

Abstract
Objective
Pharmacoresistance develops quickly during repetitive seizures, and refractory status epilepticus (RSE) remains a therapeutic challenge. The outcome of RSE is poor, with high mortality and morbidity. New treatments are needed. Deep hypothermia (20°C) is used clinically during reconstructive cardiac surgery and neurosurgery, and has proved safe and effective in those indications. We tested the hypothesis that deep hypothermia reduces RSE and its long‐term consequences.
Methods
We used a model of SE induced by lithium and pilocarpine and refractory to midazolam. Several EEG measures were recorded in both hypothermic (n = 17) and normothermic (n = 20) animals. Neuronal injury (by Fluoro‐Jade B), cell‐mediated inflammation, and breakdown of the blood–brain barrier (BBB) (by immunohistochemistry) were studied 48 h following SE onset.
Results
Normothermic rats in RSE seized for 4.1 ± 1.1 h, and at 48 h they displayed extensive neuronal injury in many brain regions, including hippocampus, dentate gyrus, amygdala, entorhinal and pyriform cortices, thalamus, caudate/putamen, and the frontoparietal neocortex. Deep hypothermia (20°C) of 30 min duration terminated RSE within 12 min of initiation of hypothermia, reduced EEG power and seizure activity upon rewarming, and eliminated SE‐induced neuronal injury in most animals. Normothermic rats showed widespread breakdown of the BBB, and extensive macrophage infiltration in areas of neuronal injury, which were completely absent in animals treated with hypothermia.
Interpretation
These results suggest that deep hypothermia may open a new therapeutic avenue for the treatment of RSE and for the prevention of its long‐term consequences.
Introduction
While we have made considerable progress in treating epilepsy, status epilepticus (SE) remains a therapeutic challenge. SE has an incidence of 10–41/100,000.1, 2 Mortality was over 50% in the VA Cooperative Study,3 27% in population‐based studies in Virginia4 and 11–24% in other studies.5 Morbidity is considerable, particularly in the elderly. Almost a quarter of survivors experience deterioration in their functional outcome,6 with 10% requiring long‐term care,7 6% developing an associated chronic encephalopathy,8 and 41% ultimately developing epilepsy.9
Drugs fail to stop SE in 31–53% of cases.3, 10, 11 During SE, pharmacoresistance develops progressively. The anticonvulsant potency of benzodiazepines can decrease 20‐fold in 30 min of seizures.12 Phenytoin and barbiturates also lose potency, but more slowly.13 In clinical studies, early treatment of SE is much more effective than late treatment, suggesting pharmacoresistance. In the VA Cooperative Study,3 four treatments were randomly rotated; the first treatment was successful in 53% of patients, the third in 2% of patients. Seizure‐induced trafficking of synaptic GABAA and glutamate receptors may in part explain the development of time‐dependent pharmacoresistance.14 Refractory SE (RSE) defined by refractoriness to at least two drugs, and super‐refractory SE (SRSE), defined by failure to respond to adequate treatment for at least 24 h, have become commonplace in Intensive Care Units, at an enormous cost and with very poor outcomes.15
We need an alternative treatment for RSE/SRSE, and our results suggest that hypothermia could be that treatment. Hypothermia acts by a completely different mechanism than anticonvulsant drugs, and may be able to stop RSE. Mild hypothermia reduces seizure activity in experimental animals,16, 17, 18 although seizures often recur upon rewarming19 and sometimes convulsive seizures stop but EEG continues to show seizure activity.20 Successful treatment of clinical SE with mild hypothermia has been reported.18, 21, 22 However, hypothermia failed to reduce the incidence of neonatal seizures following hypoxia‐ischemia.23
Deep hypothermia has not been studied extensively for the treatment of RSE, in spite of the demonstration that cooling to 23°C stops kainate seizures better than cooling to 28°C19 and of reports of partial success in preventing seizure recurrence upon rewarming.20, 24 However, it failed to prevent epileptogenesis following experimental SE.25 Recent developments in ICU technology have reduced the complications of hypothermia. Mild hypothermia has become routine treatment for neonatal hypoxic‐ischemic encephalopathy,26 and traumatic brain injury.27 It has been used extensively for postcardiac arrest encephalopathy,28 although recent studies do not support its usefulness in children29 or adults.30
Deep hypothermia is routinely used to protect the brain or spinal cord when circulatory arrest is needed in cardiac surgery,31 and neurosurgery,32 although it has significant complications, including increased risks of bleeding, coagulopathy, and infection.33, 34, 35 Today, the technology for delivering mild hypothermia is available in most hospitals, and the technology for delivering deep hypothermia is available in major surgical centers. We studied deep hypothermia in the treatment of benzodiazepine‐refractory experimental SE, and found that it is very effective in stopping seizures and reducing SE‐associated neuronal injury, cell‐mediated inflammation, and breakdown of the blood–brain barrier (BBB).
Materials and Methods
Animals
Male Wistar rat (300–400 g; Charles River, MA) were used. Rats were housed in a temperature‐ and humidity‐controlled room with 12 h light‐dark cycles (7 am–7 pm) and had free access to food and water. All experiments were conducted with the approval and in accordance with the regulations of the Institutional Animal care and Use Committee of West Los Angeles VA Medical Center.
Induction of SE and drug treatment
Rats were administered lithium chloride (3 mEq/kg, #L‐0505; Sigma, St. Louis, MO) subcutaneously and, 16 h later, SE was induced with i.p. pilocarpine hydrochloride (60 mg/kg, #P6503; Sigma). Only lithium/pilocarpine‐treated rats displaying behavioral/EEG seizures were used. All rats received scopolamine methyl bromide (1 mg/kg, #S8502; Sigma), a muscarinic antagonist that does not cross the BBB, at the same time as pilocarpine, to decrease peripheral cholinergic effects such as pulmonary secretions. The seizures occurred 8–20 min after pilocarpine injection, so that time from pilocarpine injection to midazolam treatment was ~30 min. The time between the second stage 3 or higher seizure and the onset of continuous polyspikes was both short and reproducible (1.28 ± 1.1 min, n = 16). The second stage 3 seizure was chosen as our time anchor to insure that all rats were in full SE. The animals subsequently received scopolamine (2 mg/kg i.p.; #S1013; Sigma) to remove the original seizure trigger without stopping SE, and midazolam (3 mg/kg; Caraco Pharmaceutical Laboratories Ltd, Detroit, MI) i.p. 12 min after the second major seizure (stage 3 or higher) to make sure that pharmacoresistance and self‐sustaining seizures were well established. In this study, RSE was refractory to one drug (the usual clinical definition is two drugs), which is sufficient for proof‐of‐principle of the use of hypothermia, and minimizes the complex interactions between multiple drugs and temperature homeostasis.
Deep hypothermia
In some animals, cooling was initiated after midazolam injection (“early cooling”; n = 5) (Fig. 1). In other animals, cooling was not initiated for an additional 15 min to eliminate the rats that stop seizing after drug treatment and therefore were not pharmacoresistant (“delayed cooling”; n = 17). Animals were placed in rodent restraint bags (Harvard Apparatus, Holliston, MA) so that the nose and mouth extended beyond the bag. Holes were cut in the bag to allow connection of the EEG cables to the electrode connector and placement of the thermistor rectal probe (Yellow Springs Instr., Yellow Springs, OH). The animal was then placed in an ice bag and two more ice bags were placed on the sides. The body temperature was maintained at 20°C for 30 min. At the end of the cooling period, the animal, in the bag with cables and probes still in place, was transferred to a heating pad and allowed to warm up to normal body temperature. The cables and probes were then removed and the animal released into the normal housing cage.
Figure 1.
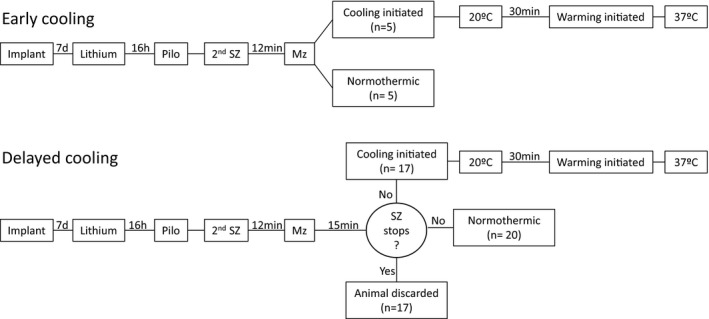
Experimental flow. Status epilepticus (SE) was induced by lithium administration followed by pilocarpine injection. Midazolam (3 mg/kg) was injected 12 min after the second “stage 3” seizure. Then the animals were placed in ice packs and cooled to 20°C for 30 min. In early cooling, hypothermia was initiated after midazolam injection. In delayed cooling, hypothermia was initiated 15 min after midazolam only in animals that were refractory to midazolam. When midazolam alone stopped SE, the animals were discarded.
Temperature monitoring
The body temperature was monitored in all animals using a rectal probe. Some animals had a dental drill hole in the skull at 4 mm anterior to bregma and 2 mm left of the medial suture for brain surface temperature measurement. A guide cannula was fixed in place over the opening 1–2 weeks before SE induction, to allow placement of a thermocouple temperature probe (Physitemp, Clifton, NJ). Analog output from the rectal probe and the thermocouple probe were fed into a BioPac MP150 analog to digital converter (BioPac Systems, Goleta, CA). The digital values were calibrated against a NIST traceable reference thermometer.
EEG monitoring
Under isoflurane anesthesia, the animals were implanted with stainless steel skull screws to serve as recording electrodes. Two electrodes were used for bipolar recording and were located 3 mm anterior to lambda and 4 mm left and right of the medial suture. The third electrode served as reference and was located 1 mm anterior to bregma and 1 mm to the right of the midline defined by the medial suture. The screw electrodes were connected to a tripolar connector (Plastics One, Roanoke, VA) and dental cement used to cover the electrodes so that just the connector was exposed. Animals were used 1–2 weeks after electrode implantation.
The BioPac Systems MP150 was used to record digital EEG using a BioPac UM100A preamplifier. Sampling rate was 200 Hz. Recording was started at pilocarpine injection and was continuous for 24 h which included an initial prepilocarpine segment of EEG, the development of SE, drug treatment, cooling, warming, and the overnight recovery period.
The EEG were processed offline to detect seizures and spikes using Stellate Systems Harmony (Natus, Pleasanton, CA) software with default parameters: amplitude threshold 2.7, minimum frequency 3 Hz, maximum coefficient of variation 40% for seizure detection and a spike amplitude threshold of six for spike detection.
The outcome measures were the ratio of EEG power at t time divided by the average baseline EEG power (before pilocarpine), the number of seizures per 24 h, the cumulative seizure time per 24 h (time spent seizing, subtracting post‐ and interictal time), the number of spikes per 24 h, the time spent in high amplitude (>2× prepilocarpine baseline) EEG discharge per 24 h, and the time needed for EEG amplitude to fall below two times the pre‐pilocarpine EEG amplitude for at least 1 min, which in this experimental paradigm is close to the time of termination of SE.
Tissue preparation for neuronal injury and immunohistochemical studies
Some animals of the normothermic (n = 9) and hypothermic group (delayed hypothermia; n = 9) were anesthetized with an overdose of pentobarbital (100 mg/kg i.p.) 48 h after induction of SE. Then, the animals underwent transcardiac perfusion with 4% phosphate‐buffered formaldehyde (#P‐6148; Sigma). Brains were kept in situ at 4°C overnight, after which they were removed and postfixed in the same perfusate for 2–3 h. Subsequently, brains were kept in Phosphate buffer (PB) 0.1 mol/L containing 30% sucrose for 48–72 h. Floating sections (30 μmol/L thickness) were obtained using a sliding microtome.
Fluoro‐Jade B staining
Coronal sections were mounted, dried, incubated in potassium permanganate solution (0.06%; w/v) for 15 min, washed and incubated in Fluoro‐Jade B staining solution for 30 min. After three rinses, slides were dried overnight at room temperature, cleared three times in xylene and coverslipped with Permount medium. In hippocampal CA1, the number of injured cells was counted by unbiased stereology using the optical dissector method. The first series of one in five sections were stained with Fluoro‐Jade B, and the analysis was performed using a microscope (Olympus AX70, Center Valley, PA) with a motorized stage connected to a computer running the Stereo Investigator software (MBF Bioscience, Williston, VT). A counting frame of 45 × 45 μm was randomly positioned in a sampling grid of 70 × 120 μm. In the other areas, distribution of Fluoro‐jade B‐positive cells was scored as follows: 0, no injury; 1, 1–30 positive cells per field; 2, 31–60 positive cells per field; 3, 61–100 positive cells per field; 4, more than 100 positive cells per field.
Immunohistochemical studies
Macrophages/monocytes were revealed using an antigen retrieval procedure (#HK080‐9K; BioGenex Laboratories, San Ramon, CA), in which the coronal floating sections were heated at 80°C for 35 min in 10 mmol/L citrate solution (pH 6.0) and cooled to room temperature. Sections were then rinsed in distilled water, washed in 0.1 mol/L PB for 10 min and incubated in a blocking buffer (5% donkey serum) in 0.1 mol/L PB at room temperature for 1 h. They were then incubated overnight at 4°C with the blocking buffer containing a primary antibody: mouse monoclonal anti‐macrophages/monocytes (clone ED‐1; Millipore, Billerica, MA; MAB1435; 1/300) or mouse monoclonal anti‐T‐cell (clone 15‐6A1; Santa Cruz Biotechnology, Dallas, TX; sc‐52711; 1/1000), then washed in PB three times and exposed to Alexa‐Fluor 488‐linked donkey anti‐mouse (#A21202; Life Technologies, Carlsbad, CA) immunoglobulins diluted 1:200 with the blocking solution for 2 h at room temperature.
For the detection of rat IgG, coronal sections were incubated in the same blocking buffer for 1 h and then with Alexa Fluor 488 donkey anti‐rat IgG (H+L) antibody (#A21208; Life Technologies) diluted 1:200 with the blocking solution for 2 h at room temperature.
Statistical analyses
EEG and cell injury data for normothermic and hypothermic groups were analyzed with the Mann–Whitney test (GraphPad version 6, La Jolla, CA). Comparison of cell survival was analyzed with Fischer exact test (GraphPad version 6). Statistical significance was defined as P < 0.05. In all graphs, data are presented as mean ± SEM.
Results
Deep hypothermia reduces EEG power and stops seizures
We first examined the therapeutic benefit of “early” cooling, initiated just after midazolam treatment. Temperature was brought down to 20°C and maintained as near to that temperature as possible for 30 min. Mean cooling time from 37°C to 20°C was 39.5 ± 2.67 min (mean ± SEM; n = 5). Mean rewarming time from 20°C to 36°C was 65.4 ± 11.8 min (mean ± SEM; n = 5). Rectal and brain surface temperatures were measured in the early treatment group, and were nearly identical, with less than half a degree of difference at any time point (n = 5 in each group).30 Only rectal temperature was measured in subsequent experiments. Cooling lowered EEG power in a dose‐dependent fashion (Table 1). EEG power in normothermic animals decreased from pretreatment SE, probably in part due to midazolam, but remained significantly above baseline for 4.1 ± 1.1 h (mean ± SEM; n = 9) after midazolam injection, indicating continuation of SE. In hypothermic animals, cooling brought power down to baseline level in 23.8 ± 3.9 min (mean ± SEM; n = 5) and significantly lowered EEG power compared to normothermia at 30 min and beyond as described in a preliminary report.36 In these animals, EEG power decreased below prepilocarpine baseline, and remained close to baseline values for the remaining of the 24 h, suggesting that SE had been terminated and did not return.
Table 1.
Effect of temperature on relative EEG power, defined as the ratio of EEG power at a given temperature divided by the average baseline EEG power (before pilocarpine)
| Normothermic group (n = 9) | Hypothermic group (n = 5) | Temperature of hypothermic animals | reduction in EEG power compared to normothermic | P‐value (Mann–Whitney) |
|---|---|---|---|---|
| 86.7 ± 20.9 | 55.5 ± 32.8 | 32°C | 36% | 0.28 |
| 77.3 ± 30.2 | 21.3 ± 15.4 | 28°C | 72% | 0.109 |
| 60.2 ± 26.1 | 19.9 ± 19.3 | 25°C | 67% | 0.142 |
| 34.7 ± 12.2 | 1.1 ± 0.7 | 20°C | 97% | 0.004a |
P < 0.01.
Altogether, these results showed that midazolam treatment followed by early cooling is a very efficient procedure to stop SE. However, the presence of false positives in the hypothermic groups (animals whose SE might have stopped from midazolam alone) could not be ruled out. In our second series of experiments (“delayed hypothermia”), the cooling was delayed 15 min after midazolam and only applied to rats which remained in SE at that time. Indeed, this study showed that midazolam alone stopped SE in 17 out of 70 animals (24.3%) and these animals were discarded. In EEG studies, normothermic (n = 20) and hypothermic (n = 17) groups were comparable until cooling was initiated: time from pilocarpine injection to treatment was similar (normothermic group: 35 ± 2.5 min; hypothermic group: 37 ± 2.5 min), as were relative EEG power at initiation of hypothermia [normothermic group: 19 ± 1, hypothermic group: 18 ± 1 times baseline, non‐significant (NS)] and progression to polyspikes (normothermic group: 1.3 ± 0.4 min, hypothermic group: 1.1 ± 0.6 min). Cooling significantly lowered EEG power compared to normothermic rats at every time point between 15 and 135 min following the initiation of cooling. EEG power decreased below pre‐pilocarpine baseline 30 min following initiation of cooling and slightly increased above this baseline at 135 min and beyond (Fig. 2). Mortality during SE or in the ensuing 48 h was 42% in normothermic rats (13/31), against 10% in hypothermic animals (2/22; early and delayed groups; P = 0.0127 by Fischer exact test, two‐tailed). Delayed hypothermia (n = 17) reduced the number of seizures by 65% (P < 0.001), the number of spikes by 91% (P < 0.0001), cumulative seizure time by 60% (P < 0.01), the time needed to reach an EEG power of twice the preseizure baseline by 94% (P < 0.0001), the time in high‐amplitude discharges by 55% (P < 0.01) during the first 24 h following the initiation of cooling (Table 2). The duration of individual seizures was not altered: 24 ± 1 sec in normothermics (n = 20) versus 26 ± 2 sec in hypothermics, (n = 17; NS). Time to terminate SE after initiation of hypothermia was 198 ± 41 min in normothermics (n = 20) and 12 ± 2 min in hypothermics (n = 17; P < 0.0001). The hypothermic group (n = 17) had 19.3 ± 5.2 seizures/24 h versus 55.5 ± 11 in normothermics (n = 20; P < 0.001). During rewarming, there were some spikes but no seizures in hypothermic animals. During the rewarming period of the hypothermic animals, normothermic animals had 12 ± 3 seizures (mean ± SEM; n = 20). However, 02:31–16:17 h after the end of rewarming, isolated seizures were found in eight out of 17 hypothermic animals (4.1 ± 1.4 seizures; mean ± SEM), and in one animal, SE returned.
Figure 2.
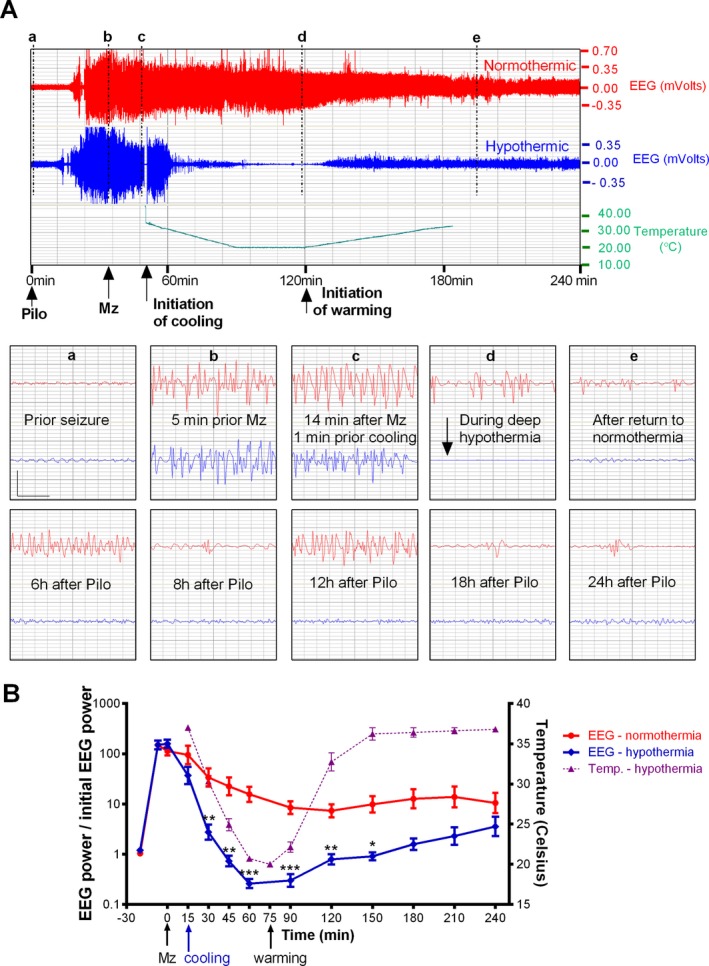
Delayed hypothermia transiently reduces relative EEG power. (A) The upper panel shows the compressed 4‐hr EEG of a normothermic (red) and a hypothermic rat (blue, 20°C for 30 min) with rectal temperature. Both rats received midazolam 3 mg/kg ip (arrow) and cooling was initiated 15 min later. The lower panel shows the magnified 4‐sec EEG traces marked by vertical lines (a‐d) (vertical bar = 0.5 mV; horizontal bar = 1 sec). (B) The left y‐axis of this graph shows the ratio of EEG power at each time points to initial EEG power at baseline, before pilocarpine injection. Note the logarithmic scale. Time points between 15 and 135 min following the initiation of cooling showed a significant difference between normothermic and hypothermic rats (***P < 0.001; **P < 0.01; *P < 0.05). The right y‐axis shows the rectal temperature scale.
Table 2.
Effect of deep hypothermia on several EEG measures recorded during the first 24 h following initiation of cooling (mean ± SEM)
| Normothermic group (n = 20) | Hypothermic group (n = 17) | Reduction | P‐value (Mann–Whitney) | |
|---|---|---|---|---|
| Number of computer‐detected seizures/24 h | 55.5 ± 11 | 19.3 ± 5.2 | 65% | 0.0005 |
| Cumulative seizure time (min) | 24.4 ± 5.4 | 9.6 ± 3.7 | 60% | 0.0016 |
| Number of computer‐detected spikes/24 h | 4898.5 ± 861.9 | 441.5 ± 109.7 | 91% | <0.0001 |
| Time in high amplitude discharge (h) | 9.2 ± 1.2 | 4.2 ± 1.4 | 55% | 0.001 |
Distribution of neuronal injury
When animals were perfused 48 h after pilocarpine injection, and neuronal injury was studied with Fluoro‐jade B, normothermic animals (n = 9) which had prolonged refractory SE showed neuronal injury in many areas, including CA1, CA3, frontoparietal cortex, piriform cortex, entorhinal cortex, caudoputmanen, thalamus, and hilus of dentate gyrus (Fig. 3). When refractory SE was treated with delayed hypothermia, none of the brains showed significant neuronal injury in any region except for three out of 9 animals which, respectively, showed 1, 4, and 7 injured cells per field in the frontoparietal cortex (Fig. 3 and Table 3). The severity of CA1 injury was positively correlated with seizure severity, as measured by the ratio of EEG power at 45 min divided by the average baseline EEG power before pilocarpine (P = 0.001, Spearman correlation).
Figure 3.
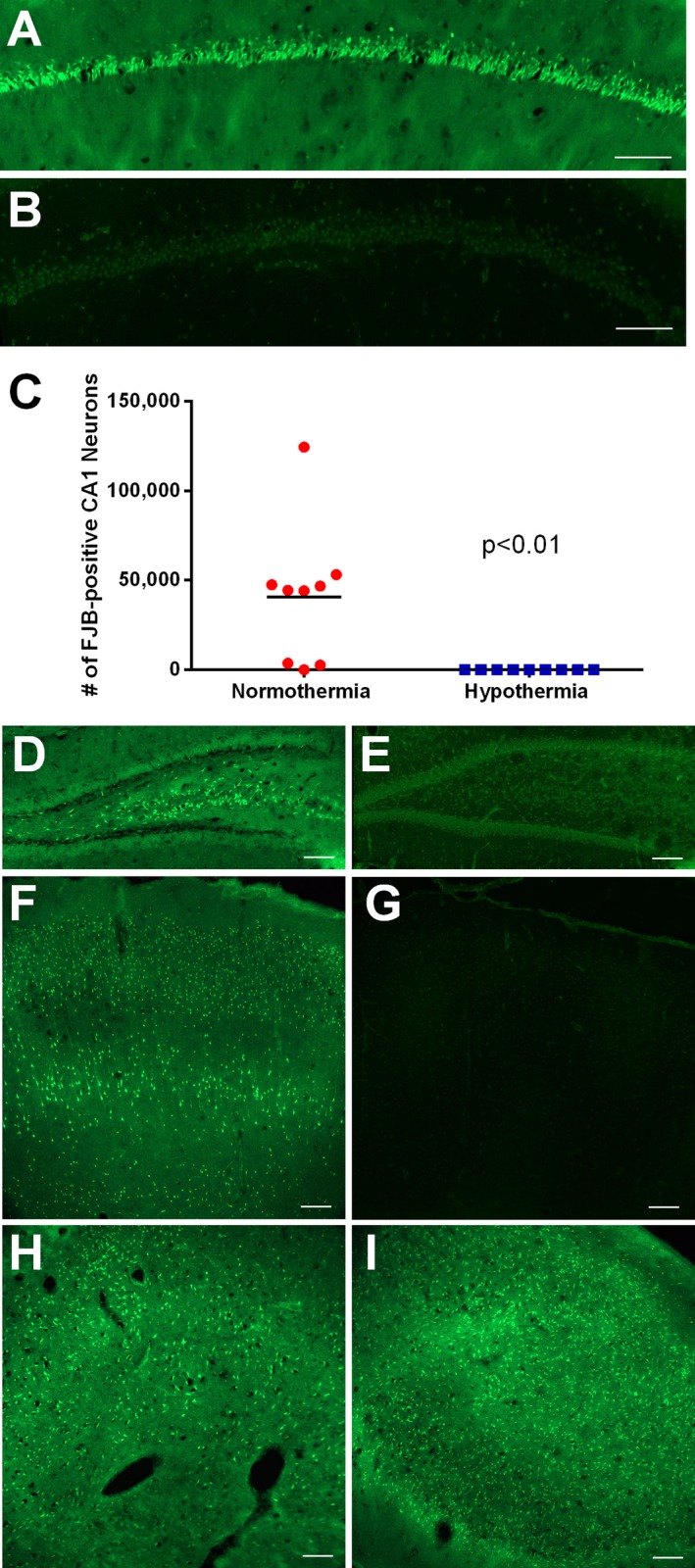
Reduction of neuronal injury in animals treated with deep hypothermia. (A and B) These images show fluoro‐jade B staining in CA1 area 48 h following status epilepticus (SE) in a normothermic (A) and hypothermic (B) rat. Note that background of hypothermic hippocampus was enhanced to show anatomy. (C) This graph shows the number of fluoro‐jade B‐ positive CA1 cells counted by an unbiased stereological method. Deep (delayed) hypothermia significantly reduced CA1 injury. (D–I) These images show fluoro‐jade B staining in the hilus of dentate gyrus (D–E), the frontoparietal neocortex (F and G), thalamus (H) and the piriform cortex (I) 48 h following SE in normothermic (D, F, H, and I) and hypothermic (E and G) animals. Bars = 100 microns.
Table 3.
Neuronal injury assessed by fluoro‐jade B staining in rats subjected to refractory SE followed by normothermia or hypothermia
| Normothermic rats (n = 9) | Hypothermic rats (n = 9) | |
|---|---|---|
| Frontoparietal cortex | 3 (1–4) [8] | 0 (1) [3] |
| Entorinal cortex | 4 (4) [8] | 0 (0) [0] |
| Piriform cortex | 4 (4) [8] | 0 (0) [0] |
| Amygdala | 4 (4) [8] | 0 (0) [0] |
| CA3 | 1 (1–2) [6] | 0 (0) [0] |
| Dentate gyrus, hilus | 1 (1–2) [8] | 0 (0) [0] |
| Thalamus | 4 (1–4) [8] | 0 (0) [0] |
| Caudoputamen | 4 (4) [6] | 0 (0) [0] |
The first numbers represent the median damage score. The numbers in parentheses indicate the range of neuronal injury. The numbers in brackets represent the numbers of animals in which neuronal injury could be observed. SE, status epilepticus.
Distribution of IgG‐like immunoreactivity
In normothermic animals (n = 9), 48 h after pilocarpine injection, antibodies against rat IgG revealed diffuse and cell staining of many areas, suggesting brain–blood barrier breakdown. All regions which showed neuronal injury also showed IgG‐like immunoreactivity (LI). This sharply contrasted with the brains of hypothermia‐treated animals (n = 9), which were devoid of IgG‐LI (Fig. 4A and B).
Figure 4.
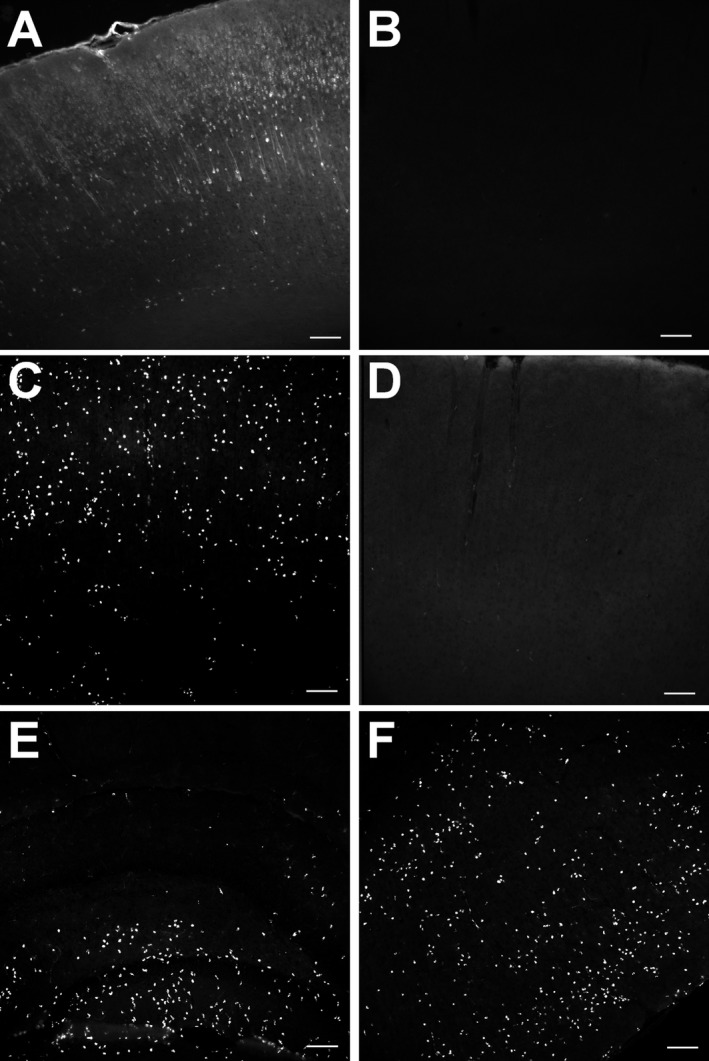
Reduction in blood–brain barrier breakdown and of macrophages in animals treated with deep hypothermia. (A and B) These images show IgG‐like immunoreactivity in the frontoparietal cortex 48 h following status epilepticus in a normothermic (A) and hypothermic (B) animal. Bars = 100 microns. (C–F) These images show staining with a macrophage marker in frontoparietal cortex (C and D), hippocampus (E) and piriform cortex (F) of normothermic (C, E, and F) and hypothermic animals (D). Bars = 100 microns.
Distribution of monocytes‐macrophages and T cells
In the normothermic animals (n = 9), using ED‐1 antibody, a marker of monocytes and macrophages, we observed 48 h after initiation of RSE many monocytes‐macrophages in many areas, including CA1, CA3, frontoparietal cortex, piriform cortex, entorhinal cortex, caudoputmanen, thalamus, and hilus of dentate gyrus. Hypothermia‐treated animals showed no macrophages in the brain (n = 9; Fig. 4C–F). There were few T cells in either group (data not shown).
Discussion
Our results show that RSE was terminated by deep hypothermia of relatively brief duration in all animals, and returned long after the rewarming period in only one of 17 animals. In hypothermic animals, EEG power was remained below preseizure baseline for 75 min after rewarming, probably reflecting a combination of postictal depression and midazolam‐induced depression. This is in sharp contrast to normothermic animals, which retained an EEG power at least an order of magnitude above preseizure baseline for 4.1 ± 1.1 h after seizure onset. Many hours after return to normothermia, however, the EEG showed a return of isolated seizures in half of the animals. By then, midazolam would be expected to have been metabolized, but pilocarpine might still be present in sufficient amount to restart seizure activity.
Mild hypothermia has been shown to reduce seizure activity in experimental animals and in humans.16, 17, 18 Moderate hypothermia19, 20 reduced the severity of SE and of SE‐associated neuronal loss. Deep hypothermia had even greater effects.19 Anecdotal clinical reports suggest that mild hypothermia inhibits seizures.18, 21, 22 Cold saline perfusion suppressed a spike focus during electrocorticography.16 Deep hypothermia has not been used for RSE, but is routinely used to protect the brain or spinal cord when circulatory arrest is needed in cardiac surgery31 and neurosurgery.32 Many of the complications reported after deep hypothermia are the result of induced circulatory arrest, not of hypothermia itself.34, 37
Deep hypothermia efficiently attenuated the long‐term consequences of RSE, including neuronal injury, BBB disruption and cell‐mediated inflammation. Hypothermia has been shown to be strongly neuroprotective in ischemia, trauma, and other pathological conditions.38 Still, it is remarkable that neuronal injury as measured by Fluoro‐Jade B staining was completely prevented by hypothermia in most animals, since hypothermia was initiated on the average 31 min after seizure onset, and in this model of SE, signs of neuronal injury are already present after 30 min of seizures.39 However, midazolam treatment, while unable to stop SE by itself, may have contributed in part to the neuroprotection provided by hypothermia.
The therapeutic efficacy of hypothermia can be explained by its biological properties. Hypothermia reduces cerebral metabolic rate by 6–7% per degree Celsius, so that at 20°C, human cerebral oxygen consumption was measured at one‐fifth of normothermic values.40 Hypothermia alters the function of ion pumps,41 intrinsic membrane properties and voltage‐gated ion channels.42 It slows release of excitatory neurotransmitters43 and modifies gene expression.44 These actions reduce excitatory drive and would be expected to inhibit seizure activity as observed in this study. They also activate several neuroprotective mechanisms: reduction in the cerebral demand for oxygen and glucose;45 preservation of ATP and energy stores and of tissue pH; reduction in the release of excitotoxic amino acids46 and of calcium influx into neurons,47 inhibition of early gene expression and stress response, induction of the expression of heat shock, and other stress proteins;48 and finally, inhibition of early molecular cascades involved in neuronal apoptosis.49 The neuroprotective role of hypothermia has been well‐documented after cardiac arrest,28, 50 stroke,51 neonatal hypoxic‐ischemic encephalopathy,52, 53 and traumatic brain injury.54 It has also been seen with seizure‐associated neuronal injury.55
In addition to its action on seizure activity and neuroprotective mechanisms, hypothermia can reduce BBB disruption and inflammation following traumatic brain injury.38 In the present study, hypothermia may have directly contributed to reduction of neuronal injury, BBB leakage and macrophage‐mediated inflammation. However, these effects could also be explained by the rapid termination of RSE.
These results suggest that deep hypothermia may open a new therapeutic avenue for the treatment of RSE and for the prevention of its long‐term consequences. The technology for carrying out mild hypothermia is available in most hospitals, and that for deep hypothermia exists in major surgical centers. In view of the high expense and poor prognosis of ICU pharmacotherapy of RSE, hypothermia might be considered as an alternative treatment of last resort of RSE and of SRSE.
Author Contributions
Conception and design of the study was done by C. W., R. B., M. G., and J. N.; acquisition and analysis of data by C. W., R. B., M. G., and J. N.); or drafting the manuscript or figures by C. W., J. N., and R. B.
Conflict of Interest
None declared.
Acknowledgments
This work was supported in part by Merit Review Award # I01 BX000273‐07 from the United States (U.S.) Department of Veterans Affairs (Biomedical Laboratory Research and Development Service) and NINDS (grant UO1 NS074926; C. W.). We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
References
- 1. DeLorenzo RJ, Hauser WA, Towne AR, et al. A prospective, population‐based epidemiologic study of status epilepticus in Richmond, Virginia. Neurology 1996;46:1029–1035. [DOI] [PubMed] [Google Scholar]
- 2. Knake S, Rosenow F, Vescovi M, et al. Incidence of status epilepticus in adults in Germany: a prospective, population‐based study. Epilepsia 2001;42:714–718. [DOI] [PubMed] [Google Scholar]
- 3. Treiman DM, Meyers PD, Walton NY, et al. A comparison of four treatments for generalized convulsive status epilepticus. Veterans Affairs Status Epilepticus Cooperative Study Group. N Engl J Med 1998;339:792–798. [DOI] [PubMed] [Google Scholar]
- 4. Towne AR, Pellock JM, Ko D, DeLorenzo RJ. Determinants of mortality in status epilepticus. Epilepsia 1994;35:27–34. [DOI] [PubMed] [Google Scholar]
- 5. Fountain NB. Status epilepticus: risk factors and complications. Epilepsia 2000;41(Suppl 2):S23–S30. [DOI] [PubMed] [Google Scholar]
- 6. Claassen J, Hirsch LJ, Emerson RG, Mayer SA. Treatment of refractory status epilepticus with pentobarbital, propofol, or midazolam: a systematic review. Epilepsia 2002;43:146–153. [DOI] [PubMed] [Google Scholar]
- 7. Lowenstein DH, Alldredge BK. Status epilepticus at an urban public hospital in the 1980s. Neurology 1993;43(3 Pt 1):483–488. [DOI] [PubMed] [Google Scholar]
- 8. Aminoff MJ, Simon RP. Status epilepticus. Causes, clinical features and consequences in 98 patients. Am J Med 1980;69:657–666. [DOI] [PubMed] [Google Scholar]
- 9. Hesdorffer DC, Logroscino G, Cascino G, et al. Risk of unprovoked seizure after acute symptomatic seizure: effect of status epilepticus. Ann Neurol 1998;44:908–912. [DOI] [PubMed] [Google Scholar]
- 10. Holtkamp M, Othman J, Buchheim K, Meierkord H. Predictors and prognosis of refractory status epilepticus treated in a neurological intensive care unit. J Neurol Neurosurg Psychiatry 2005;76:534–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mayer SA, Claassen J, Lokin J, et al. Refractory status epilepticus: frequency, risk factors, and impact on outcome. Arch Neurol 2002;59:205–210. [DOI] [PubMed] [Google Scholar]
- 12. Kapur J, Macdonald RL. Rapid seizure‐induced reduction of benzodiazepine and Zn2+ sensitivity of hippocampal dentate granule cell GABAA receptors. J Neurosci 1997;17:7532–7540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mazarati AM, Baldwin RA, Sankar R, Wasterlain CG. Time‐dependent decrease in the effectiveness of antiepileptic drugs during the course of self‐sustaining status epilepticus. Brain Res 1998;814:179–185. [DOI] [PubMed] [Google Scholar]
- 14. Naylor DE, Liu H, Niquet J, Wasterlain CG. Rapid surface accumulation of NMDA receptors increases glutamatergic excitation during status epilepticus. Neurobiol Dis 2013;54:225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ferlisi M, Shorvon S. The outcome of therapies in refractory and super‐refractory convulsive status epilepticus and recommendations for therapy. Brain 2012;135(Pt 8):2314–2328. [DOI] [PubMed] [Google Scholar]
- 16. Sartorius CJ, Berger MS. Rapid termination of intraoperative stimulation‐evoked seizures with application of cold Ringer's lactate to the cortex. Technical note. J Neurosurg 1998;88:349–351. [DOI] [PubMed] [Google Scholar]
- 17. Maeda T, Hashizume K, Tanaka T. Effect of hypothermia on kainic acid‐induced limbic seizures: an electroencephalographic and 14C‐deoxyglucose autoradiographic study. Brain Res 1999;818:228–235. [DOI] [PubMed] [Google Scholar]
- 18. Karkar KM, Garcia PA, Bateman LM, et al. Focal cooling suppresses spontaneous epileptiform activity without changing the cortical motor threshold. Epilepsia 2002;43:932–935. [DOI] [PubMed] [Google Scholar]
- 19. Liu Z, Gatt A, Mikati M, Holmes GL. Effect of temperature on kainic acid‐induced seizures. Brain Res 1993;631:51–58. [DOI] [PubMed] [Google Scholar]
- 20. Schmitt FC, Buchheim K, Meierkord H, Holtkamp M. Anticonvulsant properties of hypothermia in experimental status epilepticus. Neurobiol Dis 2006;23:689–696. [DOI] [PubMed] [Google Scholar]
- 21. Kendall GS, Mathieson S, Meek J, Rennie JM. Recooling for rebound seizures after rewarming in neonatal encephalopathy. Pediatrics 2012;130:e451–e455. [DOI] [PubMed] [Google Scholar]
- 22. Orlowski JP, Erenberg G, Lueders H, Cruse RP. Hypothermia and barbiturate coma for refractory status epilepticus. Crit Care Med 1984;12:367–372. [DOI] [PubMed] [Google Scholar]
- 23. Azzopardi DV, Strohm B, Edwards AD, et al. Moderate hypothermia to treat perinatal asphyxial encephalopathy. N Engl J Med 2009;361:1349–1358. [DOI] [PubMed] [Google Scholar]
- 24. Kowski AB, Kanaan H, Schmitt FC, Holtkamp M. Deep hypothermia terminates status epilepticus–an experimental study. Brain Res 2012;1446:119–126. [DOI] [PubMed] [Google Scholar]
- 25. Steinbrenner M, Kowski AB, Schmitt FC, Holtkamp M. Hypothermia did not prevent epilepsy following experimental status epilepticus. Brain Res 2014;1572:50–58. [DOI] [PubMed] [Google Scholar]
- 26. Shankaran S, Pappas A, McDonald SA, et al. Childhood outcomes after hypothermia for neonatal encephalopathy. N Engl J Med 2012;366:2085–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kamps M, Bisschops LA, van der Hoeven JG, Hoedemaekers CW. Hypothermia does not increase the risk of infection: a case control study. Crit Care 2011;15:R48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bernard SA, Gray TW, Buist MD, et al. Treatment of comatose survivors of out‐of‐hospital cardiac arrest with induced hypothermia. N Engl J Med 2002;346:557–563. [DOI] [PubMed] [Google Scholar]
- 29. Moler FW, Silverstein FS, Holubkov R, et al. Therapeutic hypothermia after out‐of‐hospital cardiac arrest in children. N Engl J Med 2015;372:1898–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nielsen N, Wetterslev J, Cronberg T, et al. Targeted temperature management at 33 degrees C versus 36 degrees C after cardiac arrest. N Engl J Med 2013;369:2197–2206. [DOI] [PubMed] [Google Scholar]
- 31. Gega A, Rizzo JA, Johnson MH, et al. Straight deep hypothermic arrest: experience in 394 patients supports its effectiveness as a sole means of brain preservation. Ann Thorac Surg 2007;84:759–766; discussion 66‐7. [DOI] [PubMed] [Google Scholar]
- 32. Todd MM, Hindman BJ, Clarke WR, et al. Mild intraoperative hypothermia during surgery for intracranial aneurysm. N Engl J Med 2005;352:135–145. [DOI] [PubMed] [Google Scholar]
- 33. Palmers PJ, Hiltrop N, Ameloot K, et al. From therapeutic hypothermia towards targeted temperature management: a decade of evolution. Anaesthesiol Intensive Ther 2015;47:156–161. [DOI] [PubMed] [Google Scholar]
- 34. Parissis H, Hamid U, Soo A, Al‐Alao B. Brief review on systematic hypothermia for the protection of central nervous system during aortic arch surgery: a double‐sword tool? J Cardiothorac Surg 2011;6:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Polderman KH, Herold I. Therapeutic hypothermia and controlled normothermia in the intensive care unit: practical considerations, side effects, and cooling methods. Crit Care Med 2009;37:1101–1120. [DOI] [PubMed] [Google Scholar]
- 36. Niquet J, Baldwin R, Gezalian M, Wasterlain CG. Deep hypothermia for the treatment of refractory status epilepticus. Epilepsy Behav 2015;49:313–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bickler PE, Warren DE, Clark JP, et al. Anesthetic protection of neurons injured by hypothermia and rewarming: roles of intracellular Ca2+ and excitotoxicity. Anesthesiology 2012;117:280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Darwazeh R, Yan Y. Mild hypothermia as a treatment for central nervous system injuries: Positive or negative effects. Neural Regen Res 2013;8:2677–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fujikawa DG. The temporal evolution of neuronal damage from pilocarpine‐induced status epilepticus. Brain Res 1996;725:11–22. [DOI] [PubMed] [Google Scholar]
- 40. McCullough JN, Zhang N, Reich DL, et al. Cerebral metabolic suppression during hypothermic circulatory arrest in humans. Ann Thorac Surg 1999;67:1895–1899; discussion 919‐21. [DOI] [PubMed] [Google Scholar]
- 41. Volgushev M, Vidyasagar TR, Chistiakova M, et al. Membrane properties and spike generation in rat visual cortical cells during reversible cooling. J Physiol 2000;522(Pt 1):59–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shen KF, Schwartzkroin PA. Effects of temperature alterations on population and cellular activities in hippocampal slices from mature and immature rabbit. Brain Res 1988;475:305–316. [DOI] [PubMed] [Google Scholar]
- 43. Yang XF, Ouyang Y, Kennedy BR, Rothman SM. Cooling blocks rat hippocampal neurotransmission by a presynaptic mechanism: observations using 2‐photon microscopy. J Physiol 2005;567(Pt 1):215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fuller BJ. Gene expression in response to low temperatures in mammalian cells: a review of current ideas. Cryo Letters 2003;24:95–102. [PubMed] [Google Scholar]
- 45. Soukup J, Zauner A, Doppenberg EM, et al. The importance of brain temperature in patients after severe head injury: relationship to intracranial pressure, cerebral perfusion pressure, cerebral blood flow, and outcome. J Neurotrauma 2002;19:559–571. [DOI] [PubMed] [Google Scholar]
- 46. Busto R, Globus MY, Dietrich WD, et al. Effect of mild hypothermia on ischemia‐induced release of neurotransmitters and free fatty acids in rat brain. Stroke 1989;20:904–910. [DOI] [PubMed] [Google Scholar]
- 47. Colbourne F, Grooms SY, Zukin RS, et al. Hypothermia rescues hippocampal CA1 neurons and attenuates down‐regulation of the AMPA receptor GluR2 subunit after forebrain ischemia. Proc Natl Acad Sci USA 2003;100:2906–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu L, Yenari MA. Therapeutic hypothermia: neuroprotective mechanisms. Front Biosci 2007;12:816–825. [DOI] [PubMed] [Google Scholar]
- 49. Nagel S, Papadakis M, Pfleger K, et al. Microarray analysis of the global gene expression profile following hypothermia and transient focal cerebral ischemia. Neuroscience 2012;208:109–122. [DOI] [PubMed] [Google Scholar]
- 50. Hypothermia after Cardiac Arrest Study G . Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med 2002;346:549–556. [DOI] [PubMed] [Google Scholar]
- 51. Polderman KH. Induced hypothermia and fever control for prevention and treatment of neurological injuries. Lancet 2008;371:1955–1969. [DOI] [PubMed] [Google Scholar]
- 52. Gluckman PD, Wyatt JS, Azzopardi D, et al. Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: multicentre randomised trial. Lancet 2005;365:663–670. [DOI] [PubMed] [Google Scholar]
- 53. Shankaran S, Laptook AR, Ehrenkranz RA, et al. Whole‐body hypothermia for neonates with hypoxic‐ischemic encephalopathy. N Engl J Med 2005;353:1574–1584. [DOI] [PubMed] [Google Scholar]
- 54. Dietrich WD, Bramlett HM. The evidence for hypothermia as a neuroprotectant in traumatic brain injury. Neurotherapeutics 2010;7:43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lundgren J, Smith ML, Blennow G, Siesjo BK. Hyperthermia aggravates and hypothermia ameliorates epileptic brain damage. Exp Brain Res 1994;99:43–55. [DOI] [PubMed] [Google Scholar]