

Abstract
Objective
While abnormalities in myelin in tuberous sclerosis complex (TSC) have been known for some time, recent imaging‐based data suggest myelin abnormalities may be independent of the pathognomonic cortical lesions (“tubers”). Multiple mouse models of TSC exhibit myelination deficits, though the cell types responsible and the mechanisms underlying the myelin abnormalities remain unclear.
Methods
To determine the role of alterations in mTOR signaling in myelination, we generated a conditional knockout (CKO) mouse model using Cre‐recombinase and the Olig2 promoter to inactivate the Tsc2 gene in oligodendrocyte precursor cells.
Results
Characterization of myelin and myelin constituent proteins demonstrated a marked hypomyelination phenotype. Diffusion‐based magnetic resonance imaging studies were likewise consistent with hypomyelination. Hypomyelination was due in part to decreased myelinated axon density and myelin thickness as well as decreased oligodendrocyte numbers. Coincident with hypomyelination, an extensive gliosis was seen in both the cortex and white matter tracks, suggesting alterations in cell fate due to changes in mTOR activity in oligodendrocyte precursors. Despite a high‐frequency appendicular tremor and altered gait in CKO mice, no significant changes in activity, vocalizations, or anxiety‐like phenotypes were seen.
Interpretation
Our findings support a known role of mTOR signaling in regulation of myelination and demonstrate that increased mTORC1 activity early in development within oligodendrocytes results in hypomyelination and not hypermyelination. Our data further support a dissociation between decreased Akt activity and increased mTORC1 activity toward hypomyelination. Thus, therapies promoting activation of Akt‐dependent pathways while reducing mTORC1 activity may prove beneficial in treatment of human disease.
Introduction
Investigation of neurodevelopmental diseases, such as tuberous sclerosis complex (TSC), has traditionally focused on neuronal abnormalities in the neocortex. TSC is a multisystem neurodevelopmental disorder with prominent neurological manifestations including attention deficit hyperactivity disorder, autism, and epilepsy. Recent technological advances in magnetic resonance imaging (MRI) of the brain have revealed widespread white matter changes in a variety of neurodevelopmental disorders including TSC. White matter and glial abnormalities are well characterized adjacent to the cortical tubers in TSC, but only recently with increased use of more advanced techniques for imaging myelin has an appreciation come about for abnormalities in nontuber white matter, subcortical white matter, and commissural white matter tracts.1, 2 Fractional anisotropy (FA), a MRI measure which reflects the directional organization of the brain and is influenced by the extent and orientation of white matter tracts,3 is decreased in patients with TSC and autism spectrum disorders (ASD) as compared to controls or TSC patients without ASD.4 Similar alterations in FA have been seen prospectively in patients with cryptogenic autism.5 These data implicate myelin abnormalities in autism and support such abnormalities as contributing to the severity of the TSC clinical manifestations. Recent clinical studies show improvement in the integrity of white matter in TSC patients following treatment with the mTORC1 inhibitor everolimus, suggesting that myelin abnormalities are reversible in human neurologic diseases and that myelin may serve as a much needed biomarker for therapies.6, 7, 8
Loss of either TSC1 or TSC2 can cause TSC.9 The complex of hamartin/tuberin, the protein products of TSC1 and TSC2, respectively, function within the PI3K signaling pathway as inhibitors of mTORC1 and activators of mTORC2, thus perturbation of the hamartin/tuberin complex results in increased mTORC1 and decreased mTORC2 signaling. The role of PI3K signaling in myelination has been studied extensively with several mouse models showing that decreased activity of the PI3K pathway and AKT kinase in oligodendrocytes leads to hypomyelination10, 11 and increased Akt activity in oligodendrocytes results in hypermyelination.12, 13 More recently, Lebrun‐Julien et al. reported the surprising finding of hypomyelination in the context of increased mTORC1 activity following targeted deletion of Tsc1 from oligodendrocytes in the spinal cord.14
While abnormal brain imaging findings from TSC patients support the link between clinical disease and myelin abnormalities, it is unknown if cortical myelin abnormalities are due to (1) abnormal signaling from neurons with failure to stimulate proper myelination, (2) cell autonomous oligodendrocyte dysfunction, or (3) some the combination of the two. Previous studies with neuronal‐specific conditional knockout (CKO) animal models of TSC have demonstrated noncell autonomous abnormalities in myelin.15 Tsc1 CKO animals generated in our laboratory also demonstrated myelin abnormalities with altered mTORC1 and mTORC2 signaling. However, we targeted dorsal neural progenitor cells using Emx1‐Cre transgene that is expressed in excitatory neurons, astrocytes, and a subset of oligodendrocytes.16, 17
To address cell autonomous contributions of Tsc2/tuberin and mTOR signaling in cortical oligodendrocytes, we conditionally targeted Tsc2 in oligodendrocytes using Olig2 transgenic mice. We report a marked hypomyelination phenotype following loss of Tsc2 from oligodendrocytes and reduction of oligodendrocytes numbers in the cortex and corpus callosum (CC). Our findings support that cell autonomous dysregulation of mTOR signaling in cortical oligodendrocytes contributes to human disease pathology in TSC and expands the repertoire of cells types involved in TSC disease pathogenesis.
Materials and Methods
Tsc2 CKO mice
The Tsc2 gene was conditionally targeted in oligodendrocytes utilizing the Olig2 promoter (Jackson Laboratory #011103, Sacramento, California, USA). Olig2‐Cre animals were crossed to a Tsc2 floxed knockin line we created.18 Littermate Olig2‐Cre‐negative Flox/Wt and Flox/Wt Olig2‐Cre‐positive mice were used as controls. Lineage tracing experiments utilized the Ai14 mouse line (Jackson Laboratory #007908, Sacramento, California, USA), which expresses red fluorescent protein after exposure to Cre. Genotyping was performed using PCR as described previously.19 Tsc2 homozygous floxed animals are maintained on a mixed C57/BL10 background. All procedures in which mice were used were approved by the Vanderbilt University IACUC.
Immunofluorescence
Brain tissues were extracted and dissected from Tsc2‐Olig2 CKO and littermate controls as described previously.19, 20, 21 Mice were anesthetized with ketamine/xylazine and perfused with ice‐cold phosphate buffered saline (PBS) followed by 4% paraformaldehyde in PBS. Brains were fixed overnight at 4°C in 4% paraformaldehyde/PBS and then cryoprotected for 48 h in 30% sucrose prior to sectioning. For immunofluorescence, tissue sections allowed to warm to room temperature, washed in Tris buffered saline (TBS), and blocked with 5% normal goat serum (NGS) with 0.1% Triton‐X100 in TBS for 1 h at room temperature. Antigen retrieval was used for APC with heating a 1:10 dilution of TBS in a microwave 3× for 40 sec, permeabilization with 10% Triton‐X100 in TBS, then blocking with superblock (10% bovine serum albumin (BSA), 10% NGS, 0.03% NaN3 in TBS).22 Sections were probed with primary antibodies in blocking solution overnight at 4°C followed by species appropriate secondary antibodies (anti‐mouse, anti‐rabbit, or anti‐rat Alexa 488 and 555 fluorochromes, Invitrogen, Waltham, MA, USA) for 1 h at room temperature. Photomicrographs were obtained using a Zeiss epifluorescence microscope (Oberkochen, Germany) or an AMG Evos epifluorescence microscope (ThermoFisher, Waltham, MA, USA). Image analysis was performed with Image J (National Institutes of Health, Bethesda, MA, USA) and Adobe Photoshop CS5 (Adobe Systems, San Jose, CA, USA). Experiments were performed in triplicate to ensure reproducibility. Primary antibodies and dilutions were as follows: glial fibrillary acidic protein (GFAP) 1:1000, phospho‐S6 (Ser240‐244), myelin‐associated glycoprotein (MAG) 1:100 (all Cell Signaling, Danvers, MA, USA), Olig2 1:500 (Millipore, Temecula, CA, USA), myelin basic protein (MBP) 1:200 (Abcam, Cambridge, MA, USA), and APC 1:100 (Calbiochem, San Diego, CA, USA).
Sudan black staining
Slides were warmed to room temperature and washed with PBS followed by 70% EthOH. Slides were incubated in filtered 0.4% Sudan black in 70% EthOH for 30 min, washed in 70% EthOH and then with water. Photomicrographs were obtained using a Zeiss bright field microscope.
Magnetic resonance imaging
Following anesthesia with ketamine/xylazine, mice underwent intracardiac perfusion with 2% paraformaldehyde/2.5% glutaraldehyde/1 mmol/L Gd‐DTPA in PBS followed by postfixing for 1 week at 4°C. 3D MRI was performed on a 15.2T 11‐cm horizontal bore Bruker Biospec scanner (Bruker BioSpin, Billerica, MA) with field of view = 19.2 × 14.4 × 10.8 mm3 and matrix size = 128 ×96 × 72 for a nominal isotropic resolution of 150 × 150 × 150 μm3. Diffusion tensor imaging (DTI) data were acquired using a 3D diffusion weighted fast spin‐echo sequence with TR/TE/ESP = 200/19.0/7.1 msec and ETL = 4. Diffusion weighting was achieved with δ/Δ = 5/12 msec, b‐value = 3000 sec/mm2, 30 directions, and two signal averages with gradient polarity reversal for a total scan time of ~12 h. Multiexponential T2 (MET2) data were acquired using a 3D multiple spin‐echo sequence with TR/TE/NE/BW/NEX = 520 msec/5.8 msec/18/38.5 kHz/6 and a total scan time of ~6 h.
For DTI analysis, diffusion tensors were estimated voxel‐wise using a linear least‐squares approach. From the tensors, DTI indices FA, mean, axial, and radial diffusivity (MD, AD, and RD, respectively) were calculated on a voxel‐wise basis. For MET2 analysis, data were fit by non‐negative least squares (NNLS)23 to the sum of signals from 100 logarithmically spaced T 2 components, as defined by the extended phase graph algorithm, similar to previous work.24, 25 T 2 spectra were regularized using a minimum curvature constraint with a conservative regularization held constant across all voxels. Myelin water fraction (MWF) was defined as the percentage of signal with T 2 < 17 msec over total signal.26, 27 For quantitation, the average of regions of interest (ROIs) from the splenium, mid, and genu of the CC for each animal were averaged to generate a single data point per animal.
Transmission electron microscopy
Specimens were processed for TEM per standard protocol and imaged in the Vanderbilt Cell Imaging Shared Resource‐Research Electron Microscopy facility (Vanderbilt University, Nashville, TN, USA). Briefly, following anesthesia with ketamine/xylazine, mice underwent intracardiac perfusion as above for MRI and stored at 4°C. The samples were washed in 0.1 mol/L cacodylate buffer then incubated for 1 h in 1% osmium tetraoxide at room temperature, washed with 0.1 mol/L cacodylate buffer, and dehydrated through a graded ethanol series followed by propylene oxide. Samples were infiltrated with graded concentrations of Epon 812 resin and propylene oxide, exchanged into pure epoxy resin, and polymerized at 60°C for 48 h. Ultrathin sections (70–80 nm) were then cut from the block and collected on 300‐mesh copper grids. The copper grids were postsection stained at room temperature with 2% uranyl acetate (aqueous) and then with lead citrate. Samples were subsequently imaged on the Philips/FEI Tecnai T12 electron microscope (Hillsboro, Oregon, USA) at various magnifications.
Determination of axon density and calculation of G ratios
Number of myelinated and nonmyelinated axons per 15,000× field were determined and axon density calculated. G ratio, the ratio of axon diameter to the myelinated axon diameter were calculated via determination of the average axon and average myelinated axon diameter. G ratios were then plotted on the ordinate with axon diameter on the abscissa. A minimum of 50 axons per animal with an n > 3 animals were used for generation of the G‐ratio plot.
Immunoblotting
Control and CKO animals were anesthetized with isoflurane and tissues were extracted on ice, flash frozen in liquid nitrogen, and stored at −80°C until use. Cortical lysates included the CC. Lysate preparation, sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE), and western blotting were performed as described previously.19 Primary antibodies included pS6 (Ser235/236), pS6 (Ser240/244), S6, pAkt (Ser473), pAkt (Thr308), Akt, pNDRG1‐Thr346, NDRG1, MAG, GFAP, 2′,3′‐cyclic nucleotide 3′‐phosphodiesterase (CNPase), and actin (Cell Signaling, 1:1000 dilution). Additional antibodies were MBP 1:1000 (rat, Abcam), PGP9.5 1:2000 (Serotec, Kidlington, UK), and actin 1:2000 (mouse, Sigma, St. Louis, MO, USA).
Primary astrocyte cultures
Primary mixed glial cultures were prepared from P0 to P2 mouse brains with modification of published protocols.28 Following euthanasia, brains were placed in ice‐cold minimum essential media (MEM) and cortex isolated from ventral structures and meninges. Dissociated tissues were subsequently plated into poly‐l‐lysine (PLL)‐coated flasks and incubated at 37°C and 8.5% CO2. Oligodendrocyte precursors were isolated by shaking overnight at 220 rpm to make both enriched oligodendrocyte and astrocyte cultures. Following shaking, oligodendrocyte precursors were plated into PLL‐coated six‐ or 24‐well plates in OL media (Dulbecco's modified Eagle's medium (DMEM) supplemented with BSA, progesterone, putrescine, sodium selinite, 3,3′,5‐triiodo‐l‐thyronine, insulin, glutamine, holo‐transferrin, B27, and fetal bovine serum (FBS) and subsequently harvested at DIV 9. Astrocytes remaining in the T25 flask were harvested, washed with PBS, and dissociated with 0.25% trypsin in Hanks' balanced salt solution (HBSS). Dissociated cells were plated into PLL‐coated six‐ or 24‐well culture dishes until day of harvest.
For IF microscopy, cells in 24‐well dishes were washed with PBS followed by fixation with 4% paraformaldehyde for 10 min. Following PBS washes, cells were labeled as above for tissue sections and image obtained with an Evos fluorescent microscope (AMG).
Behavioral studies
All behavior experiments were conducted in the Vanderbilt Murine Neurobehavioral Core and approved by the Vanderbilt IACUC. Animals were housed on a 12‐h light–dark cycle with free access to food and water and were allowed to acclimate the environment for a minimum of 7 days prior to experiments.
Elevated zero maze
The maze apparatus was used as described previously.19 Animals were placed in the open arm of the maze and activity was recorded for 5 min. Data were acquired and analyzed using the ANY‐Maze video‐tracking software program (SDI, San Diego, CA).
Rota rod
Balance and coordination were measured using a Rota rod (Ugo Basile, Varese, Italy). Animals were placed on rotating rod at 5 rpm with graded increases in rotational velocity over 5 min to a maximum of 49 rpm. Time to animals fall from the rod or to complete two complete circles on the rod was recorded. Animals were trained on the apparatus for three trials daily for 2 days prior to testing on day 3. The average time for the three trials on day 3 was recorded.
Ultrasonic vocalizations
Male and female P4/5‐, P6/7‐, and P10/11‐day‐old pups were removed from the dam and placed in a sound‐proofed container. Ultrasonic vocalizations were recorded with the Avisoft bioacoustics recorder for 5 min and counted. Pups were immediately returned to their home cage upon completion of the recording.
Results
To determine the cell autonomous role of Tsc2 in oligodendrocytes and myelination, we targeted the GAP‐encoding exons of Tsc2 using a floxed knockin allele18 crossed to a transgenic Olig2‐Cre mouse.29 Mice with deletion of both copies of Tsc2 in oligodendrocytes (Tsc2 F/F; Olig2‐Cre, referred to hereafter as Olig2‐Tsc2 CKO) were viable and fertile and tended to be slightly smaller that wild‐type litter mages but with larger brains (Fig. S1). By 30 days of age, CKO mice were noted to walk with a splayed gait (Fig. S2) and demonstrated a high‐frequency appendicular and vibrissal tremor, similar to that reported in other mouse models of hypomyelination.30 No clinical seizures were observed during handling or with routine care. Photomicrographs of whole brain sections stained with the myelin marker sudan black demonstrated reduced staining in major white matter tracks and throughout the brains of Olig2‐Tsc2 CKO P70‐day‐old mice (Fig. 1). Similar reductions in cortical myelin were seen using immunofluorescence staining against MBP. Quantitation of protein expression from P70 day cortical extracts demonstrated significant reductions in content of the myelin constituent proteins consistent with hypomyelination. Phosphorylation of the mTORC1 target S6 ribosomal protein (S6) and of N‐Myc downstream regulated 1 (NDRG1), a downstream target of mTORC2, was increased and decreased, respectively, consistent with increased mTORC1 and decreased mTORC2 signaling activity following loss of tuberin.31
Figure 1.
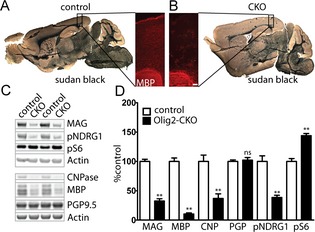
Hypomyelination secondary to loss of Tsc2 from oligodendrocytes. Sudan black and MBP immunofluorescence staining of P70 control and Olig2‐CKO brains demonstrates diffuse decreases in myelin (A, B). Cortical expression of the myelin proteins MBP, MAG, and CNPase are significantly decreased in P70 cortex (C, D). Activation of mTORC1 signaling (pS6) and attenuation of mTORC2 (pNDRG1) signaling is seen with increased S6 phosphorylation and decreased NDRG1 phosphorylation, respectively. n = 3–6 animals per group. **P < 0.01. MBP, myelin basic protein; CKO, conditional knockout; MAG, myelin‐associated glycoprotein; CNPase, 2′,3′‐cyclic nucleotide 3′‐phosphodiesterase; NDRG1, N‐Myc downstream regulated 1.
MRI‐based studies have demonstrated changes in diffusion‐weighted parameters such as FA in patients with TSC as well as in patients with autism, suggesting that myelin abnormalities may contribute to disease morbidity. To determine if similar myelin abnormalities exist following loss of Tsc2 in oligodendrocytes, we imaged P60 Olig2‐Tsc2 CKO and littermate control mouse brains ex vivo with a 15.2T MRI scanner. FA was significantly reduced in the CC in the Olig2‐Tsc2 CKO brains compared to littermate controls, suggesting decreased directional organization of white matter tracts (Fig. 2). As both RD (perpendicular to the axon) and AD (following the direction of the axon) contribute to FA (cartoon inset, Fig. 2), analysis of directionality demonstrated that the reduction in FA in the CC was predominantly due to increased RD but not AD (data not shown, P = 0.0436), supporting that FA reduction is more likely due to a myelin abnormality than an axonal pathology. Characterization of the MWF,26, 27 a recently developed and more specific MRI‐based technique for quantitation of myelin, showed global reductions in myelin content, similar to that seen with immunoblot.
Figure 2.
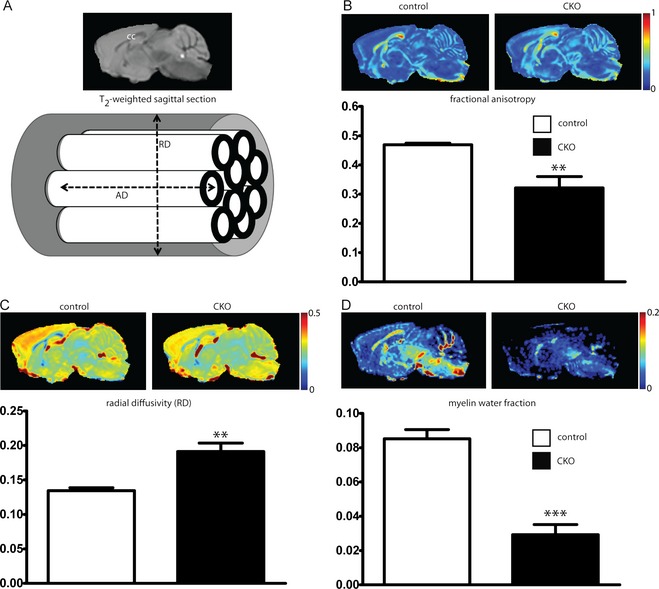
Decreased fractional anisotropy and myelin total water fraction following loss of Tsc2. P60 control and Tsc2‐Olig2 CKO mice were imaged ex vivo with a 15.2T MRI scanner. Traditional T2‐weighted imaging for anatomical reference (A). Fractional anisotropy was decreased (B) secondary to increases in radial diffusivity (C). The myelin water fraction, a more specific marker for myelin quantity, is also significantly reduced, consistent with hypomyelination (D). n = 5 animals per group. **P < 0.01, ***P < 0.001. CKO, conditional knockout; MRI, magnetic resonance imaging.
Electron microscopy was used to evaluate whether the observed hypomyelination and MRI changes were due to decreased total axonal myelin or alterations in myelin thickness. The overall density of myelinated axons was significantly decreased in the CC of the Olig2‐Tsc2 CKO mice at P60 (Fig. 3), with an associated decrease in the overall myelin fraction. G ratios (ratio of axon diameter to myelinated axon diameter) were calculated in the CC and were increased in the Olig2‐Tsc2 CKO mice compared to littermate controls, consistent with decreased myelin thickness. Binning of the axons by diameter demonstrates that the increased G ratio is significant in axons less than 1 μmol/L in diameter. These data suggest a reduced ability to myelinate axons in the CKO animals, with a bias toward myelination of larger diameter axons. The PI3K signaling pathway is involved in oligodendrocyte proliferation and differentiation, thus we sought to determine if changes in proliferation or differentiation could be a contributing mechanism to the observed hypomyelination. We identified cortical oligodendrocytes and myelin using immunostaining for the oligodendrocyte marker, Olig2, and MBP. There was a marked overall reduction in MBP expression seen diffusely throughout the cortex as well as the CC in P17 Olig2‐Tsc2 CKO brains and ~50% reduction in Olig2+ cells (Fig. 4). A similar reduction in Olig2+ cell number was seen at 5 days of age (data not shown, P = 0.0048, n = 5–7). The number of MBP/Olig2 double‐labeled mature cortical oligodendrocytes was also significantly decreased. To determine the density of mature oligodendrocytes in white matter tracts, APC+ oligodendrocytes were counted in the CC. Consistent with that seen in the cortex, a 50% reduction in APC+ oligodendrocytes was seen in the CC. These data suggest that both decreased oligodendrocyte number and impaired maturation of oligodendrocytes may be contributing to the decrease in myelination.
Figure 3.
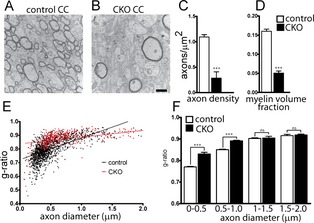
Decreased myelin density and myelin thickness in Tsc2‐Olig2 CKO. Electron micrographs from P60 littermate control (A) and Tsc2‐Olig2 CKO (B) CC demonstrate a decreased density of myelinated axons in CKO (C) brains as well as a reduction in the myelin volume fraction (D). Scale bar = 500 nm. Myelin thickness is decreased as demonstrated by an increased G ratio in Tsc2‐Olig2 CKO CC (E). G ratio plotted as a function of axon diameter bins (F). n > 50 axons from three animals per genotype. ***P < 0.001 by Student's t‐test. CKO, conditional knockout; CC, corpus callosum.
Figure 4.
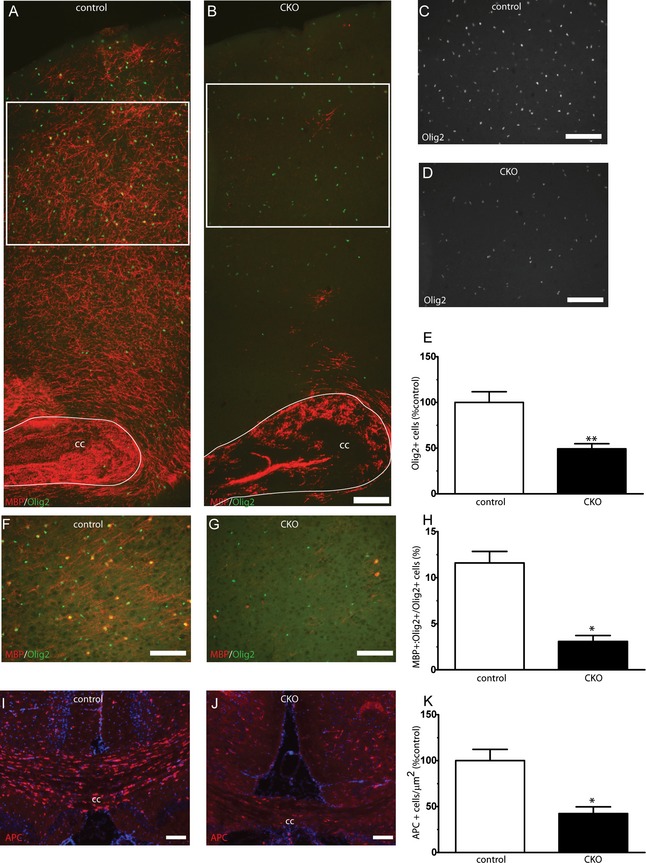
Hypomyelination and decreased oligodendrocyte numbers in CKO cortex and white matter tracts. Olig2‐positive oligodendrocyte precursors were identified with immunofluorescence. Decreased numbers of cortical Olig2+ cells were seen in CKO cortex (B, D‐E) versus wild‐type control cortex (A, C, E). Cortical regions from where cell counts occurred in (C, D) are noted with white inset boxes on (A, B). Reduced MBP+/Olig2+ mature cortical oligodendrocyte number (F–H). Reduced APC+ oligodendrocyte numbers were seen from the corpus callosum of CKO animals versus littermate controls (I–K). Scale bar = 100 μm. N = 3–6 animals per group. *P < 0.05, **P < 0.01. CKO, conditional knockout; MBP, myelin basic protein.
Reduced oligodendrocyte numbers could be due to changes in proliferation or differentiation. Data from spinal cord injury models demonstrate a link between mTOR activity and cell fate, with activation of PI3K signaling stimulating differentiation of preoligodendrocytes to astrocytes.32 As cortical tubers in TSC patients have prominent astrocyte pathology, we hypothesized that the decrease in oligodendrocyte number results from altered differentiation of oligodendrocyte precursors into astrocytes rather than mature oligodendrocytes. Increased expression of the astrocyte marker GFAP was seen diffusely in white matter tracts and cortex in P17 Olig2‐Tsc2 CKO (Fig. 5) animals and persisted throughout adulthood. We questioned whether this astrogliosis was a nonspecific response to altered structure or whether it was secondary to cellular fate change. To address this hypothesis, primary astrocyte cultures were generated from P0 to P2 mouse pups and evaluated with immunofluorescence and immunoblotting. In culture, astrocytes from CKO mouse brains were larger with a more diffuse shape as compared to a stellate shape of the controls. This was also reflected with both a slight but significant increase in spherical cell size as measured with a Cellometer during plating (control 14.55 ± 0.29 μm, CKO 16.218 ± 0.30 μm, n = 7–13 independent cultures, P = 0.0019 by two‐tailed t‐test) as well as an 80% increase protein yield (control 6.56 ± 0.61 μg/μL, CKO 11.92 ± 0.84 μg/μL, n = 7–18 independent cultures, P < 0.0001 by two‐tailed t‐test). Protein analysis demonstrated that the cortical astrocytes were targeted with a near complete ablation of tuberin as well as expected increase in S6 phosphorylation and reduction in Akt phosphorylation, consistent with deletion of Tsc2 and subsequent mTORC1 activation and suppression of mTORC2 signaling.
Figure 5.
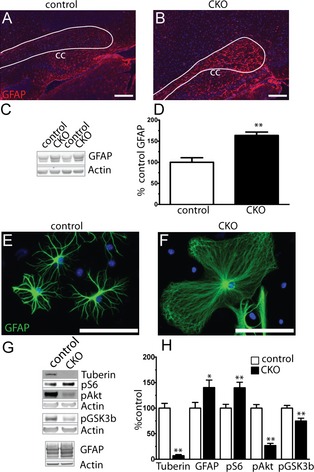
Diffuse cortical and white matter astrogliosis following loss of Tsc2. Sagittal sections from P17 CKO brains (B) demonstrated a qualitative increase in expression of the astrocyte marker GFAP in the corpus callosum and cortex versus controls (A). Scale bar = 100 μm. GFAP expression level is significantly increased in P17 cortical extracts (C, D). n = 4–5 animals per group. **P < 0.01. Astrocytes from primary cultures of CKO cortex demonstrated an abnormal morphology and increase in size relative to control astrocytes (E, F). Western blot analysis of primary astrocyte culture extracts (G, H). Data represent mean ± SEM, n = 4–7 extracts per group. *P < 0.05, **P < 0.01. CKO, conditional knockout; GFAP, glial fibrillary acidic protein.
We sought to determine the contribution of increased mTORC1 activity to Olig2‐Tsc2 CKO hypomyelination through inhibition with rapamycin. Two treatment paradigms were used: an early treatment paradigm with 0.1 mg/kg rapamycin initiated prior to the maximal rate of myelination, P3 to P1733; and a late paradigm with treatment started after myelination is nearly complete, with 0.6 mg/kg rapamycin daily from P30 to P60. At P17, following the early treatment paradigm, despite the expected reduction in S6 phosphorylation, cortical hypomyelination was not rescued (Table 1). Consistent with published reports following mTORC1 inhibition, overall expression of myelin proteins decreased in rapamycin‐treated controls. Olig2+ cell expression in the cortex was not changed with rapamycin treatment (data not shown), suggesting the possibility that the timing of treatment was too late to correct the reduction in cell number. In contrast to the early treatment paradigm, the late treatment paradigm did significantly increase MAG expression, but did not rescue the reductions in MBP or CNPase. The increased expression of GFAP persisted after rapamycin treatment, suggesting insensitivity of the gliosis to mTORC1 inhibition (Fig. 6).
Table 1.
Early RAPA treatment fails to rescue myelination
| Antibody | Control V | CKO V | Control RAPA | CKO RAPA |
|---|---|---|---|---|
| pS6 (ser240/244) | 100.00 ± 1.96 | 221.20 ± 19.57 | 32.81 ± 4.77 | 49.80 ± 5.67 |
| MBP | 100.00 ± 0.48 | 34.63 ± 5.78a | 33.23 ± 9.95a | 34.80 ± 7.29a |
| MAG | 100.00 ± 3.39 | 43.20 ± 8.31a | 59.58 ± 9.96a | 67.49 ± 6.67 |
| CNPase | 100.00 ± 3.44 | 13.48 ± 3.55a | 49.81 ± 16.93 | 31.38 ± 10.48a |
| GFAP | 100.00 ± 13.08 | 227.30 ± 23.62a | 87.88 ± 11.42 | 158.80 ± 20.44 |
| PGP9.5 | 100.00 ± 2.37 | 95.61 ± 14.08 | 83.20 ± 7.77 | 81.28 ± 2.98 |
Expression of myelin proteins in P17 cortical extracts from mice treated with 0.1 mg/kg rapamycin or vehicle from P3 to P17. Data represent mean ± SEM, n = 3–6 animals per group. CKO, conditional knockout; V, vehicle; RAPA, rapamycin; MBP, myelin basic protein; MAG, myelin‐associated glycoprotein; CNPase, 2′,3′‐cyclic nucleotide 3′‐phosphodiesterase; GFAP, glial fibrillary acidic protein; ANOVA, analysis of variance.
P < 0.05 by one‐way ANOVA versus control vehicle.
Figure 6.
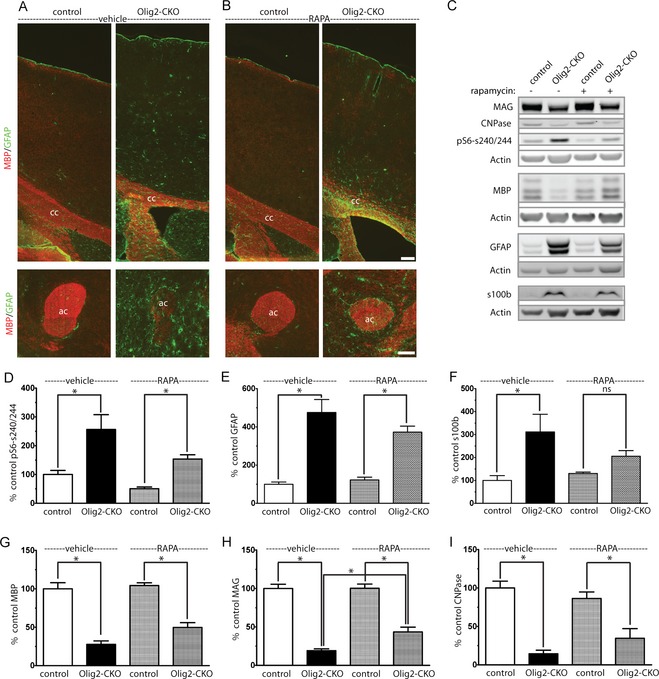
Improved myelination but persistent gliosis following rapamycin treatment. CKO and littermate control animals were treated with rapamycin 3 mg/kg, 5 days per week from P30 to P60. P60 sagittal brain sections were stained with MBP to identify myelin and GFAP to identify astroctyes (A, B) (cc – corpus callosum, ac – anterior commissure) Scale bar = 100 μm. Gliosis was increased as identified GFAP and s100β expression (C, E, and F) and remained significantly elevated despite rapamycin treatment (D). Expression of MAG improved with rapamycin treatment but did not completely normalize (C, G–I). Data represent mean ± SEM, n = 4 animals per group. *P < 0.05 by one‐way ANOVA followed by Tukey's multiple comparison test. CKO, conditional knockout; MBP, myelin basic protein; GFAP, glial fibrillary acidic protein; MAG, myelin‐associated glycoprotein; ANOVA, analysis of variance.
As the late rapamycin treatment paradigm led to slight improvement in myelin quantity as well as an improvement in the tremor seen in Olig2‐Tsc2 CKO mice (data not show), we questioned whether myelin abnormalities may contribute to behavior abnormalities and whether they may be ameliorated with rapamycin treatment (Fig. 7). Despite the clear tremor, the latency to falling during rotarod testing was not different between Olig2‐Tsc2 CKO and littermate controls (Figs. 7, S3). Furthermore, while rapamycin treatment did qualitatively reduce the tremor, it did not alter mean fall latency in either group or normalize gait abnormalities (Fig. S2). As human myelination abnormalities have been associated with ASD,5 we expanded our studies to address autism relevant behaviors. Ultrasonic vocalizations generated by mouse pups when separated from their mothers can serve as a communication surrogate in mice.34, 35 Vocalizations were determined in male and female control and Olig2‐Tsc2 CKO pups at P4/5, P6/7, and P10/11 derived from control and heterozygous mothers. Vocalization quantity was not significantly different between groups at any time point, though a significant decrease in the frequency of vocalizations in both groups was seen from P4 to P10. When subdivided by maternal genotype or pup gender, similar trends were seen between groups though a significant difference in vocalizations at P6/7 was seen in male control versus CKO pups (P < 0.05 by one‐way ANOVA, Fig. S3). To determine if hypomyelination contributes to anxiety‐related behavior, mice were tested with the elevated zero maze. No significant differences were noted with respect to the time spent in the open arm of the maze between Olig2‐Tsc2 CKO and controls, though Olig2‐Tsc2 CKO mice tended to be less active and had fewer, but longer visits to the open arm. The overall distance travelled, number of entries, and duration of visits returned to that of control values after treatment with rapamycin.
Figure 7.
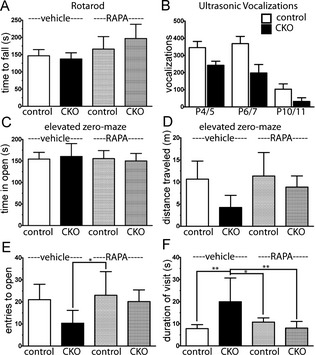
Absence of motor deficits, anxiety phenotype, or communication deficits with loss of Tsc2 in oligodendrocytes. To determine the presence of a motor defects, time to fall during rotarod testing was determined in P60‐ to P70‐day‐old CKO and control mice treated with vehicle or RAPA from P30 until day of testing (A). Ultrasonic vocalizations were used to determine the presence of a communication deficit in the Tsc2 Olig2 CKO mice pups from P4 to P11 (B). The presence of an anxiety phenotype was analyzed with the elevated zero maze in P60–70 mice treated with vehicle or rapamycin from P30. Time in the open arm (C), as well as overall distance traveled (D), entries to the open (E), and duration of visit to the open arm (F) were determined in control and CKO mice following vehicle and rapamycin treatment. n = 8–15 animals per group. *P < 0.05, **P < 0.01 by one‐way ANOVA versus the control vehicle group. CKO, conditional knockout; RAPA, rapamycin; ANOVA, analysis of variance.
Discussion
Myelin abnormalities have recently been reported in a wide array of neurodevelopmental disorders including TSC, Angelman's syndrome, and autism in part to advances in MRI techniques.4, 5, 36 Consistent with these clinical findings, animal studies in TSC have demonstrated myelin abnormalities which though have been largely attributed to the neuronal abnormalities. We have demonstrated a marked hypomyelination phenotype following loss of Tsc2 from oligodendrocytes and demonstrated MRI findings which recapitulate that seen in humans with TSC.
An extensive literature exists to support a role for PI3K/Akt/mTOR signaling in myelination, and in the context of this literature, our findings and those of Lebrun‐Julien et al.14 are unexpected. A variety of mouse models have shown a positive correlation between Akt/mTORC1 activity and the degree of myelination. Whether from loss of upstream signaling through deletion of IGF1 or direct deletion of Akt isoforms, myelin quantity is decreased.37, 38 The effect of rapamycin on mutant and normal tissue along with more recent data with oligodendrocyte‐specific deletion of mTOR and Raptor, supports that mTOR activity downstream of Akt is important in the myelination process.22, 39, 40 Conversely, hyperactivation of Akt signaling through deletion of PTEN or insertion of a constitutively active Akt results in a diffuse hypermyelination phenotype13, 29, 39, 41, 42 which is sensitive to rapamycin. In contrast to these studies, despite increased mTORC1 activity, the brains of Olig2‐Tsc2 CKO mice are markedly hypomyelinated. The observed tremor phenotype is similar to that of other models of hypomyelination including the Cnp1 CKO mouse.30 The decreased number of cortical and callosal oligodendrocytes suggests that altered proliferation of oligodendrocyte precursors contributes to the hypomyelination. The demonstration that both the G ratios of myelinated CKO axons were increased and that the proportion of Olig2+/MBP+ oligodendrocytes was reduced supports that oligodendrocyte maturation may also be impaired. The apparent increase in axon diameter in addition to decreased myelin thickness may be due in part to a bias toward myelination of larger axons versus a direct targeting of cortical excitatory neurons (Fig. S4).
Increased mTORC1 activity can lead to reductions in mTORC2 activity likely through a feedback loop involving IRS‐1. The decrease in NDRG1 phosphorylation in our model supports that mTORC2 signaling is decreased. We have hypothesized that decreased mTORC2 activity may contribute to myelination abnormalities based on our previous work with the Emx1‐Rictor CKO.19 Preliminary studies in our Olig2‐Rictor CKO mouse (unpublished observation) and two recent reports from the Macklin and Wood labs with oligodendrocyte specific ablation of mTOR, Rictor and Raptor, provide additional support for a role for mTORC2 signaling in myelination.22, 40 While reduced mTORC2 activity is likely contributing to the hypomyelination in our model, the degree of hypomyelination seen is more dramatic than that seen with the Rictor CKO models, suggesting that this is not the key mechanism for the hypomyelination. Similarly, feedback inhibition of Akt activity is likely contributing to the hypomyelination through decreased phosphorylation of downstream effectors. One likely candidate is GSK3, which when inhibited by phosphorylation by Akt is associated with oligodedrogenesis.43 Additional studies in primary cultures of oligodendrocytes will be required to fully delineate the specific signaling changes in oligodendrocyte precursors.
The extensive cortical and callosal gliosis is of interest as cortical tubers in TSC contain a mix of neuronal and glial components.20 While the gliosis on our model may be a nonspecific response to hypomyelination, our data demonstrate that astrocytes are targeted. Data from a spinal cord injury models have demonstrated that increased BMP signaling through mTOR can change the fate of oligodendrocyte precursors from oligodendrocytes to an astrocytic lineage.44 This, combined with the demonstration in culture that astrocytes are targeted in our mouse model in concert with a reduction in cortical oligodendrocytes, suggests the possibility of a fate change. While direct targeting of astrocytes by the Olig2‐Cre driver may serve as a potential confounder, the stark contrast between our Olig2‐Tsc2 model versus the Olig2‐PTEN model of Harrington,29 which at P20 demonstrates normal to increased expression of myelin proteins as well as gliosis (Fig. S5), supports a cell autonomous role for oligodendrocyte dysfunction in the hypomyelination. Additionally, the similarity of our results to Lebrun‐Julien et al.,14 who used the Cre‐CNP driver to study the spinal cord, also supports a cell autonomous mechanism for hypomyelination. Additional fate mapping studies and generation of primary oligodendrocytes cultures will be of benefit in elucidating mechanisms for the oligodendrocyte reduction.
Given the known association of mTORC1 activity in myelin production, the lack of a rescue with the early rapamycin treatment paradigm is not surprising, especially when the detrimental effect of rapamycin on control pups is seen. The lack of efficacy and the reduction of Olig2+ cells seen as early as P5 suggests that loss of oligodendrocyte precursors may occur at an earlier time in development. While these data differ from the data of Way et al.,33 it is likely that the degree of mTORC1 activity in neurons, while likely normalized by rapamycin in the Way et al. model, is expected to be suppressed by rapamycin in the Olig2 model. The improvements in expression of myelin proteins in the late rapamycin paradigm supports a continued process by which altered mTORC1, and likely mTORC2, activity inhibit proper myelination. The persistence of increased S6 phosphorylation after rapamycin may suggest that the inhibition of mTORC1 activity was incomplete, though given the decreased numbers of oligodendrocyte precursors seen prior to P17 and that rapamycin can decrease expression of myelin proteins, a complete rescue may not be possible as a deficiency of functional oligodendrocytes is likely.
The lack of a dramatic behavioral phenotype other than the gait abnormality and tremor is surprising given the degree of myelin impairment. The animals had no clear phenotype on rotarod testing and only a subtle, though rapamycin‐sensitive alterations in activity with elevated zero maze testing with no clear anxiety phenotype. The lack of a rotarod phenotype is even more surprising in the context of the gait abnormality and that Olig2 may additionally target a subset of spinal cord motor neurons. Using both an Ai14‐RFP reporter mouse line and analysis of spinal cord extracts, we do see targeting of spinal cord motor neurons and reductions in expression of spinal cord myelin proteins, respectively (Fig. S6), similar to that demonstrated by Lebrun‐Julien et al.14 Likewise, there were no changes in the overall number of ultrasonic vocalizations with the exception of male pups only at P6/7. The behavior findings may be complicated by the fact that the animals used for the behavior studies were not fully back‐crossed (generation 4–5) which likely contributed to variability in activity and vocalizations. More studies in fully back‐crossed (generation 10+) animals are required to better determine if indeed autism‐relevant behaviors exist in this model.
These findings demonstrate that cell autonomous loss of Tsc2 in oligodendrocytes leads to abnormal myelination and supports that abnormal myelination in TSC is likely due to both intrinsic abnormalities in oligodendrocytes as well as altered communication between neurons and oligodendrocytes.15 In addition, we utilized both standard (DTI) and novel (myelin water imaging) MRI techniques which may serve as important biomarkers in monitoring disease progression or response of myelin to therapeutic interventions. Characterization of the developmental changes attributed to oligodendrocytes and myelination is important for a better understanding of TSC as well as other neurodevelopmental disorders, including cryptogenic autism, for which myelination abnormalities may contribute to disease pathology. Importantly, given that myelination is largely a postnatal process, a window may exist for potential therapeutic interventions, thus further identification of relevant targets is of key importance. Future studies with this model focused on ASD‐relevant behaviors will need be conducted to better determine the applicability of this model as a tool to study pharmacologic interventions directed both at rescue of myelination and improvement in neuropsychologic sequela of autism.
Author Contributions
R. P. C. conceived experiments, performed experiments, interpreted data, and crafted the manuscript. N. C. K., K. L. W., and M. D. D. performed MRI experiments, data analysis, and reviewed the manuscript. B. P., G. W., M. D. G., and E. M. performed experiments, generated primary astrocyte cultures, performed western blots, generated photomicrographs, and reviewed the manuscript. C. F. generated the mouse with the targeted deletion of Tsc2 used in these studies and critically reviewed the manuscript. K. C. E. conceived experiments, aided in data interpretation, and critically reviewed the manuscript.
Conflict of Interest
None declared.
Supporting information
Figure S1. Weight over time in Tsc2 CKO mice (A, B). Average body (C), brain (D), and brain/body ratio (E) in a cohort of P60‐ to P97‐day‐old mice. n = 6, *P < 0.05, **P < 0.01, ***P = 0.0001 by paired two‐tailed t‐test.
Figure S2. Abnormal gait in Tsc2 CKO mice. To characterize gait in control (A) and CKO (B) mice, mice were run on a motorized treadmill (TreadScan 2.0, Clever Sys, Inc) and front and rear track width measured manually by a blinded examiner. CKO mice demonstrated a splayed gait of the rear legs which was evident by P30 and was not improved with rapamycin treatment from P30 to P60 (C). No significant differences were noted with front track width at P30–P45, though the width was significantly increased by P60 in CKO mice and was unaffected by rapamycin treatment. n = 6–7, *control versus CKO P < 0.05, #control RAPA versus CKO RAPA P < 0.05 by one‐way ANOVA.
Figure S3. Absence of communication deficits based on gender or maternal genotype or learning effect on rotarod. Ultrasonic vocalizations were used to determine the presence of a communication deficit in the Tsc2 Olig2 CKO mice pups from P4 to P11. Vocalizations tended to be slightly, but not significantly decreased in CKO pups from both control (A) and heterozygous (B) dams. Similar trends were seen with both male (C) and female (D) pups, though a significant difference in vocalizations was seen at P6/7 in male pups only, likely attributable to an increase in vocalizations in the control pups. n = 7–11 animals per group. *P < 0.05 by one‐way ANOVA versus the control vehicle group. In P60 control and CKO mice treated with vehicle or rapamycin, no differences in latency to fall from the rotarod were seen during training or on the day 3 testing (E). n = 8–15 animals per group. *P < 0.05 by one‐way ANOVA versus the control vehicle group.
Figure S4. Lineage tracing of Olig2+ cells in cortex in primary culture. Olig2+ targeted cells were identified with Ai14 (RFP) labeling and costaining will neuronal and glial markers in cortex of P20‐day‐old mice. Clear costaining with the neuronal marker NeuN (A) is not appreciated in cortex, whereas double labeling is seen with Gad67‐positive neurons (B, yellow arrow inset). Perinuclear RFP signal is seen wrapped by MBP+ and MAG (C, D). GFAP+ projections are seen surrounding the perinuclear RFP signal in cortex (E). Scale bar = 100 μm. Mixed neuronal and glial cultures from Olig2+ Ai14 mice were costained with NeuN, Gad67, and GFAP. No RFP signal is seen to colocalize with the pyramidal appearing NeuN+ cells (E, green circle), whereas as noted in cortex, smaller NeuN+ and Gad67+ cells do demonstrate a faint perinuclear RFP signal (E, F, yellow circle). Extensive RFP signal is seen diffusely in GFAP+ astrocytes with a more intense perinuclear signal (G). Scale bar = 200 μm.
Figure S5. Increased GFAP expression in Olig2‐PTEN CKO cortex and primary culture. To address the hypothesis that gliosis in the Olig2‐Tsc2 CKO mouse is responsible for the hypomyelination was asked whether gliosis was also present in the Olig2‐PTEN CKO which in contrast to the Tsc2 model has been shown to be hypermyelinated. Immunofluorsescence staining with GFAP and MBP in P20 Olig2‐PTEN CKO cortex demonstrates increased GFAP (B, E) expression and maintained MBP expression (A, D) in the CKO. Analysis of P20 Olig2‐PTEN cortical extracts shown in comparison to P30 Olig2‐Tsc2 cortical extracts (G), demonstrates increase GFAP expression along with preserved (MAG, MBP, CNPase) to increased (PLP) expression of myelin proteins. A similar increase in GFAP expression was seen in primary cultures of astrocytes derived from the Olig2‐PTEN mice (H).
Figure S6. Spinal cord hypomyelination and targeting of spinal cord motor neurons by Olig2. Similar to data by Lebrun‐Julien et al., with the Cre‐CNP‐Tsc1 mouse, expression of the myelin proteins MAG, CNPase, MBP, and PLP were reduced in extracts from P30 Olig2‐Tsc2 CKO spinal cord. Similar to findings in cortex, GFAP expression and S6 phosphorylation were significantly increased. n = 4 animals per group. *P < 0.05, **P < 0.01 (A, B). As has been reported previously, spinal motor neurons are targeted by Olig2 as demonstrated by the Ai14 reporter line (C, inset yellow arrow) with expression of RFP extending to a subset of myelinated axons in the sciatic nerve (D, inset red arrow). Scale bar = 100 μm.
Acknowledgments
We thank Sherry Syed and Peggy Winzenburger for their technical assistance. This research was supported by the National Institute of Neurological Disorders and Stroke; National Institutes of Health (K08NS083710 to R. P. C. and 1R01 NS078289 to K. C. E.) and from a Tuberous Sclerosis Alliance Postdoctoral Fellowship award to C. F. Electron microscopy performed in part through the use of the VUMC Cell Imaging Shared Resource with special thanks to Janice Williams for assistance.
References
- 1. Arulrajah S, Ertan G, Jordan L, et al. Magnetic resonance imaging and diffusion‐weighted imaging of normal‐appearing white matter in children and young adults with tuberous sclerosis complex. Neuroradiology 2009;51:781–786. [DOI] [PubMed] [Google Scholar]
- 2. Simao G, Raybaud C, Chuang S, et al. Diffusion tensor imaging of commissural and projection white matter in tuberous sclerosis complex and correlation with tuber load. AJNR Am J Neuroradiol 2010;31:1273–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Basser PJ, Pierpaoli C. Microstructural and physiological features of tissues elucidated by quantitative‐diffusion‐tensor MRI. J Magn Reson B 1996;111:209–219. [DOI] [PubMed] [Google Scholar]
- 4. Peters JM, Sahin M, Vogel‐Farley VK, et al. Loss of white matter microstructural integrity is associated with adverse neurological outcome in tuberous sclerosis complex. Acad Radiol 2012;19:17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wolff JJ, Gu HB, Gerig G, et al. Differences in white matter fiber tract development present from 6 to 24 months in infants with autism. Am J Psychiatry 2012;169:589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Franz DN, Care MM, Holland‐Bouley K, et al. Everolimus reduced seizure activity and altered the microarchitecture of CNS white matter in patients with tuberous sclerosis complex. Ann Neurol 2011;70:S118–S118. [Google Scholar]
- 7. Ess KC, Roach ES. New therapies for tuber‐less sclerosis: white matter matters? Neurology 2012;78:520–521. [DOI] [PubMed] [Google Scholar]
- 8. Tillema JM, Leach JL, Krueger DA, Franz DN. Everolimus alters white matter diffusion in tuberous sclerosis complex. Neurology 2012;78:526–531. [DOI] [PubMed] [Google Scholar]
- 9. Kwiatkowska J, Jozwiak S, Hall F, et al. Comprehensive mutational analysis of the TSC1 gene: observations on frequency of mutation, associated features, and nonpenetrance. Ann Hum Genet 1998;62(Pt 4):277–285. [DOI] [PubMed] [Google Scholar]
- 10. Tschopp O, Yang ZZ, Brodbeck D, et al. Essential role of protein kinase B gamma (PKB gamma/Akt3) in postnatal brain development but not in glucose homeostasis. Development 2005;132:2943–2954. [DOI] [PubMed] [Google Scholar]
- 11. D'Ercole AJ, Ye P, O'Kusky JR. Mutant mouse models of insulin‐like growth factor actions in the central nervous system. Neuropeptides 2002;36:209–220. [DOI] [PubMed] [Google Scholar]
- 12. Flores AI, Narayanan SP, Morse EN, et al. Constitutively active Akt induces enhanced myelination in the CNS. J Neurosci 2008;28:7174–7183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Goebbels S, Oltrogge JH, Kemper R, et al. Elevated phosphatidylinositol 3,4,5‐trisphosphate in glia triggers cell‐autonomous membrane wrapping and myelination. J Neurosci 2010;30:8953–8964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lebrun‐Julien F, Bachmann L, Norrmen C, et al. Balanced mTORC1 activity in oligodendrocytes is required for accurate CNS myelination. J Neurosci 2014;34:8432–8448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Meikle L, Talos DM, Onda H, et al. A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J Neurosci 2007;27:5546–5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Richardson WD, Kessaris N, Pringle N. Oligodendrocyte wars. Nat Rev Neurosci 2006;7:11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Carson RP, Van Nielen DL, Winzenburger PA, Ess KC. Neuronal and glia abnormalities in Tsc1‐deficient forebrain and partial rescue by rapamycin. Neurobiol Dis 2012;45:369–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fu C, Ess KC. Conditional and domain‐specific inactivation of the Tsc2 gene in neural progenitor cells. Genesis 2013;51:284–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Carson RP, Fu C, Winzenburger P, Ess KC. Deletion of Rictor in neural progenitor cells reveals contributions of mTORC2 signaling to tuberous sclerosis complex. Hum Mol Genet 2013;22:140–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ess KC, Kamp CA, Tu BP, Gutmann DH. Developmental origin of subependymal giant cell astrocytoma in tuberous sclerosis complex. Neurology 2005;64:1446–1449. [DOI] [PubMed] [Google Scholar]
- 21. Fu C, Cawthon B, Clinkscales W, et al. GABAergic interneuron development and function is modulated by the Tsc1 gene. Cereb Cortex 2011;22:2111–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wahl SE, McLane LE, Bercury KK, et al. Mammalian target of rapamycin promotes oligodendrocyte differentiation, initiation and extent of CNS myelination. J Neurosci 2014;34:4453–4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lawson CL, Hanson RH. Solving least squares problems. Englewood Cliffs, NJ: Prentice‐Hall, 1974. [Google Scholar]
- 24. Lebel RM, Wilman AH. Transverse relaxometry with stimulated echo compensation. Magn Reson Med 2010;64:1005–1014. [DOI] [PubMed] [Google Scholar]
- 25. Prasloski T, Madler B, Xiang QS, et al. Applications of stimulated echo correction to multicomponent T2 analysis. Magn Reson Med 2012;67:1803–1814. [DOI] [PubMed] [Google Scholar]
- 26. MacKay A, Whittall K, Adler J, et al. In vivo visualization of myelin water in brain by magnetic resonance. Magn Reson Med 1994;31:673–677. [DOI] [PubMed] [Google Scholar]
- 27. Maedler B, Drabycz SA, Kolind SH, et al. Is diffusion anisotropy an accurate monitor of myelination? Correlation of multicomponent T‐2 relaxation and diffusion tensor anisotropy in human brain. Magn Reson Imaging 2008;26:874–888. [DOI] [PubMed] [Google Scholar]
- 28. O'Meara RW, Ryan SD, Colognato H, Kothary R. Derivation of enriched oligodendrocyte cultures and oligodendrocyte/neuron myelinating co‐cultures from post‐natal murine tissues. J Vis Exp 2011;54:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Harrington EP, Zhao CO, Fancy SPJ, et al. Oligodendrocyte PTEN is required for myelin and axonal integrity, not remyelination. Ann Neurol 2010;68:703–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lappe‐Siefke C, Goebbels S, Gravel M, et al. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet 2003;33:366–374. [DOI] [PubMed] [Google Scholar]
- 31. Huang JX, Manning BD. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem Soc Trans 2009;37:217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cheng XX, Wang YP, He Q, et al. Bone morphogenetic protein signaling and olig1/2 interact to regulate the differentiation and maturation of adult oligodendrocyte precursor cells. Stem Cells 2007;25:3204–3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Way SW, Rozas NS, Wu HC, et al. The differential effects of prenatal and/or postnatal rapamycin on neurodevelopmental defects and cognition in a neuroglial mouse model of tuberous sclerosis complex. Hum Mol Genet 2012;21:3226–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Crawley JN. Designing mouse behavioral tasks relevant to autistic‐like behaviors. Ment Retard Dev Disabil Res Rev 2004;10:248–258. [DOI] [PubMed] [Google Scholar]
- 35. Scattoni ML, Crawley J, Ricceri L. Ultrasonic vocalizations: a tool for behavioural phenotyping of mouse models of neurodevelopmental disorders. Neurosci Biobehav Rev 2009;33:508–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Peters SU, Kaufmann WE, Bacino CA, et al. Alterations in white matter pathways in Angelman syndrome. Dev Med Child Neurol 2011;53:361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ye P, Li LQ, Richards RG, et al. Myelination is altered in insulin‐like growth factor‐I null mutant mice. J Neurosci 2002;22:6041–6051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dummler B, Tschopp O, Hynx D, et al. Life with a single isoform of Akt: mice lacking Akt2 and Akt3 are viable but display impaired glucose homeostasis and growth deficiencies. Mol Cell Biol 2006;26:8042–8051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Narayanan SP, Flores AI, Wang F, Macklin WB. Akt signals through the mammalian target of rapamycin pathway to regulate CNS myelination. J Neurosci 2009;29:6860–6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bercury KK, Dai J, Sachs HH, et al. Conditional ablation of raptor or Rictor has differential impact on oligodendrocyte differentiation and CNS myelination. J Neurosci 2014;34:4466–4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Goebbels S, Oltrogge JH, Wolfer S, et al. Genetic disruption of Pten in a novel mouse model of tomaculous neuropathy. EMBO Mol Med 2012;4:486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fraser MM, Bayazitov IT, Zakharenko SS, Baker SJ. Phosphatase and tensin homolog, deleted on chromosome 10 deficiency in brain causes defects in synaptic structure, transmission and plasticity, and myelination abnormalities. Neuroscience 2008;151:476–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Azim K, Butt AM. GSK3 beta negatively regulates oligodendrocyte differentiation and myelination in vivo. Glia 2011;59:540–553. [DOI] [PubMed] [Google Scholar]
- 44. Sabo JK, Cate HS. Signalling pathways that inhibit the capacity of precursor cells for myelin repair. Int J Mol Sci 2013;14:1031–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Weight over time in Tsc2 CKO mice (A, B). Average body (C), brain (D), and brain/body ratio (E) in a cohort of P60‐ to P97‐day‐old mice. n = 6, *P < 0.05, **P < 0.01, ***P = 0.0001 by paired two‐tailed t‐test.
Figure S2. Abnormal gait in Tsc2 CKO mice. To characterize gait in control (A) and CKO (B) mice, mice were run on a motorized treadmill (TreadScan 2.0, Clever Sys, Inc) and front and rear track width measured manually by a blinded examiner. CKO mice demonstrated a splayed gait of the rear legs which was evident by P30 and was not improved with rapamycin treatment from P30 to P60 (C). No significant differences were noted with front track width at P30–P45, though the width was significantly increased by P60 in CKO mice and was unaffected by rapamycin treatment. n = 6–7, *control versus CKO P < 0.05, #control RAPA versus CKO RAPA P < 0.05 by one‐way ANOVA.
Figure S3. Absence of communication deficits based on gender or maternal genotype or learning effect on rotarod. Ultrasonic vocalizations were used to determine the presence of a communication deficit in the Tsc2 Olig2 CKO mice pups from P4 to P11. Vocalizations tended to be slightly, but not significantly decreased in CKO pups from both control (A) and heterozygous (B) dams. Similar trends were seen with both male (C) and female (D) pups, though a significant difference in vocalizations was seen at P6/7 in male pups only, likely attributable to an increase in vocalizations in the control pups. n = 7–11 animals per group. *P < 0.05 by one‐way ANOVA versus the control vehicle group. In P60 control and CKO mice treated with vehicle or rapamycin, no differences in latency to fall from the rotarod were seen during training or on the day 3 testing (E). n = 8–15 animals per group. *P < 0.05 by one‐way ANOVA versus the control vehicle group.
Figure S4. Lineage tracing of Olig2+ cells in cortex in primary culture. Olig2+ targeted cells were identified with Ai14 (RFP) labeling and costaining will neuronal and glial markers in cortex of P20‐day‐old mice. Clear costaining with the neuronal marker NeuN (A) is not appreciated in cortex, whereas double labeling is seen with Gad67‐positive neurons (B, yellow arrow inset). Perinuclear RFP signal is seen wrapped by MBP+ and MAG (C, D). GFAP+ projections are seen surrounding the perinuclear RFP signal in cortex (E). Scale bar = 100 μm. Mixed neuronal and glial cultures from Olig2+ Ai14 mice were costained with NeuN, Gad67, and GFAP. No RFP signal is seen to colocalize with the pyramidal appearing NeuN+ cells (E, green circle), whereas as noted in cortex, smaller NeuN+ and Gad67+ cells do demonstrate a faint perinuclear RFP signal (E, F, yellow circle). Extensive RFP signal is seen diffusely in GFAP+ astrocytes with a more intense perinuclear signal (G). Scale bar = 200 μm.
Figure S5. Increased GFAP expression in Olig2‐PTEN CKO cortex and primary culture. To address the hypothesis that gliosis in the Olig2‐Tsc2 CKO mouse is responsible for the hypomyelination was asked whether gliosis was also present in the Olig2‐PTEN CKO which in contrast to the Tsc2 model has been shown to be hypermyelinated. Immunofluorsescence staining with GFAP and MBP in P20 Olig2‐PTEN CKO cortex demonstrates increased GFAP (B, E) expression and maintained MBP expression (A, D) in the CKO. Analysis of P20 Olig2‐PTEN cortical extracts shown in comparison to P30 Olig2‐Tsc2 cortical extracts (G), demonstrates increase GFAP expression along with preserved (MAG, MBP, CNPase) to increased (PLP) expression of myelin proteins. A similar increase in GFAP expression was seen in primary cultures of astrocytes derived from the Olig2‐PTEN mice (H).
Figure S6. Spinal cord hypomyelination and targeting of spinal cord motor neurons by Olig2. Similar to data by Lebrun‐Julien et al., with the Cre‐CNP‐Tsc1 mouse, expression of the myelin proteins MAG, CNPase, MBP, and PLP were reduced in extracts from P30 Olig2‐Tsc2 CKO spinal cord. Similar to findings in cortex, GFAP expression and S6 phosphorylation were significantly increased. n = 4 animals per group. *P < 0.05, **P < 0.01 (A, B). As has been reported previously, spinal motor neurons are targeted by Olig2 as demonstrated by the Ai14 reporter line (C, inset yellow arrow) with expression of RFP extending to a subset of myelinated axons in the sciatic nerve (D, inset red arrow). Scale bar = 100 μm.