, in the calyx of Cln1 −/− mice, the ΔC m_train was significantly smaller (999 ± 56 fF, n = 8, P = 0.023, t‐test), indicating that Ppt1 deficiency may have led to ~25% reduction in exocytosis induced by the stimulation train. Because the stimulation train depletes the readily releasable vesicle pool (RRP), the newly generated SVs are mobilized during the stimulation.26 Thus, the observed decrease in the ΔC m train may result from either a smaller RRP, or slower SV mobilization, or both.
Figure 2.
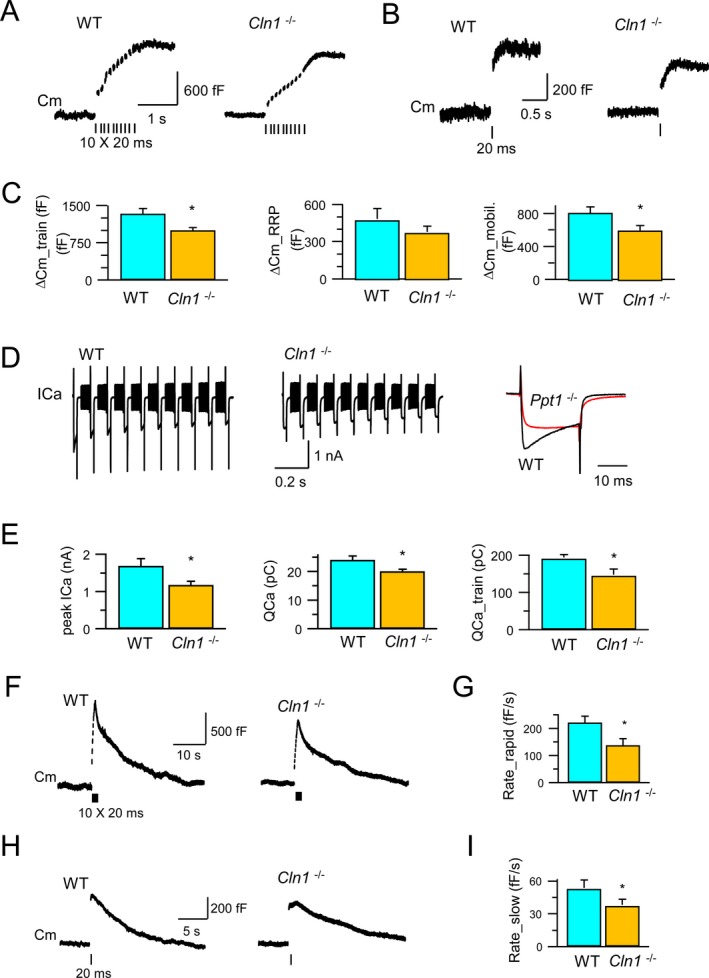
Exocytosis, endocytosis and Ca2+ currents at the calyces of Held. Exocytosis of synaptic vesicles and RRP in the calyx of Held from both WT and Cln1 −/− mice were measured by monitoring the whole‐cell membrane capacitance (C m) (A and B). Compared to its WT littermate, Cln1 −/− mice had 25% reduction in exocytosis (upper panel), a small, but insignificant decrease in the RRP size (middle panel), and significant inhibition of vesicle mobilization of releasable vesicles (C). The depolarization‐evoked Ca2+ currents were also detected. (D) The peak amplitude (ICa) was reduced from 1.7 nA (WT) to 1.2 nA (Cln1 −/−); while the charge of Ca2+ influx was reduced from 24 pC (WT) to 20 pC (Cln1 −/−) for a single 20 msec pulse, and from 190 pC (WT) to 144 pC (Cln1 −/−) for a train of 10 pulses (E). Compared to the WT mice (F), Cln1 −/− littermates showed substantial reduction in rapid endocytosis (221 fF/sec vs. 136 fF/sec) (F and G) and in slow endocytosis (52 fF/sec vs. 37 fF/sec) (H and I). RRP, readily releasable vesicle pool; WT, wild‐type; ICa, calcium current. *means that the results are significantly different between wt and Ppt1 −/−, at the significance level: P < 0.05 (two‐tailed student t‐test).
To determine which of these possibilities are operating here, we first investigated whether Ppt1 deficiency adversely affected the size of RRP, which can be measured as a C m increase (ΔC m_RRP) following a single 20 msec depolarization pulse.34 The results showed that the ΔC m_RRP in the WT calyx (482 ± 86 fF; n = 5) was not significantly different from that in the Cln1 −/− calyx (380 ± 43 fF; n = 9; P = 0.27) (Fig. 2B and C, middle panel). By subtracting ΔC m_RRP from ΔC m_train, we estimated the C m increase contributed by the mobilized SVs over the stimulation train (ΔC m_mobil.) to be 825 ± 72 fF (n = 5) in the WT calyx, and 613 ± 59 fF (n = 8; P = 0.04) in Cln1 −/− calyx, which was significantly lower (Fig. 2B and C, right panel). Taken together, these results suggested that Ppt1 deficiency adversely affected SV exocytosis most likely due to the inhibition of SV mobilization essential for continuously generating fresh SVs to maintain the RRP of SVs under sustained stimulation.
Suppressed calcium influx inhibits SV‐mobilization in the brain of Cln1 −/− mice
It has been reported that the synchronous SV fusion is essential for neurotransmission in the central nervous system and it is evoked by Ca(2+) influx induced by the action potential.33 Since depolarization induces the ICa, which regulates fusion and mobilization of the SVs, we investigated whether depolarization‐evoked ICa is suppressed in Cln1 −/− mice. The results showed that during depolarization induced by −80 mV to +10 mV, ICa in the WT calyx (n = 5) peaked at 1.7 ± 0.2 nA and led to 24 ± 1.4 pC of Ca2+ influx (QCa) over a 20 msec pulse. By contrast, in the Cln1 −/− calyx (n = 9), both the peak of ICa and Ca2+ influx were significantly reduced to 1.2 ± 0.1 nA (P = 0.048), and 20 ± 0.8 pC (P = 0.049), respectively (Fig. 2D and E, left panel). Furthermore, the total amount of Ca2+ influx during the train stimulation (QCa_train) decreased from 190 ± 8 pC (n = 5) in the WT calyx to 144 ± 15 pC (n = 9) in the Cln1 −/− calyx (P = 0.016) (Fig. 2D and E, middle and right panels). These results suggested that in Cln1 −/− mice, depolarization‐induced Ca2+ influx was suppressed, which most likely inhibited SV mobilization.
SV endocytosis in the nerve terminals is suppressed in Cln1 −/− mice
Previously, we reported that Ppt1 deficiency caused persistent retention of the SVs on presynaptic membrane.35 This may have been caused by disruption of SV endocytosis due to impaired depalmitoylation of the SV‐associated proteins, which require dynamic palmitoylation (palmitoylation–depalmitoylation) for recycling. Notably, it has been shown that S‐palmitoylation regulates large conductance calcium‐ and voltage‐activated potassium (BK) channels.36 Because varying factors may regulate endocytosis, the retrieval of SV membranes occurs at different rates. Indeed, it has been reported that C m measurement in the calyx causes both rapid and slow C m decay after stimulation, which correspond to rapid and slow endocytosis,34 respectively. Since rapid and slow endocytosis may be subject to differential regulation, we sought to determine whether they were selectively affected in Cln1 −/− mice. We found that stimulation with ten 20 msec pulses at 10 Hz triggered rapid C m decay at an initial rate of 221 ± 24 fF/sec in the WT calyx (n = 4) (Fig. 2F and G), which reflected rapid endocytosis. However, this rate decreased significantly to 136 ± 25 fF/sec in the calyx of Cln1 −/− mice (n = 6) (P = 0.047), suggesting that rapid endocytosis was inhibited by Ppt1 deficiency. Moreover, milder stimulation with a 20 msec depolarization pulse induced slow endocytosis, which was indicated as C m decay with an initial rate of 52 ± 7 fF/sec in the WT calyx (n = 4). Notably, in the Cln1 −/− calyx, this rate declined significantly to 37 ± 5 fF/sec (n = 6; P = 0.03) (Fig. 2H and I) strongly suggesting that Ppt1‐ deficiency inhibited slow endocytosis. Taken together, our results showed that the lack of Ppt1 impaired both rapid and slow endocytosis of SVs in Cln1 −/− mouse brain disrupting neurotransmission.
Decreased cleavage of agrin in brain tissues of Cln1 −/− mice
Emerging evidence suggests that activity‐regulated proteolysis at neuronal synapses mediates synaptic strength and structural remodeling of neuronal circuits.12 Moreover, it has been reported that agrin‐22 is the active fragment of agrin and that agrin cleavage is catalyzed specifically by neurotrypsin‐generating agrin‐22, which is essential for the induction of dendritic filopodia.37 Thus, we measured agrin‐22 levels in brain cortices of Cln1 −/− mice and in those of their WT littermates by western blot analysis. Our results showed that the levels of high‐molecular‐weight, full‐length agrin in the brains of Cln1 −/− mice were markedly higher compared with those of their WT littermates (Fig. 3A). Moreover, compared with WT littermates, the levels of agrin‐22 in the brains of Cln1 −/− mice were disproportionately low (Fig. 3A, lower panel). These results raised the possibility that the cleavage of full‐length agrin required for generating agrin‐22 may be impaired in Cln1 −/− mice.
Figure 3.
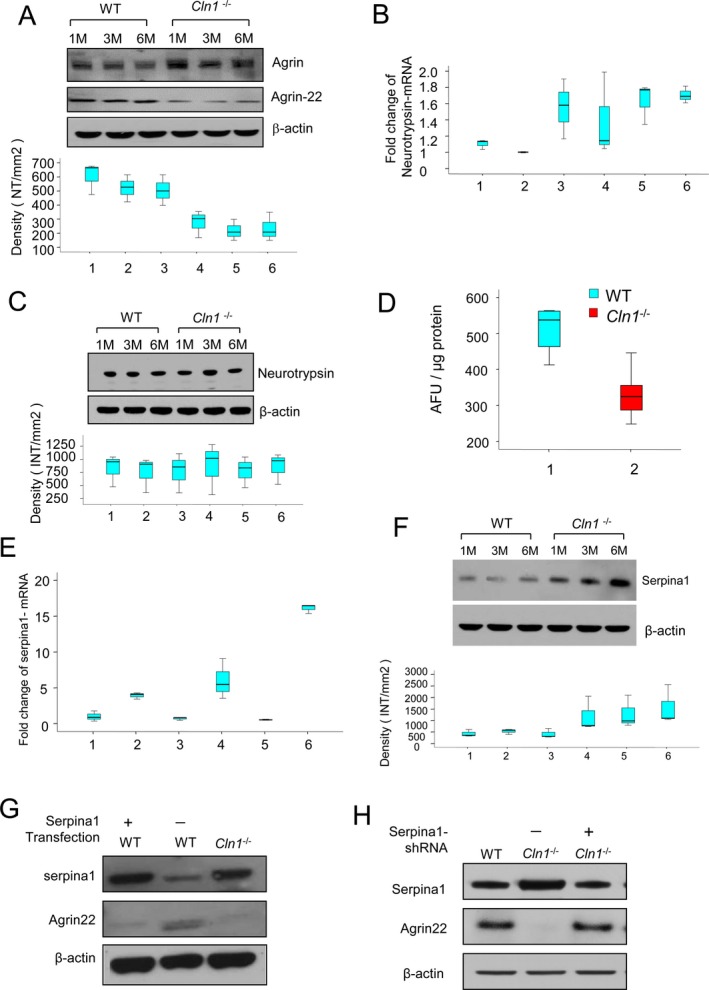
Neurotrypsin‐mediated agrin‐22 production is disrupted in Cln1 −/− mouse brain. Western blots analysis of intact and cleaved agrin (agrin‐22) proteins in wild‐type (WT) and Cln1 −/− mouse brains (A). The lower panel showed that the mean densitometry of each agin‐22 from WT and Cln1 −/− mouse brains. Compared with WT mice the levels of agrin‐22 in the brain of Cln1 −/− littermates were markedly reduced (P < 0.05, Excel student t‐test) (A). Neurotrypsin‐mRNA levels measured by qRT‐PCR showed no significant difference between WT mice and their Cln1 −/− littermates (1 and 2 for 1‐month‐old WT and Cln1 −/− mice, respectively; 3 and 4 for 3‐month‐old WT and Cln1 −/− mice, respectively; 5 and 6 for 6‐month old WT and Cln1 −/− mice, respectively) (B). Western blot analysis showed that neurotrypsin protein levels between Cln1 −/− and their WT littermates were virtually no difference (C). Enzymatic activities of neurotrypsin in brain tissues of 6‐month old Cln1 −/− mice was significantly lower compared with those of their WT littermates (D). Quantitative RT‐PCR and Western blot analyses showed increased levels of serpina1‐mRNA (E; 1 and 2 for 1‐month‐old WT and Cln1 −/− mice; 3 and 4 for 3‐month‐old WT and Cln1 −/− mice; 5 and 6 for 6‐month‐old WT and Cln1 −/− mice, P < 0.05, Excel student t‐test) and protein (F) from cortical tissues of Cln1 −/− mice compared with those of their WT littermates. Serpina1 overexpression in WT mouse glial cells significantly reduced the level of agrin‐22 (G). To further confirm the inhibition of serpina1 to neurotrypsin activity, serpina1 knockdown in Cln1 −/− glial cells showed that agrin‐22 protein level was increased (H).
Neurotrypsin‐like protease activity is reduced in the brain tissues of Cln1 −/− mice
Agrin is a large proteoglycan and the only known substrate of neurotrypsin,14, 16 a serine protease localized to the neuronal synapses in the brain. Mutations in the neurotrypsin gene have been reported to cause myoclonus, seizures, and mental retardation.15 Since INCL patients also develop synaptic dysfunction, manifested as psychomotor retardation, myoclonus, and seizures, we reasoned that Cln1 −/− mice may have reduced neurotrypsin activity in the brain, which impairs agrin‐22 production leading to synaptic pathology. Accordingly, we first determined the neurotrypsin‐mRNA levels in the brain of WT and Cln1 −/− mice by quantitative RT‐PCR. The results showed no significant differences in brain neurotrypsin‐mRNA levels between Cln1 −/− mice and those of their WT littermates (Fig. 3B). Western blot analysis also showed that there were virtually no differences in the brain neurotrypsin‐protein levels between Cln1 −/− mice and those of their WT littermates (Fig. 3C). Next, we determined brain neurotrypsin‐like protease activity in Cln1 −/− mice and in their WT littermates. The results showed that protease activity in the Cln1 −/− mouse brain was significantly lower compared with that of their WT littermates (Fig. 3D). These results demonstrate that the catalytic activity of neurotrypsin, but not the level of neurotrypsin‐protein, is adversely affected by disruption of the Cln1 gene.
The enzymatic activity of serine proteases in the brain is regulated by endogenous inhibitors called serpins.18 Previously, in a cDNA microarray analysis, we found that the level of serine protease inhibitor clade A (commonly known as serpina1) was elevated several fold in Cln1 −/− mouse brain. Thus, we performed quantitative real‐time RT‐PCR using total RNA from the brain cortices of Cln1 −/− mice and those of their WT littermates to confirm the microarray findings. The results showed that serpina1‐mRNA levels were significantly elevated in brain tissues of Cln1 −/− mice in an age‐dependent manner (Fig. 3E). Notably, in Cln1 −/− mice the increased serpina1‐mRNA levels were appreciable as early as 1 month of age. The total serpina1‐protein levels were also markedly elevated in Cln1 −/− mouse brain (Fig. 3F). These results raised the possibility that overproduction of serpina1 may suppress neurotrypsin activity in Cln1 −/− mouse brain.
Serpina1 suppresses agrin‐22 production
To delineate whether there is a relationship among the levels of neurotrypsin, serpina1, and agrin‐22, we transfected cultured astroglia from WT mice with either vector alone (control) or serpina1‐cDNA construct and determined enzymatic activity of neurotrypsin. The results showed that serpina1‐overexpression markedly suppressed neurotrypsin‐like protease activity as attested by reduced cleavage of intact agrin, which caused reduced agrin‐22 levels (Fig. 3G). As expected, the transfection of the cells with vector alone (control) did not alter agrin‐22 levels suggesting that neurotrypsin activity was not altered. To further confirm these results, we transfected cultured astroglia from Cln1 −/− mice with serpina1‐shRNA and measured the level of agrin‐22. Remarkably, the results showed a substantial increase in agrin‐22 level compared with that in the scrambled shRNA‐treated cells (Fig. 3H). Taken together, these data suggested that serpina1 overexpression in WT astroglia inhibited neurotrypsin activity and consequently, agrin‐22 levels were reduced in these cells, whereas shRNA suppression of serpina1 expression in Cln1 −/− cells increased agrin‐22 levels.
Serpina1 is expressed predominantly in astroglia from Cln1 −/− mice
The results of our experiments have shown that the levels of high‐molecular‐weight agrin in Cln1 −/− mice were substantially higher compared with those of their WT littermates. One possible explanation is that the increase in high‐molecular‐weight agrin level is due to the overexpression of the agrin gene. To evaluate this possibility, we first determined the agrin‐mRNA levels by real‐time RT‐PCR using total RNA from the brain tissues of Cln1 −/− mice and those of their WT littermates. The results showed that there was virtually no difference in the brain agrin‐mRNA levels between Cln1 −/− mice and those of their WT littermates (data not shown). To determine if specific cell types in the brain expressed serpina1 and neurotrypsin, we assessed serpina1‐ and neurotrypsin‐mRNA levels, respectively, using primary cultures of neurons and astroglia by quantitative RT‐PCR. The results showed that while neurons from both WT and Cln1 −/− mice expressed serpina1‐mRNA, the expression levels were virtually identical (Fig. 4A). However, in the astroglia from Cln1 −/− mice, the level of serpina1‐mRNA was markedly higher than in those of their WT littermates (Fig. 4B). These results were confirmed by higher serpina1‐protein level in western blot analysis (Fig. 4C and D). To further confirm these results, we used immunocytochemical analysis of cultured neurons and astroglia to determine the expression of neurotrypsin and serpina1. The results showed that while serpina1 immunostaining in cultured neurons from WT and Cln1 −/− mice was virtually undetectable, that in cultured astroglia from Cln1 −/− mice was strongly positive (Fig. 4E and F). Notably, cultured astroglia from WT and Cln1 −/− mice were also stained positive for agrin and neurotrypsin (Fig. 4G). Taken together, these results suggested possible interaction among the three proteins (i.e., neurotrypsin, serpina1, and agrin).
Figure 4.
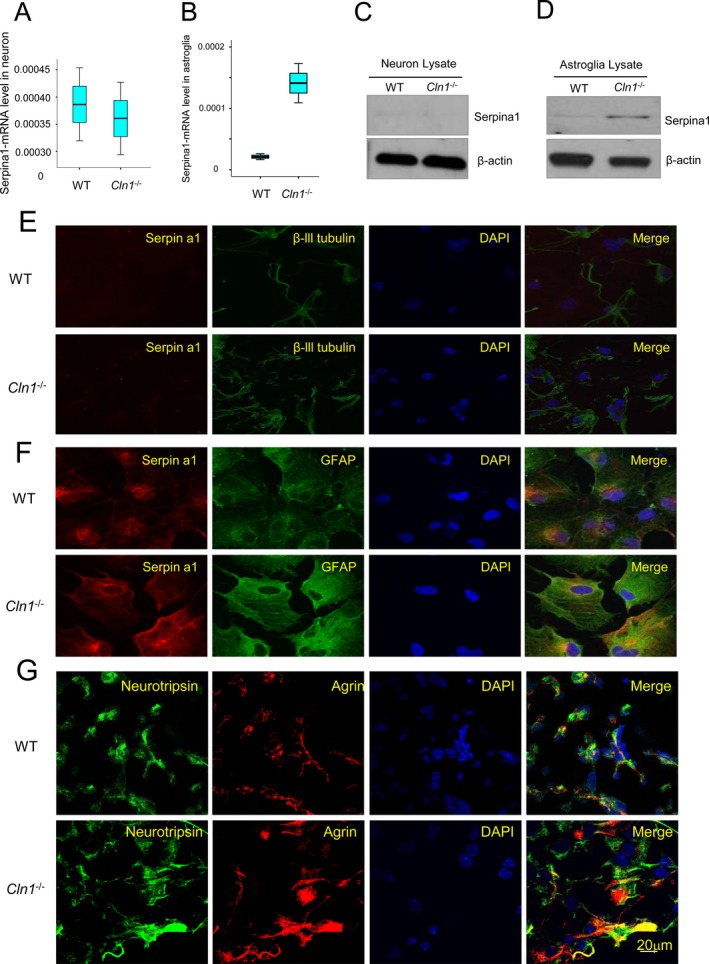
Agrin and Serpina1 expression in brain tissues and cultured astroglia. Serpina1‐mRNA levels in cultured neurons and astroglia (A and B) (P < 0.01; Excel student t‐test); (C) Western blot analysis from cultured neurons from wild‐type (WT) and Cln1 −/− mice. β‐actin was used as loading controls. (D) Western blot analysis cultured astroglia from WT and Cln1 −/− mice. (E) Cytochemical localization of serpina1 in cultured neuron from WT and Cln1 −/− mice. Cultured neurons were stained with anti‐serpina1 and beta‐3‐tublin (neuronal marker) antibodies, respectively; (F) Cultured astroglia stained with anti‐serpina1, and anti‐GFAP (astrocyte marker), respectively. (G) We performed experiments to cytochemically detect agrin and neurotrypsin in cultured astroglia and the results showed that cultured astroglia from WT mice (upper panel) and their Cln1 −/− littermates (lower panel) were stained positive for agrin as well as neurotrypsin.
Neurotrypsin and serpina1 are colocalized in synaptosomes
Emerging evidence suggests a bidirectional communication between astroglia and neurons at neuronal synapses.38 Since we found that neurotrypsin is predominantly expressed in neurons and serpina1 is expressed primarily by astroglia, serpina1‐mediated inhibition of neurotrypsin activity is likely to require their colocalization in the synapses where agrin is also present. Thus, we performed pull‐down assays using antibodies against either serpina1 or neurotrypsin in synaptosomal fractions from the brain cortices of WT and Cln1 −/− mice. The results showed that serpina1 antibody pulled down neurotrypsin (Fig. 5A) and anti‐neurotrypsin pulled down serpina1 (Fig. 5B). To further confirm the presence of both serpina1 and neurotrypsin in or near the synapses, we performed confocal microscopic analysis using cocultures of astroglia and neurons to determine whether serpina1‐ and neurotrypsin immunoreactivities colocalized at the synapses. Our results showed that both serpina1 and neurotrypsin colocalized with the synaptic marker, synaptophysin (Fig. 5C and D). Importantly, colocalization of serpina1 with synaptophysin was markedly pronounced in cocultures of astroglia and neurons from Cln1 −/− mice (Fig. 5C, lower panel).
Figure 5.
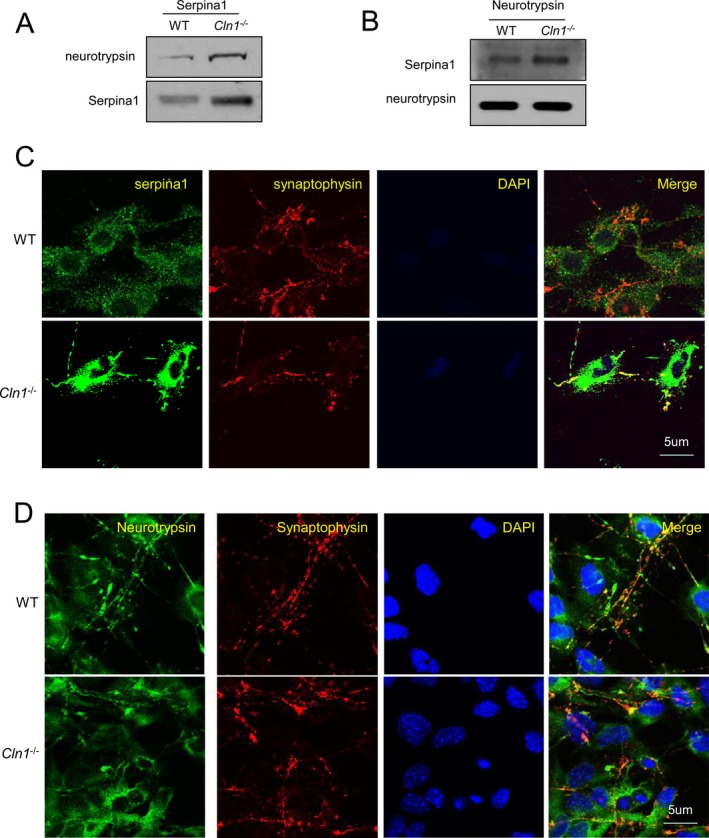
Colocalization of neurotrypsin with serpina1 in astrocyte‐neuron cocultures. (A) Pull‐down assay using serpina1 antibody showed that there was much more neurotrypsin in immunoprecipitates of synaptosome preparations from Cln1 −/− mouse brains compared with those of its wild‐type (WT) littermates; (B) Western blot analysis of the immunoprecipitates with neurotrypsin antibody showed serpina1 is pulled down in synaptosomal preparations from WT mice and their Cln1 −/− littermates; (C) Astroglia and neurons were cocultured and stained with anti‐serpina1 and anti‐synaptophysin (a synaptosome marker); (D) Cocultured astrocyte with neurons from WT and Cln1 −/− mice were stained with anti‐neurotrypsin and synaptophysin.
Oxidative stress stimulates serpina1 expression via upregulation of C/EBP‐δ
Oxidative stress in the brain has been linked to many neurodegenerative disorders although it is difficult to ascertain whether it is a major cause or a consequence of neurodegeneration.1 We previously reported that high levels of Endoplasmic Reticulum‐ and oxidative stress in the brain of Cln1 −/− mice mediate neuronal apoptosis contributing to neurodegeneration.19, 20 Moreover, the demonstration that transcription factor, CCAAT enhancer‐binding protein (C/EBP‐δ), is sensitive to oxidative stress39 prompted us to analyze whether the 5′ promoter region of the serpina1 gene contains C/EBP‐δ‐binding site(s). We identified two C/EBP‐δ‐binding sites in the 5′ promoter region of the serpina1 gene. We reasoned that oxidative‐stress may mediate C/EBP‐δ upregulation, thereby stimulating serpina1 production in the brain of Cln1 −/− mice. To test this hypothesis, we used cultured astroglia from WT mice in which oxidative stress was induced by H2O2 treatment, and determined the levels of C/EBP‐δ‐mRNA and C/EBP‐δ‐protein by real‐time RT‐PCR and western blot analysis, respectively. The results showed that compared with the untreated cells, those treated with H2O2 expressed markedly elevated levels of both C/EBP‐δ‐mRNA (Fig. 6A) and C/EBP‐δ‐protein (Fig. 6B). Consistent with these results, oxidative stress also caused marked elevation in serpina1‐mRNA (Fig. 6C) and protein (Fig. 6D) levels. Densitometric analysis (Fig. 6E) of the protein bands in the western blot confirmed the mRNA results. To further confirm that oxidative stress‐mediated C/EBP‐δ overexpression‐stimulated serpina1 production, we treated cultured astroglia from WT mice and those of their Cln1 −/− littermates with C/EBP‐δ‐siRNA and subjected these cells to oxidative stress by H2O2 treatment. The results showed that compared with the untreated and scrambled siRNA‐treated astroglia from Cln1 −/− mice, the cells treated with C/EBP‐δ‐siRNA showed substantially decreased levels of serpina1‐mRNA (Fig. 6F) and serpina1 protein (Fig. 6G). Taken together, these results suggested that the oxidative stress stimulated serpina1 gene expression via upregulation of C/EBP‐δ.
Figure 6.
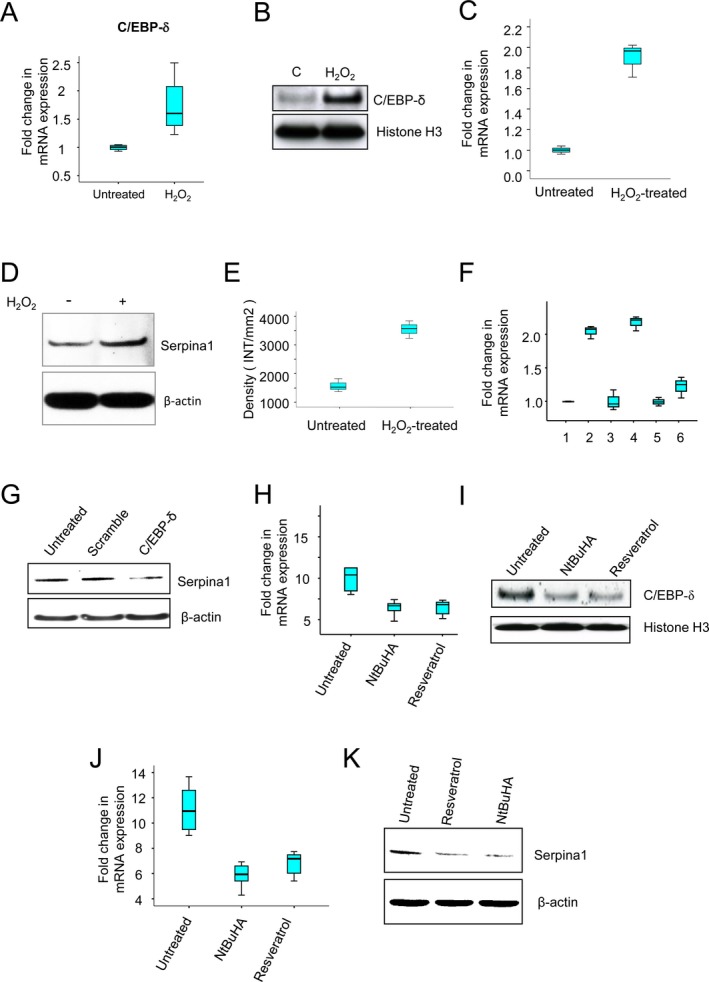
Oxidative stress via C/EBP‐δ upregulates serpina1 expression: effect of anti‐oxidants. The levels of mRNA (A) and protein (B) of oxidative‐stress sensitive transcription factor, C/EBP‐δ, were significantly higher in the WT glia cells treated with H2O2 compared with those of untreated WT glia cells. H2O2 treatment markedly increased levels of both serpina1‐mRNA (C) and serpina1‐protein (D and E) in cultured WT glia cells. Compared to the untreated and scramble siRNA‐treated controls, C/EBP‐δ‐mRNA knockdown significantly reduced the levels of sperpina1‐mRNA (F): 1, untreated control; 2, H2O2‐treated; 3, scramble siRNA‐treated; 4, siRNA‐ + H2O2‐treated; 5, C/EBP‐δ‐siRNA‐treated; 6, C/EBP‐δ‐siRNA‐ + H2O2‐treated; and serpina1‐protein (G) in cultured Cln1 −/−astroglia that were treated with H2O2 (400 μmol/L for 1 h), (n = 3, P < 0.05). Administration of either NtBuHA or RSV to Cln1 −/− mice for 3 months significantly lower levels of C/EBP‐δ ‐mRNA (H) and C/EBP‐δ‐protein (I) compared with untreated Cln1 −/− mice (P < 0.05, Excel student t‐test). Levels of serpina1‐mRNA (J) and serpina1‐protein (K) were reduced in the anti‐oxidant treated mice compared with those of untreated Cln1 −/− mice. C/EBP‐δ, CCAAT enhancer binding protein; WT, wild‐type; RSV, resveratrol.
Antioxidants reduce serpina1 production in the brain of Cln1 −/− mice
Persistent ER‐ and oxidative stress causes neuronal apoptosis,22 which contributes to neuropathology in INCL.23 Moreover, antioxidants have been reported to reduce the oxidative stress. Thus, we treated 3‐month‐old Cln1 −/− mice with several compounds that are known to manifest antioxidant property including NtBuHA24 and RSV.23 The results showed that compared with untreated Cln1 −/− mice, their antioxidant‐treated littermates showed significantly lower levels of C/EBP‐δ‐mRNA (Fig. 6H) and C/EBP‐δ‐protein (Fig. 6I) suggesting that C/EBP‐δ upregulation is sensitive to oxidative stress. Furthermore, antioxidant‐treated mice showed reduced levels of serpina1‐mRNA (Fig. 6J) and serpina1‐protein (Fig. 6K). Together, these results strongly suggested that oxidative stress‐mediated serpina1 overexpression via upregulation of C/EBP‐δ and antioxidants reduce serpina1‐expression via downregulation of C/EBP‐δ in cultured astroglia derived from Cln1 −/− mice.
Early loss of hippocampal interneurons in the brain of Cln1 −/− mice
The results of our electrophysiological studies using calyx of Held revealed abnormal synaptic function at an early postnatal age of Cln1 −/− mice. This was unexpected as the accumulation of autofluorescent storage material and increased neuronal apoptosis, characteristic pathological findings in INCL, are not detectable until these animals are 5–6 months old. Thus, our results suggest that even though the children with INCL and Cln1 −/− mice are apparently normal at birth, the pathologic process is most likely initiated at birth although it remains insidious until overt manifestation of the disease symptoms occurs later in life. Indeed, we did not detect significant alterations in dendritic filopodia until 6 months of age. However, degeneration within discrete neuronal populations, including thalamic projection neurons and cortical interneurons, have been reported to precede general cortical atrophy and seizure phenotype in Cln1 −/− mice.40 This raised the possibility that the synaptic dysfunction in Cln1 −/− mice may precipitate the loss of such discrete neuronal populations leading to an imbalance in circuit excitation–inhibition (E–I) dynamics promoting seizure activity. One potential candidate population critical for controlling circuit E–I balance is the cohort of local circuit GABAergic interneurons. Accordingly, we compared the density of several classes of inhibitory hippocampal interneurons in WT and Cln1 −/− mice. We used immunohistochemical analysis of markers that cover the vast majority of medial ganglionic eminence‐derived interneurons (SOM and PV) as well as those derived from the caudal ganglionic eminence (CCK and CoupTFII).41 Remarkably, 1‐month‐old Cln1 −/− mice exhibited reduced numbers of hippocampal CCK+, CoupTFII+, SOM+, and PV+ interneurons (Fig. 7A and C) compared with those in WT littermates. Notably, the loss of interneurons persisted and escalated through 6 months of age, when general neuronal degeneration was observed (Fig. 7B and D). These findings suggest that the vast majority of hippocampal interneurons including perisomatic (PV and CCK)‐ and dendrite (SOM)‐targeting interneurons are particularly susceptible to degeneration in young Cln1 −/− mice prior to the onset of general neuronal degeneration. It is likely that the ensuing disruption of inhibitory networks contributed to the epilepsy phenotype in Cln1 −/− mice.
Figure 7.
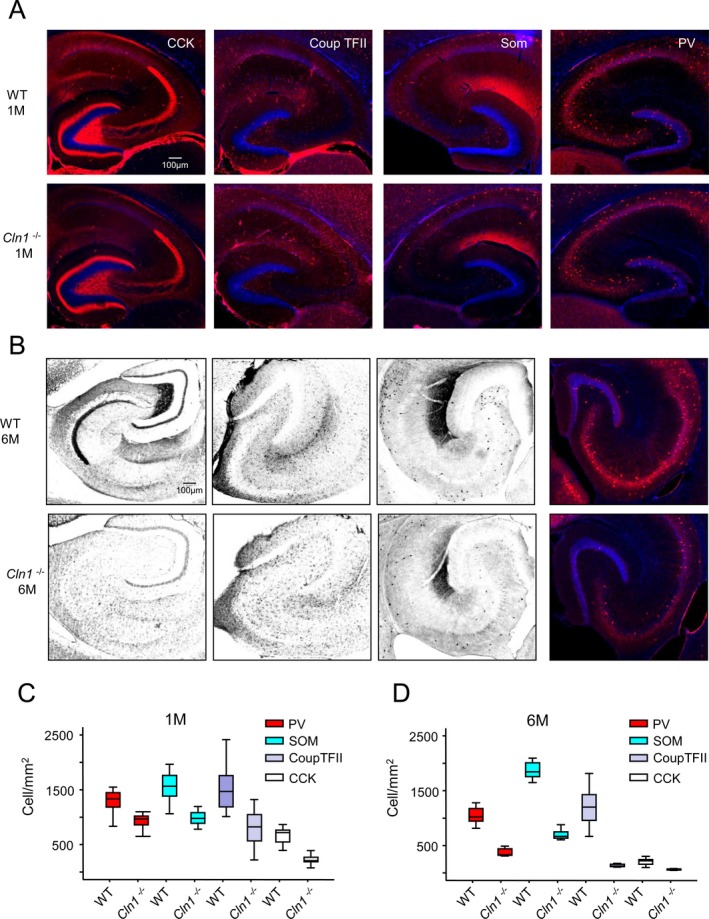
Loss of hippocampal interneurons during early development of Cln1 −/− mice. Representative images of hippocampal sections from WT mice (upper panels) and those from their Cln1 −/− littermates (lower panels) stained for GABAergic interneuron markers at 1 month (A and C) and 6 months of age (B and D). Markers examined were CCK, CoupTFII, SOM, and PV. Pooled group data for cell density counts of the labeled interneurons in WT and Cln1 −/− are provided in (C) and (D) for mice aged 1 and 6 months, respectively; (C) a total of 24 sections per marker from 3 different WT and Cln1 −/− mice were processed and counted parallel; (D) a total of 8–12 sections per marker from a WT mouse and those from a Cln1 −/− littermate were examined. Excel student t‐test was used for statistical analysis of the data, P < 0.01. WT, wild‐type; CCK, cholecystokinin; CoupTFII, COUP transcription factor 2; SOM, somatostatin; PV, parvalbumin.
Discussion
Neurodegeneration is a devastating manifestation in the majority of >50 LSDs. The mechanisms of neurodegeneration in various types of LSDs are likely to be varied and complex. In this study, we uncovered a novel mechanism by which Ppt1 deficiency contributes to synaptic dysfunction in a mouse model of a childhood neurodegenerative LSD, INCL. We previously reported that Cln1 −/− mice manifested high levels of oxidative stress in the brain.23 The results of our present study showed that increased oxidative stress upregulated C/EBP‐δ, which mediated overexpression of serpina1, thereby inhibiting neurotrypsin activity. Our results also showed that serpina1‐mediated inhibition of neurotrypsin activity led to the decline in agrin‐22 production. Since agrin‐22 has an essential role in synaptic homeostasis,12, 37 it is reasonable to believe that this abnormality, at least in part, contributed to synaptic dysfunction in Cln1 −/− mice. Immunoelectron microscopic analyses localized Ppt1 in the neuronal synapses35 and its deficiency may have prevented dynamic palmitoylation of SV‐associated proteins essential for SV recycling, which caused progressive decline in SV pool size both in cellula 42 and in vivo.35 The results of our electrophysiological studies using the calyx of Held preparations from 7‐ to 10‐day‐old pups enabled us to demonstrate that the neuropathological changes in the brain of Cln1 −/− mice are initiated much earlier than previously reported. Our results suggest that while the pathologic process starts early, it is insidious until the mice are 6–7 months old when overt clinical manifestations of neurodegeneration are clearly appreciable.
In this study, we have evaluated the pathological changes in different regions of the brain (i.e., cortex, hippocampus, and brain stem). The very nature of the experiments used in this study necessitated the use of tissues or cells from these regions. For example, in the electrophysiological studies we used the calyx of Held preparations derived from the auditory brain stem isolated from 7–10 day old pups. Interestingly, the results uncovered that electrophysiological abnormalities in Cln1 −/− mice can be detected very early in development. Therefore, even though for this study we used a different area of the Central nervous system (i.e., the brain stem) the results uncovered that the disease pathology may be initiated very early although overt disease symptoms are not detectable until the mice are 5–6 months of age. We are mindful of the facts that our study has some limitations in that functional properties of neurons depend on the location and even the age of the animal as previously reported.43 Thus, our results do not allow generalizations, which may be applicable to the whole brain. Nevertheless, these results clearly showed that compared with their WT littermates (controls), the Cln1 −/− mice manifested pathological changes that are consistent with the inactivation of the Cln1 gene.
It is unclear from the results of our study as to how Ppt1 deficiency in Cln1 −/− mice might lead to the inhibition of Ca2+ currents. However, the recent reports may provide an explanation for the decreased Ca2+ influx in the calyx of Held in Cln1 −/− mice. It has been demonstrated that the β‐subunits of the voltage‐gated Ca2+ channels (Cav β2a) undergo post‐translational S‐palmitoylation and modulate channel inactivation kinetics.44, 45 Cav β2a also undergoes dynamic palmitoylation, which requires the action of both palmitoyl‐acyl‐transferases as well as thioesterase‐like PPT1. Thus, it is likely that in Cln1 −/− mice, lack of dynamic palmitoylation of Cav β2a led to reduced Ca2+ influx due to the disruption of normal Ca2+ channel inactivation. In bovine chromaffin cells, dynamic palmitoylation of Cav β2 modulates both N‐type Ca2+ channel as well as P/Q‐type Ca2+ channel.44 Because N‐ and P/Q type Ca2+ channels are the two most abundant Ca2+ channels both in the calyx and in other synapses, our finding that Cln1 mutations contributes to the suppression of vesicle exocytosis and endocytosis may be applicable to other synapses. Notably, consistent with our findings, a recent study in Drosophila has shown that mutations in the Cln1/Ppt1 gene caused altered exocytosis and endocytosis at the synapses46 although the status of agrin‐22 levels was not determined.
Synaptic dysfunction in Cln1 −/− mice may also have contributed to the death of hippocampal interneurons. These findings are consistent with the report that discrete neuronal populations, including thalamic projection neurons and cortical interneurons precede general cortical atrophy in Cln1 −/− mice.40 The interneuron loss we found in the Cln1 −/− mouse brain is consistent with previous reports.47 Previous studies have also shown that in NCL patients as well as in animal models of NCLs show dysfunction of selective inhibitory (GABAergic) circuits causing loss and hypertrophy of remaining neurons. Moreover, emerging evidence indicates that interneurons play an active role in information processing and impairment of these neurons, which may underlie the deficit in cognitive function characteristically found in neurodegenerative disorders.48 Emerging evidence indicates that excitation and inhibition in the cerebral cortex are regulated by two distinct cell types arising from two distinct areas of the developing brain. These cell types are: excitatory projection neurons generated in the developing cortex and inhibitory interneurons, which are generated outside the cortex in the ventral forebrain.49 Furthermore, in the neocortex of patients with NCL there is degeneration of ultrastructurally identifiable inhibitory synapses50 suggesting that impaired inhibitory mechanisms may account for the development of focal and generalized seizures51, 52 as characteristically found in INCL. Inactivating mutations in the CLN1 gene causes deficiency of PPT1, a lysosomal thioesterase, which leads to oxidative stress in the brain. Thus, an ideal therapeutic agent should ameliorate the defects arising from PPT1 deficiency as well as oxidative stress. In this regard, we have recently reported the identification and characterization of a small molecule that possesses both of these properties.25 Taken together, the findings in our present study provide insight into a novel mechanism that links oxidative stress with suppression of agrin‐22 production, which may explain the synaptic dysfunction in a specific neurodegenerative disease, INCL, and suggest that a thioesterase‐mimetic antioxidant, NtBuHA, which elevated agrin‐22 levels, may have therapeutic implications for this devastating disease.
Author Contributions
S. P., Z. Z., and A. B. M. conceived the project, and designed the experiments. S. P. carried out the majority of the experiments. J. X. conducted the electrophysiological experiments using the calyx of Held preparations. K. P. and X. Y. performed the immunohistochemical analyses of the interneurons in the hippocampal tissues. G. C. conducted the experiments on transcriptional activation of C/EBP‐δ by oxidative stress. M. B. B. cultured neurons and astroglia and isolated synaptosomes. C. J. M. and L. G. W. provided guidance and suggestions on the studies involving the interneuron staining and electrophysiology, respectively, and contributed to the editing of the final manuscript. A. B. M. and S. P. together with Z. Z., K. P. and J. X. prepared the first draft of the manuscript. All authors reviewed the draft manuscript, made revisions and approved the final draft for submission.
Conflict of Interest
None declared.
Acknowledgments
We thank S. W. Levin, J. Chou and I. Owens for critical review of the manuscript and helpful suggestions. We are grateful to W. E. Fibbe and P. Sonderegger, respectively, for the generous gifts of anti‐serpina1 and anti‐agrin antibodies. This research was supported in full by the intramural program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development of the National Institutes of Health, USA.
References
- 1. Andersen JK. Oxidative stress in neurodegeneration: cause or consequence? Nat Med 2004;10(Suppl):S18–S25. [DOI] [PubMed] [Google Scholar]
- 2. Keller JN, Schmitt FA, Scheff SW, et al. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology 2005;64:1152–1156. [DOI] [PubMed] [Google Scholar]
- 3. Gilgun‐Sherki Y, Melamed E, Offen D. Oxidative stress induced‐neurodegenerative diseases: the need for antioxidants that penetrate the blood brain barrier. Neuropharmacology 2001;40:959–975. [DOI] [PubMed] [Google Scholar]
- 4. Cotman SL, Staropoli KA, Sims KB. Neuronal ceroid lipofuscinosis: impact of recent genetic advances and expansion of the clinicopathologic spectrum. Curr Neurol Neurosci Rep 2013;13:366. doi: 10.1007/s11910‐013‐0366‐z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rider JA, Rider DL. Batten disease, past, present and future. Am J Med Genet (Suppl) 1988;5:21–26. [DOI] [PubMed] [Google Scholar]
- 6. Platt FM, Boland B, van der Spoel AC. The cell biology of disease: lysosomal storage disorders: cellular impact of lysosomal dysfunction. J Cell Biol 2012;199:723–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Warrier V, Vieira M, Mole SE. Genetic basis and phenotypic correlations of the neuronal ceroid lipofuscinoses. Biochim Biophys Acta 2013;1832:1827–1830. [DOI] [PubMed] [Google Scholar]
- 8. Palo J, Santavuori P, Haltia M. Recent findings on some “new” neurometabolic diseases. Riv Patol Nerv Ment 1976;97:191–198. [PubMed] [Google Scholar]
- 9. Vesa J, Hellsten E, Verkruyse LA, et al. Mutations in the palmitoyl protein thioesterase gene causing infantile neuronal ceroid lipofuscinosis. Nature 1995;376:584–587. [DOI] [PubMed] [Google Scholar]
- 10. Camp LA, Verkruyse LA, Afendis SJ, et al. Molecular cloning and expression of palmitoyl‐protein thioesterase. J Biol Chem 1994;269:23212–23219. [PubMed] [Google Scholar]
- 11. Haltia M, Rapola J, Santavuori P. Infantile type of so‐called neuronal ceroid‐lipofuscinosis. Histological and electron microscopic studies. Acta Neuropathol 1973;26:157–170. [DOI] [PubMed] [Google Scholar]
- 12. Sonderegger P, Matsumoto‐Miyai K. Activity‐controlled proteolytic cleavage at the synapse. Trends Neurosci 2014;37:413–423. [DOI] [PubMed] [Google Scholar]
- 13. Gschwend TP, Krueger SR, Kozlov SV, et al. Neurotrypsin, a novel multidomain serine protease expressed in the nervous system. Mol Cell Neurosci 1997;9:207–219. [DOI] [PubMed] [Google Scholar]
- 14. Proba K, Gschwend TP, Sonderegger P. Cloning and sequencing of the cDNA encoding human neurotrypsin. Biochim Biophys Acta 1998;1396:143–147. [DOI] [PubMed] [Google Scholar]
- 15. Stephan A, Mateos JM, Kozlov SV, et al. Neurotrypsin cleaves agrin locally at the synapse. FASEB J 2008;22:1861–1873. [DOI] [PubMed] [Google Scholar]
- 16. Molinari F, Rio M, Meskenaite V, et al. Truncating neurotrypsin mutation in autosomal recessive nonsyndromic mental retardation. Science 2002;298:1779–1781. [DOI] [PubMed] [Google Scholar]
- 17. Bezakova G, Ruegg MA. New insights into the roles of agrin. Nat Rev Mol Cell Biol 2013;4:295–308. [DOI] [PubMed] [Google Scholar]
- 18. Reif R, Sales S, Hettwer S, et al. Specific cleavage of agrin by neurotrypsin, a synaptic protease linked to mental retardation. FASEB J 2007;21:3468–3478. [DOI] [PubMed] [Google Scholar]
- 19. Silverman GA, Bird PI, Carrell RW, et al. The serpins are an expanding superfamily of structurally similar but functionally diverse proteins. Evolution, mechanism of inhibition, novel functions, and a revised nomenclature. J Biol Chem 2001;276:33293–33296. [DOI] [PubMed] [Google Scholar]
- 20. Gupta P, Soyombo AA, Atashband A, et al. Disruption of PPT1 or PPT2 causes neuronal ceroid lipofuscinosis in knockout mice. Proc Natl Acad Sci USA 2001;98:13566–13571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bible E, Gupta P, Hofmann S, Cooper JD. Regional and cellular neuropathology in the palmitoyl protein thioesterase‐1 null mutant mouse model of infantile neuronal ceroid lipofuscinosis. Neurobiol Dis 2004;16:346–359. [DOI] [PubMed] [Google Scholar]
- 22. Dufey E, Sepúlveda D, Rojas‐Rivera D, Hetz C. Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. 1. An overview. Am J Physiol Cell Physiol 2014;307:C582–C594. [DOI] [PubMed] [Google Scholar]
- 23. Wei H, Kim SJ, Zhang Z, et al. ER‐ and oxidative‐stresses are common mediators of apoptosis in both neurodegenerative and non‐neurodegenerative lysosomal storage disorders and are alleviated by chemical chaperones. Hum Mol Genet 2008;17:469–477. [DOI] [PubMed] [Google Scholar]
- 24. Atamna H, Robinson C, Ingersoli R, et al. N‐t‐butyl hydroxylamine is an antioxidant that reverses age‐related changes in mitochondria in vivo and in vitro. FASEB J 2001;15:2196–2204. [DOI] [PubMed] [Google Scholar]
- 25. Sarkar C, Chandra G, Peng S, et al. Neuroprotection and lifespan extension in Ppt1(−/−) mice by NtBuHA: therapeutic implications for INCL. Nat Neurosci 2013;16:1608–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Friedland DR, Los JG, Ryugo DK. A modified Golgi staining protocol for use in the human brain stem and cerebellum. J Neurosci Methods 2006;150:90–95. [DOI] [PubMed] [Google Scholar]
- 27. Wu W, Xu J, Wu XS, Wu LG. Activity‐dependent acceleration of endocytosis at a central synapse. J Neurosci 2005;25:11676–11683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Azari H, Sharififar S, Rahman M, et al. Establishing embryonic mouse neuronal stem cell culture using the neurosphere assay. J Vis Exp 2011;2457. pii: 2457. doi: 10.3791/2457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lesuisse C, Martin LJ. Long‐term culture of mouse cortical neurons as a model for neuronal development, aging and death. J Neurobiol 2002;51:9–23. [DOI] [PubMed] [Google Scholar]
- 30. Dunkley PR, Jarvie PE, Heath JW, et al. A rapid method for isolation of synaptosomes on Percoll gradients. Brain Res 1986;372:115–129. [DOI] [PubMed] [Google Scholar]
- 31. Purpura DP. Dendritic spine “dysgenesis” and mental retardation. Science 1974;186:1126–1128. [DOI] [PubMed] [Google Scholar]
- 32. Schneggenburger R, Forsythe ID. The calyx of Held. Cell Tissue Res 2006;326:311–337. [DOI] [PubMed] [Google Scholar]
- 33. Sun J, Pang ZP, Qin D, et al. A dual‐Ca2+‐sensor model for neurotransmitter release in a central synapse. Nature 2007;450:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sun JY, Wu LG. Fast kinetics of exocytosis revealed by simultaneous measurements of presynaptic capacitance and postsynaptic currents at a central synapse. Neuron 2001;30:171–182. [DOI] [PubMed] [Google Scholar]
- 35. Kim SJ, Zhang Z, Sarkar C, et al. Palmitoyl‐protein thioesterase‐1 deficiency impairs synaptic vesicle recycling at nerve terminals contributing to neuropathology in humans and mice. J Clin Invest 2008;118:3075–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shipston MJ. Regulation of large conductance calcium‐ and voltage‐activated potassium (BK) channels by S‐palmitoylation. Biochem Soc Trans 2013;41:67–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Matsumoto‐Miyai K, Sokolowska E, Zurlinden A, et al. Coincident pre‐ and postsynaptic activation induces dendritic filopodia via neurotrypsin‐dependent agrin cleavage. Cell 2009;136:1161–1171. [DOI] [PubMed] [Google Scholar]
- 38. Santello M, Calì C, Bezzi P. Gliotransmission and the tripartite synapse. Adv Exp Med Biol 2012;970:307–331. [DOI] [PubMed] [Google Scholar]
- 39. Pessler‐Cohen D, Pekala PH, Kovsan J, et al. GLUT4 expression in response to oxidative stress is associated with reciprocal alterations in the C/EBP alpha and delta isoforms in 3T3‐L1 adipocytes. Arch Physiol Biochem 2006;112:3–12. [DOI] [PubMed] [Google Scholar]
- 40. Kielar C, Maddox L, Bible E, et al. Successive neuron loss in the thalamus and cortex in a mouse model of infantile neuronal ceroid lipofuscinoses. Neurobiol Dis 2007;25:150–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tricoire L, Pelkey KA, Erkkila BE, et al. A blueprint for the spatiotemporal origins of mouse hippocampal interneuron diversity. J Neurosci 2011;31:10948–10970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Virmani T, Gupta P, Liu X, et al. Progressively reduced synaptic vesicle pool size in cultured neurons derived from neuronal ceroid lipofuscinosis‐1 knockout mice. Neurobiol Dis 2005;20:314–323. [DOI] [PubMed] [Google Scholar]
- 43. Woolley CS, Gould E, Frankfurt M, McEwen BS. Naturally occurring fluctuation in dendritic spine density on adult hippocampal pyramidal neurons. J Neurosci 1990;10:4035–4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hurley JH, Cahill AL, Currie KP, Fox AP. The role of dynamic palmitoylation in Ca2+ channel inactivation. Proc Natl Acad Sci USA 2000;97:9293–9298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Heneghan JF, Mitra‐Ganguli T, Stanish LF, et al. The Ca2+ channel beta subunit determines whether stimulation of Gq‐ coupled receptors enhances or inhibits N current. J Gen Physiol 2009;134:369–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Aby E, Gumps K, Roth A, et al. Mutations in palmitoyl‐protein thioesterase 1 alter exocytosis and endocytosis at synapses in Drosopjila larvae. Fly (Austin) 2013;7:267–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cooper JD, Messer A, Feng AK, et al. Apparent loss and hypertrophy of interneurons in a mouse model of neuronal ceroid lipofuscinosis: evidence for partial response to insulin‐like growth factor‐1 treatment. J Neurosci 1999;19:2556–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Paulsen O, Moser EI. A model of hippocampal memory encoding and retrieval: GABAergic control of synaptic plasticity. Trends Neurosci 1998;21:273–278. [DOI] [PubMed] [Google Scholar]
- 49. Southwell DG, Nicholas CR, Basbaum AI, et al. Interneurons from embruonic development to cell‐based therapy. Science 2014;344:1240622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Williams RS, Lott IT, Ferrante RJ, Caviness VS Jr. The cellular pathology of neuronal ceroid‐ lipofuscinosis. A golgi‐electronmicroscopic study. Arch Neurol 1977;34:298–305. [DOI] [PubMed] [Google Scholar]
- 51. Roberts E. Metabolic and nervous system disease: a challenge for our times. Part II. Metab Brain Dis 1986;1:91–117. [DOI] [PubMed] [Google Scholar]
- 52. Roberts E. Failure of GABAergic inhibition: a key to local and global seizures. Adv Neurol 1986;44:319–341. [PubMed] [Google Scholar]