

Abstract
OBJECTIVE
We performed a whole-genome expression study to clarify the nature of the biological processes mediating between inherited genetic variations and cognitive dysfunction in schizophrenia.
METHOD
Gene expression was assayed from peripheral blood mononuclear cells using Illumina Human WG6 v3.0 chips in twins discordant for schizophrenia or bipolar disorder and control twins. After quality control, expression levels of 18,559 genes were screened for association with California Verbal Learning Test (CVLT) performance, and any memory-related probes were then evaluated for variation by diagnostic status in the discovery sample (N = 190), and in an independent replication sample (N = 73). Heritability of gene expression using the twin design was also assessed.
RESULTS
After Bonferroni correction (p < 2.69 × 10−6), CVLT performance was significantly related to expression levels for 76 genes, 43 of which were differentially expressed in schizophrenia patients, with comparable effect sizes in the same direction in the replication sample. For 41 of these 43 transcripts, expression levels were heritable. Nearly all identified genes contain common or de novo mutations associated with schizophrenia in prior studies.
CONCLUSION
Genes increasing risk for schizophrenia appear to do so in part via effects on signaling cascades influencing memory. The genes implicated in these processes are enriched for those related to RNA processing and DNA replication and include genes influencing G-protein coupled signal transduction, cytokine signaling, and oligodendrocyte function.
Keywords: schizophrenia, gene expression, cognition, endophenotype, genome-wide association study
Schizophrenia Schizophrenia is a highly heritable, genetically complex, and heterogeneous psychiatric syndrome. Twin and family studies estimate 80–85% of variance in disease liability can be accounted for by genetic factors (Cannon, Kaprio, Lonnqvist, Huttunen, & Koskenvuo, 1998). Thus, substantial efforts have been dedicated towards uncovering genetic loci increasing risk for schizophrenia. Though many methodologies have been employed including genome-wide surveys of common genetic polymorphisms (Purcell et al., 2009; Ripke et al., 2013; Schizophrenia Working Group of the Psychiatric Genomics, 2014), exome sequencing investigating rare variants (Fromer et al., 2014; Purcell et al., 2014), gene expression analyses (Gardiner et al., 2013; Guillozet-Bongaarts et al., 2014; Gulsuner et al., 2013), and integration of multiple methods (Hertzberg, Katsel, Roussos, Haroutunian, & Domany, 2015; Luo et al., 2015; van Eijk et al., 2014), nearly all gene-finding efforts in schizophrenia to date have employed diagnostic level classification as the phenotype of interest. However, there is substantial evidence that risk-increasing genes confer risk via impacting intermediate levels of impairment, such as cognitive dysfunction, suggesting that these may also be useful phenotypic targets in gene discovery studies (Cannon & Keller, 2006; Greenwood, Light, Swerdlow, Radant, & Braff, 2012; Lencz et al., 2014; Tan, Callicott, & Weinberger, 2008; Toulopoulou et al., 2007).
Compared to the diagnosis of schizophrenia, which could imply dysfunction across a host of brain systems and signaling pathways, a given endophenotype provides a more constrained framework within which to interpret the functional-biological relevance of statistically identified genes (Heck et al., 2014; Ibrahim-Verbaas et al., 2015; Lencz et al., 2014). One of the most reliable endophenotypes in schizophrenia – and most profound areas of cognitive impairment – is verbal memory (Cannon et al., 2000; Greenwood et al., 2013; Schaefer, Giangrande, Weinberger, & Dickinson, 2013; van Erp et al., 2008). The California Verbal Learning Test (CVLT), a list-learning exercise, is a robust measure of memory impairment in schizophrenia (Haut, in prep; Stone et al., 2011; van Erp et al., 2008), is heritable (Carmelli, Swan, DeCarli, & Reed, 2002; Greenwood et al., 2007; Kremen et al., 2014; Panizzon et al., 2011), and shows intermediate levels of affection among relatives of patients with schizophrenia in twin and family studies (Greenwood et al., 2013; van Erp et al., 2008), supporting its role as a candidate endophenotype in genetic studies.
Initial molecular genetic investigations of endophenotypes for schizophrenia have provided evidence that genes associated with putative cognitive endophenotypes at least partially overlap with those associated with schizophrenia (Heck et al., 2014; Lencz et al., 2014). However, so far this approach has been used largely to demonstrate the broad, shared genetic etiology between these phenotypes, rather than to identify specific genetic loci impinging on both, which is critical for elucidating the functional-physiologic significance of the underlying genetic associations. Furthermore, all of these studies have assayed common polymorphisms. Over 90% of common polymorphisms previously related to psychiatric illness are located in regulatory regions of DNA, rather than in coding regions where mutations more directly affect protein structure, suggesting that many of these loci may exert their effects via regulation of gene expression (Kim et al., 2014; Maurano et al., 2012). Thus, examining gene expression in relation to a cognitive endophenotype for schizophrenia may yield insights into genotypic effects on disease status and help to elucidate mechanisms by which cognitive dysfunction manifests in this disease.
In the current study, we used a discordant twin design to identify genes differentially expressed in relation to verbal memory performance, a cognitive endophenotype for schizophrenia. Gene expression was assayed from peripheral blood mononuclear cells (PBMCs) – the most feasible way to assess expression levels in living patients. Gene expression in PBMCs is broadly heritable and correlates with central nervous system expression patterns (Cheung et al., 2003). While it is likely that some genes influencing memory performance will not be schizophrenia-related, given the endophenotypic pattern of the CVLT, which suggests shared genetic etiology between memory and schizophrenia, we expected some proportion of these memory-related genes to show differential expression by diagnostic status (Glahn et al., 2007; Greenwood et al., 2007; van Erp et al., 2008). This subset would represent a selection of genes potentially involved in those systems underlying memory impairment in schizophrenia. A twin design further allowed us to test the heritability of expression for each of these genes, dissociating potentially etiologically relevant genetic alterations from disease-related secondary effects.
Methods
Participants
Twins born between 1940–1975 were identified on a population basis through the Swedish Twin Registry and screened for psychiatric status by linkage with the Swedish Hospital Discharge Registry (Lichtenstein et al., 2002). Pairs in which one or both co-twins had a diagnosis of schizophrenia or bipolar disorder were considered potential index pairs, and pairs in which neither twin had a history of hospitalization for either disorder were considered potential control pairs. Once recruited, all subjects were assessed by a psychiatrist using the Structured Clinical Interview for DSM-IV (SCID-IV) (Spitzer, Williams, Gibbon, & First, 1992). Diagnoses were assigned according to Diagnostic and Statistical Manual for Mental Disorders, Fourth Edition (DSM-IV) criteria, using a consensus procedure taking into account the interview results as well as hospitalization and treatment records (American Psychiatric Association, 1994). Exclusion criteria included neurological disorder, history of head injury with loss of consciousness, mental retardation, substance dependence within the past six months, and inability to read or comprehend Swedish. Control twins were additionally excluded if they had a personal history of schizophrenia, schizoaffective disorder, or bipolar disorder. Patient twin pairs were included if the proband was given a consensus diagnosis of either schizophrenia / schizoaffective disorder or bipolar I disorder. Individuals with schizoaffective disorder were included in the schizophrenia group. All included probands were clinically stable at the time of testing and the blood draw, which occurred contemporaneously. Control twin pairs matched to the index pairs for age, sex, and zygosity were recruited from the Swedish Twin Registry. Zygosity was determined by DNA using a 46 single nucleotide polymorphism panel. In total, 190 individuals who were interviewed, underwent cognitive testing, and provided blood samples that were assayed for gene expression, were included in the discovery sample for the present report. Of these individuals, 36 had a diagnosis of schizophrenia, 34 were unaffected co-twins of patients with schizophrenia, 23 individuals had a diagnosis of bipolar disorder, 16 were unaffected co-twins of patients with bipolar disorder, and 81 individuals were control twins. Included in the control twin group were 20 pairs (N = 39) where one or both individuals had a lifetime history of major depressive disorder. Across the sample, 46% of participants were male and subjects were on average 49.9 years old (95% CIs [48.5, 51.4]). No significant differences between diagnostic groups in age, sex, or zygosity were observed (p > .05). Group-specific demographic characteristics for schizophrenia patients, co-twins, and controls are listed in Table 1.
Table 1.
Demographic Information for Swedish and Finnish Twins
| Swedish Sample | ||||||
|---|---|---|---|---|---|---|
| Patient Group | N | Age (years) | Sex (% male) | Zygosity (% MZ) | SANS | SAPS |
| SZ | 36 | 50.2 [47.1, 53.4] | 56% | 42% | 40.7 [32.7, 48.7] | 21.6 [14.5, 28.6] |
| SZ Co-twins | 34 | 51.1 [47.8, 54.4] | 53% | 26% | 9.6 [4.4, 14.7] | 0.6 [0, 1.3] |
| Controls | 81 | 49.6 [47.4, 51.9] | 49% | 46% | 5.2 [3.1, 7.4] | 0.4 [0.1, 0.8] |
| Statistic | -- | 1.091 | 0.409 | 3.701 | 73.25 | 58.299 |
| Significance | -- | 0.343 | 0.815 | 0.157 | <.0001 | <.0001 |
| Finnish Sample | ||||||
| Patient Group | N | Age (years) | Sex (% male) | Zygosity (% MZ) | SANS | SAPS |
| SZ | 18 | 47.3 [44.1, 50.6] | 28% | 33% | 31.8 [13.2, 50.4] | 27.1 [17.9, 36.3] |
| SZ Co-twins | 18 | 47.3 [44.1, 50.6] | 28% | 33% | -- | -- |
| Controls | 37 | 49.4 [47.3, 51.4] | 32% | 41% | -- | -- |
| Statistic | -- | 0.498 | 0.188 | 7.619 | -- | -- |
| Significance | -- | 0.490 | 0.910 | 0.107 | -- | -- |
Note. Group means, 95% confidence intervals, and F statistics from linear mixed effect models of diagnosis on age, SANS, and SAPS, with family ID as a random variable, were reported when possible. Chi-square values were reported for sex and zygosity. SZ = schizophrenia patients; SANS = Scale for the Assessment of Negative Symptoms; SAPS = Scale for the Assessment of Positive Symptoms.
An independent twin sample from Finland was used to test for replication. Information about recruitment, clinical evaluation, and cognitive testing employed for this study was described in detail elsewhere (Oresic et al., 2012). From this study, 18 schizophrenia patients, 18 co-twins, and 37 control twins provided blood samples for gene expression (N = 73). Although cognitive test data were available on many of these subjects, the testing had been performed 2–10 years prior to the blood draw, and thus the test data were not used in the present analyses. Demographic information for these subjects is summarized in Table 1.
Cognitive Assessment
Swedish participants underwent a standard neuropsychological battery including the Vocabulary and Block Design subtests of the Wechsler Abbreviated Scale of Intelligence Scale (WASI) and the California Verbal Learning Test (CVLT). These measures had previously been translated into Swedish. The CVLT is a measure of verbal learning and memory. Individuals were read a list of 16 words and then asked to recall as many of the words as they could remember. This was repeated across four subsequent trials. The sum of words recalled across all five learning trials was used as the performance metric in all analyses. Though previous studies have demonstrated that CVLT performance is both heritable and related to schizophrenia (Glahn et al., 2007; Greenwood et al., 2007; Stone et al., 2015; van Erp et al., 2008), we conducted analyses to confirm these patterns in our sample as well. Diagnostic effects on CVLT performance were ascertained using a mixed effect ANOVA model, where family ID was included as a random variable, as programmed in R using the nlme package (Pinheiro, Bates, DebRoy, Sarkar, & Team, 2015). Structural equation modeling was performed to assess genetic and environmental contributions to CVLT performance in Mx (Neale, Boker, Xie, & Maes, 2003) including all subjects within the sample (69 monozygotic twin pairs, 89 dizygotic twin pairs), rather than the subset with RNA expression data (for a full description of the sample, see Higier et al., 2014). Details of this procedure were identical to those for evaluating heritability of gene expression described below, substituting CVLT as the variable of interest.
We additionally calculated a measure of general cognitive ability to determine whether any effects were specific to memory. Z-scores based on the means and standard deviations of control subjects were generated for the Vocabulary and Block Design subtests of the WASI. The average of these scores was used as our measure of global cognitive ability. Diagnostic effects on general ability, as well as heritability, were assessed as described for CVLT performance.
RNA Microarray Analysis
For both Swedish and Finnish samples, RNA was extracted from PBMCs drawn from a 10 ml blood sample (ABI Tempus system). RNA aliquots at 100 ng/µl were sent to the UCLA Biological Samples Processing Core for analysis. Technical replicates were run on all samples using the Illumina Human WG6 v3.0 chip, and correlations between these samples for each person were evaluated. All samples had a useable quantity of RNA, and all subjects had good correlations between their technical replicate samples (ICCs ranged from 0.902 – 0.998). Samples were preprocessed using the Illumina Bead Studio package. Technical replicates were averaged to produce one vector per person, and samples were log-transformed to ensure normalization of the sample distributions. The Illumina chip indexed a total of 24,526 markers. After rank normalization and background subtraction, 5,967 probes were excluded based on low signal strength (not significantly greater than 0 at a Bonferroni-corrected α < 0.05), leaving 18,559 genes to be carried forward for analysis of association with CVLT performance, general ability, and diagnosis.
Genome-Wide Association between Expression Levels and Memory Performance
All patients, co-twins, and controls from the Swedish sample (N = 190) were examined to test associations between peripheral gene expression and CVLT performance. Mixed effect linear regression models of gene expression on CVLT performance were run for each probe. Family identity was included as a random variable, and age and sex were included as covariates. Gene markers that met the Bonferroni-corrected significance level of α = 2.69 × 10−6 (i.e., .05/18,559) were considered hits. We conducted a genome-wide assay of global cognitive ability using the same design to ascertain if any effects were specific to memory. The same Bonferroni-corrected alpha level was considered here. Analyses were conducted in R using the lme4 package (Bates, Maechler, Bolker, & Walker, 2014) and pbkrtest package (Halekoh & Hojsgaard, 2014).
Associations between Expression Levels and Diagnostic Status
Genes found to be related to memory performance were further examined to determine if their expression patterns varied by diagnostic group. Expression levels for schizophrenia patients, schizophrenia co-twins, and controls (N = 151) were analyzed using mixed effect ANOVAs with diagnosis as the predictor, and family included as a random variable. Bipolar patients and co-twins were not included, as these groups were not represented in the Finnish twin sample available for use in testing replication of the associations. For genes whose expression varied significantly by diagnosis, fixed effect comparisons between schizophrenia patients and each of the non-patient groups (i.e., unaffected co-twins and controls) were examined. The significance threshold for the analyses by diagnosis was kept at α = 0.05, with no correction, as we had previously applied a conservative Bonferroni threshold to limit the scope to those transcripts relating to memory performance, and we had a second, independent sample in which to test replication of any diagnostic differences.
Genes differentially expressed with respect to diagnosis were examined in an independent sample of schizophrenia patients, co-twins, and controls from Finland (N = 73). Mixed effect ANOVAs, including family as a random variable, were conducted to examine whether expression of these genes varied by diagnostic status. As we predicted that the diagnostic effects would mirror the discovery sample with respect to direction, we used a one-tailed α = 0.05 significance threshold (with no correction for multiple comparisons at this replication stage). Statistical analyses were conducted in R using the nlme package (Pinheiro et al., 2015). Power analyses, when relevant, were conducted using G*Power 3.1 (Faul, Erdfelder, Lang, & Buchner, 2007) to estimate requisite sample sizes for detecting observed effect sizes given 80% power (1 - β) at α = .05.
Heritability Analyses of Gene Expression
For those RNA probes found to be related to both memory and schizophrenia, we used structural equation modeling to compare gene expression covariance in monozygotic (MZ) twin pairs (N = 26 pairs) to dizygotic (DZ) twin pairs (N = 37 pairs) in order to assess the relative contribution of additive genetics (A), dominant genetics (D), common environment (C), and unique environment (E) (Plomin, DeFries, Knopik, & Neiderhiser, 2013). Models were run in Mx, which uses the maximum likelihood estimate to fit models to covariance matrices (Neale et al., 2003). Best fitting models were selected based on the Root Mean Square Error of Approximation (RMSEA) and the Akaike Information Criterion (AIC), for both of which lower values reflect better fit (Akaike, 1974). AIC also penalizes the addition of parameters, so best fit models by this metric are also relatively more parsimonious (Kline, 2011). We also tested the significance of the genetic and familial environmental contributions to each model by means of chi-square difference tests (i.e., directly comparing models in which each of these parameters was present versus absent).
Gene Classification
Illumina probe IDs were converted to standard gene symbols using the Bioconductor R package illuminaHumanv4.db (Dunning, Lynch, & Eldridge). Gene symbols for all probes significantly associated with both CVLT performance and diagnostic status were input into the Database for Annotation, Visualization, and Integrated Discovery (DAVID) for functional annotation clustering (Huang da, Sherman, & Lempicki, 2009). This function can group genes together based on classification terms from multiple gene ontology databases; for the current study, we included the default sources and clustering criteria. Clusters identified by this tool were assigned enrichment values and corresponding significance statistics based on the number of genes in the input list matching the included annotation terms relative to the number expected by chance.
Results
Cognitive Assessment
Consistent with previous studies (Glahn et al., 2007; Greenwood et al., 2007; Stone et al., 2015; van Erp et al., 2008), we found that CVLT performance was impaired in schizophrenia, correlated with general cognitive ability, and partially heritable. CVLT performance (total words recalled across the 5 learning trials; maximum score = 80) was significantly associated with diagnostic status, F(5, 77) = 10.66, p < .0001, marginal R2 = .220. Schizophrenia patients on average scored significantly lower (M = 38.8, 95% CIs [34.5, 43.2]) than all other groups (schizophrenia co-twins: M = 50.8, 95% CIs [46.8, 54.8]; bipolar patients: M = 51.6, 95% CIs [46.9, 56.2]; bipolar co-twins: M = 57.8, 95% CIs [53.9, 61.6]; controls (history of depression): M = 50.4, 95% CIs [47.0, 53.8]; controls (no history of depression): M = 55.7, 95% CIs [52.7, 58.7]). CVLT performance also accounted for a significant portion of the variance in general cognitive ability, F(1, 71) = 26.65, p < .0001, marginal R2 = .108. The best fitting model of genetic and environmental contributions to variance in CVLT performance included a significant genetic component, accounting for 53% of the variance (χ2 (4, N = 316) = 1.885, p = .757, RMSEA = 0, AIC = −6.115), which is consistent with the correlations within zygosity groups (MZ: r(67) = .566, p = 3.95e−7; DZ: r(87) = .237, p = .025).
General cognitive ability was also significantly associated with diagnostic status, though schizophrenia patients showed a relatively less profound deficit compared to that observed for verbal memory, F(5, 67) = 3.11, p = .0138, marginal R2 = .061. Schizophrenia patients on average scored significantly lower (M = −.49, 95% CIs [−.89, −.10]) than schizophrenia co-twins (M = −.27, 95% CIs [−.67, −.12]) and controls (history of depression: M = −.01, 95% CIs [−.34, .33]; no history of depression: M = .01, 95% CIs [−.23, .25]). No other groups scored significantly differently than controls (bipolar patients: M = −.47, 95% CIs [−.98, .03]; bipolar co-twins: M = .14, 95% CIs [−.50, .78]). The best fitting model of genetic and environmental contributions to variance in general ability included a significant genetic component, accounting for 73% of the variance (χ2 (4, N = 292) = 0.626, p = .960, RMSEA = 0, AIC = −7.374), consistent with within zygosity correlations (MZ: r(60) = .719, p = 4.77e−11; DZ: r(82) = .434, p = 3.53e−5).
Genome-Wide Association between Expression Levels and Memory Performance
The expression levels for 76 of the 18,559 probes retained following quality control were significantly associated with CVLT performance after Bonferroni correction (p < 2.69 × 10−6). For all of these probes, higher expression was associated with higher CVLT performance. For the subset of these genes also related to diagnostic status, the t and p values for the fixed effect of each probe on CVLT are reported in Table 2. Effects for all 76 are reported in the Supplementary Materials (S1). The average effect size for the 76 probes significantly associated with CVLT (partial R2 = .131, 95% CIs [.128, .134]) was reduced only moderately when excluding schizophrenia and bipolar patients (partial R2 = .095, 95% CIs [.090, .099]), indicating that for the most part, the effects are independent of factors secondary to disease expression, such as chronicity (S1). Additionally, among the schizophrenia probands, gene expression for all markers associated with CVLT was not significantly correlated with positive symptoms, as measured by the Scale for the Assessment of Positive Symptoms, p’s > 0.25, nor did it differ between probands on (N = 19) or off (N = 17) antipsychotic medication, p’s > 0.25, suggesting these effects are not secondary to clinical state or medication exposure either. No genes were significantly related to global cognitive ability after correcting for multiple comparisons.
Table 2.
Heritability Estimates and Statistical Associations between Gene Expression, CVLT, and Diagnostic Status
| Swedish Discovery Sample | Finnish Replication Sample | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Association with CVLT | Association with Diagnosis | Heritability | Association with Diagnosis | |||||||||||
| Illumina Probe | Gene Symbol | t | df | pBon | Partial R2 | F | df | p | Partial R2 | h2 | F | df | p | Partial R2 |
| ILMN_1752111 | SMARCAL1 | 5.15 | 171.82 | 0.013 | 0.134 | 6.26 | 2, 61 | 0.003 | 0.078 | 0.47 | 1.95 | 2, 42 | 0.155 | 0.046 |
| ILMN_2224143 | MCM3 | 5.35 | 167.21 | 0.005 | 0.146 | 6.25 | 2, 61 | 0.003 | 0.080 | 0.47 | 2.61 | 2, 42 | 0.085 | 0.062 |
| ILMN_1728298 | SBK1 | 5.03 | 178.74 | 0.022 | 0.124 | 6.07 | 2, 61 | 0.004 | 0.075 | 0.33 | 2.78 | 2, 42 | 0.073 | 0.063 |
| ILMN_1777740 | THEM6 | 5.02 | 183.32 | 0.023 | 0.121 | 5.99 | 2, 61 | 0.004 | 0.075 | 0.47 | 2.25 | 2, 42 | 0.118 | 0.055 |
| ILMN_1750800 | ACO1 | 5.33 | 174.07 | 0.006 | 0.140 | 5.81 | 2, 61 | 0.005 | 0.075 | 0.54 | 1.98 | 2, 42 | 0.151 | 0.047 |
| ILMN_2073012 | TMEM203 | 5.06 | 173.37 | 0.020 | 0.128 | 5.75 | 2, 61 | 0.005 | 0.075 | 0.35 | 2.19 | 2, 42 | 0.124 | 0.049 |
| ILMN_1676745 | ZNF142 | 5.32 | 187.98 | 0.005 | 0.131 | 5.74 | 2, 61 | 0.005 | 0.074 | 0.48 | 2.06 | 2, 42 | 0.141 | 0.048 |
| ILMN_1787461 | RUNX3 | 5.46 | 186.37 | 0.003 | 0.138 | 5.49 | 2, 61 | 0.006 | 0.068 | 0.00 | 2.59 | 2, 42 | 0.087 | 0.059 |
| ILMN_1775522 | MAGED1 | 6.14 | 178.43 | < .0001 | 0.174 | 5.32 | 2, 61 | 0.007 | 0.073 | 0.52 | 3.02 | 2, 42 | 0.060 | 0.072 |
| ILMN_2412549 | GAR1 | 5.31 | 173.46 | 0.006 | 0.140 | 5.26 | 2, 61 | 0.008 | 0.070 | 0.45 | 2.74 | 2, 42 | 0.076 | 0.064 |
| ILMN_1689953 | CD81 | 5.49 | 183.50 | 0.002 | 0.141 | 5.22 | 2, 61 | 0.008 | 0.066 | 0.41 | 1.45 | 2, 42 | 0.245 | 0.034 |
| ILMN_1701731 | AKR1B1 | 5.04 | 182.99 | 0.021 | 0.122 | 4.86 | 2, 61 | 0.011 | 0.065 | 0.41 | 1.88 | 2, 42 | 0.165 | 0.042 |
| ILMN_1686871 | PARP1 | 5.70 | 168.60 | 0.001 | 0.161 | 4.68 | 2, 61 | 0.013 | 0.060 | 0.40 | 2.49 | 2, 42 | 0.095 | 0.060 |
| ILMN_1786024 | POLR3H | 5.01 | 182.79 | 0.024 | 0.121 | 4.56 | 2, 61 | 0.014 | 0.049 | 0.58 | 2.41 | 2, 42 | 0.102 | 0.058 |
| ILMN_1733696 | IMP3 | 5.32 | 164.72 | 0.006 | 0.147 | 4.56 | 2, 61 | 0.014 | 0.058 | 0.24 | 1.82 | 2, 42 | 0.174 | 0.042 |
| ILMN_1721978 | CARD11 | 5.27 | 183.58 | 0.007 | 0.131 | 4.44 | 2, 61 | 0.016 | 0.050 | 0.46 | 2.86 | 2, 42 | 0.069 | 0.068 |
| ILMN_1700604 | RBM14 | 4.88 | 172.74 | 0.045 | 0.121 | 4.41 | 2, 61 | 0.016 | 0.052 | 0.31 | 1.38 | 2, 42 | 0.262 | 0.034 |
| ILMN_2097793 | ZBTB4 | 5.32 | 177.36 | 0.006 | 0.138 | 4.38 | 2, 61 | 0.017 | 0.056 | 0.25 | 2.52 | 2, 42 | 0.092 | 0.059 |
| ILMN_1673991 | ATIC | 5.92 | 187.94 | < .0001 | 0.157 | 4.28 | 2, 61 | 0.018 | 0.056 | 0.34 | 3.09 | 2, 42 | 0.056 | 0.071 |
| ILMN_1766010 | YARS | 5.31 | 172.45 | 0.006 | 0.140 | 4.21 | 2, 61 | 0.019 | 0.053 | 0.44 | 2.67 | 2, 42 | 0.081 | 0.063 |
| ILMN_2162989 | TMEM189 | 4.88 | 187.02 | 0.042 | 0.113 | 4.12 | 2, 61 | 0.021 | 0.049 | 0.53 | 1.69 | 2, 42 | 0.197 | 0.041 |
| ILMN_1665943 | MAP4K1 | 5.29 | 187.88 | 0.006 | 0.130 | 4.10 | 2, 61 | 0.021 | 0.054 | 0.38 | 2.00 | 2, 42 | 0.149 | 0.046 |
| ILMN_2334296 | IL18BP | 5.18 | 187.67 | 0.011 | 0.125 | 4.08 | 2, 61 | 0.022 | 0.046 | 0.50 | 2.15 | 2, 42 | 0.129 | 0.047 |
| ILMN_2044453 | LPAR5 | 4.96 | 187.39 | 0.029 | 0.116 | 4.05 | 2, 61 | 0.022 | 0.053 | 0.51 | 1.22 | 2, 42 | 0.305 | 0.030 |
| ILMN_2153787 | ARRDC5 | 4.87 | 185.73 | 0.043 | 0.113 | 4.03 | 2, 61 | 0.023 | 0.055 | 0.54 | 1.85 | 2, 42 | 0.169 | 0.046 |
| ILMN_1788538 | NCALD | 4.99 | 187.95 | 0.025 | 0.117 | 3.89 | 2, 61 | 0.026 | 0.050 | 0.47 | 2.23 | 2, 42 | 0.120 | 0.055 |
| ILMN_1670948 | TSR2 | 4.98 | 185.88 | 0.026 | 0.118 | 3.77 | 2, 61 | 0.029 | 0.048 | 0.42 | 1.67 | 2, 42 | 0.201 | 0.039 |
| ILMN_1671257 | DKC1 | 5.33 | 169.07 | 0.006 | 0.144 | 3.69 | 2, 61 | 0.031 | 0.050 | 0.45 | 1.87 | 2, 42 | 0.167 | 0.045 |
| ILMN_1663954 | NELFCD | 5.18 | 185.13 | 0.011 | 0.127 | 3.68 | 2, 61 | 0.031 | 0.053 | 0.49 | 2.32 | 2, 42 | 0.111 | 0.054 |
| ILMN_1761479 | ZC3HC1 | 5.49 | 156.95 | 0.003 | 0.161 | 3.60 | 2, 61 | 0.033 | 0.046 | 0.52 | 2.23 | 2, 42 | 0.120 | 0.053 |
| ILMN_2183687 | LIME1 | 4.98 | 183.23 | 0.027 | 0.119 | 3.60 | 2, 61 | 0.033 | 0.045 | 0.00 | 1.97 | 2, 42 | 0.152 | 0.050 |
| ILMN_1801348 | GOT2 | 5.15 | 178.21 | 0.013 | 0.129 | 3.54 | 2, 61 | 0.035 | 0.044 | 0.51 | 2.78 | 2, 42 | 0.073 | 0.062 |
| ILMN_1779185 | SPECC1L | 4.99 | 162.99 | 0.028 | 0.133 | 3.46 | 2, 61 | 0.038 | 0.042 | 0.42 | 2.28 | 2, 42 | 0.115 | 0.052 |
| ILMN_1753885 | YTHDF1 | 5.05 | 185.90 | 0.019 | 0.121 | 3.38 | 2, 61 | 0.040 | 0.046 | 0.29 | 1.91 | 2, 42 | 0.161 | 0.046 |
| ILMN_1740265 | ACOT7 | 4.96 | 185.07 | 0.029 | 0.117 | 3.38 | 2, 61 | 0.041 | 0.044 | 0.49 | 2.32 | 2, 42 | 0.111 | 0.054 |
| ILMN_2306189 | MAGED1 | 4.99 | 187.34 | 0.025 | 0.117 | 3.36 | 2, 61 | 0.041 | 0.050 | 0.50 | 2.66 | 2, 42 | 0.082 | 0.065 |
| ILMN_1748894 | GTPBP3 | 4.87 | 173.16 | 0.047 | 0.120 | 3.35 | 2, 61 | 0.042 | 0.044 | 0.49 | 2.48 | 2, 42 | 0.096 | 0.059 |
| ILMN_1685580 | CBLB | 5.47 | 184.39 | 0.003 | 0.140 | 3.33 | 2, 61 | 0.042 | 0.043 | 0.57 | 2.34 | 2, 42 | 0.109 | 0.058 |
| ILMN_2343097 | NCALD | 5.12 | 188.00 | 0.014 | 0.122 | 3.33 | 2, 61 | 0.043 | 0.044 | 0.43 | 2.47 | 2, 42 | 0.097 | 0.062 |
| ILMN_1708328 | RAB11FIP3 | 5.01 | 185.32 | 0.023 | 0.119 | 3.32 | 2, 61 | 0.043 | 0.044 | 0.52 | 2.11 | 2, 42 | 0.134 | 0.050 |
| ILMN_1751338 | NUP133 | 5.20 | 167.84 | 0.011 | 0.139 | 3.31 | 2, 61 | 0.043 | 0.047 | 0.49 | 2.01 | 2, 42 | 0.146 | 0.047 |
| ILMN_1744068 | ELP3 | 5.25 | 173.50 | 0.008 | 0.137 | 3.20 | 2, 61 | 0.048 | 0.043 | 0.51 | 1.90 | 2, 42 | 0.162 | 0.043 |
| ILMN_1763460 | NHP2L1 | 5.02 | 183.07 | 0.022 | 0.121 | 3.19 | 2, 61 | 0.048 | 0.041 | 0.27 | 1.80 | 2, 42 | 0.179 | 0.039 |
Note. Seventy-six genes were associated with CVLT after correcting for multiple comparisons (p < 2.69 × 10−6) and, of those, 43 were also associated with diagnostic status (p < .05). For CVLT- and diagnosis-associated genes, the effect of each probe on CVLT adjusted for age and sex in the discovery sample, as well as the overall effect of diagnosis on each probe in both samples, is reported. Broad heritability estimates from best fitting models are listed as well. Italics denote heritability models where the chi-square was not significant, indicating good model fit. For all but four of the heritable genes (IMP3, ZBTB4, NHP2L1, YTHDF1), removal of the genetic term resulted in a significant decrement in model fit, p < .05.
Associations between Expression Levels and Diagnostic Status
Of the 76 probes whose expression correlated with CVLT performance, 43 showed expression patterns that varied significantly in relation to diagnosis (Figure 1; Table 2). In fixed effect comparisons between schizophrenia patients and each of the non-patient groups (i.e., unaffected co-twins and controls), patients showed significantly lower expression than controls at all 43 probes, and significantly lower expression than co-twins at 19 of the 43 probes (p < 0.05; S3). As all 76 probes were positively correlated with CVLT performance, reduced expression in patients is consistent with behavioral performance deficits observed in this group. Unaffected co-twins were intermediate between probands and controls on mean expression values for most of these genes, but did not differ significantly from controls for any. See Supplementary Materials (S2 and S3) for further descriptive information on the distributions of expression levels by diagnostic group.
Figure 1.
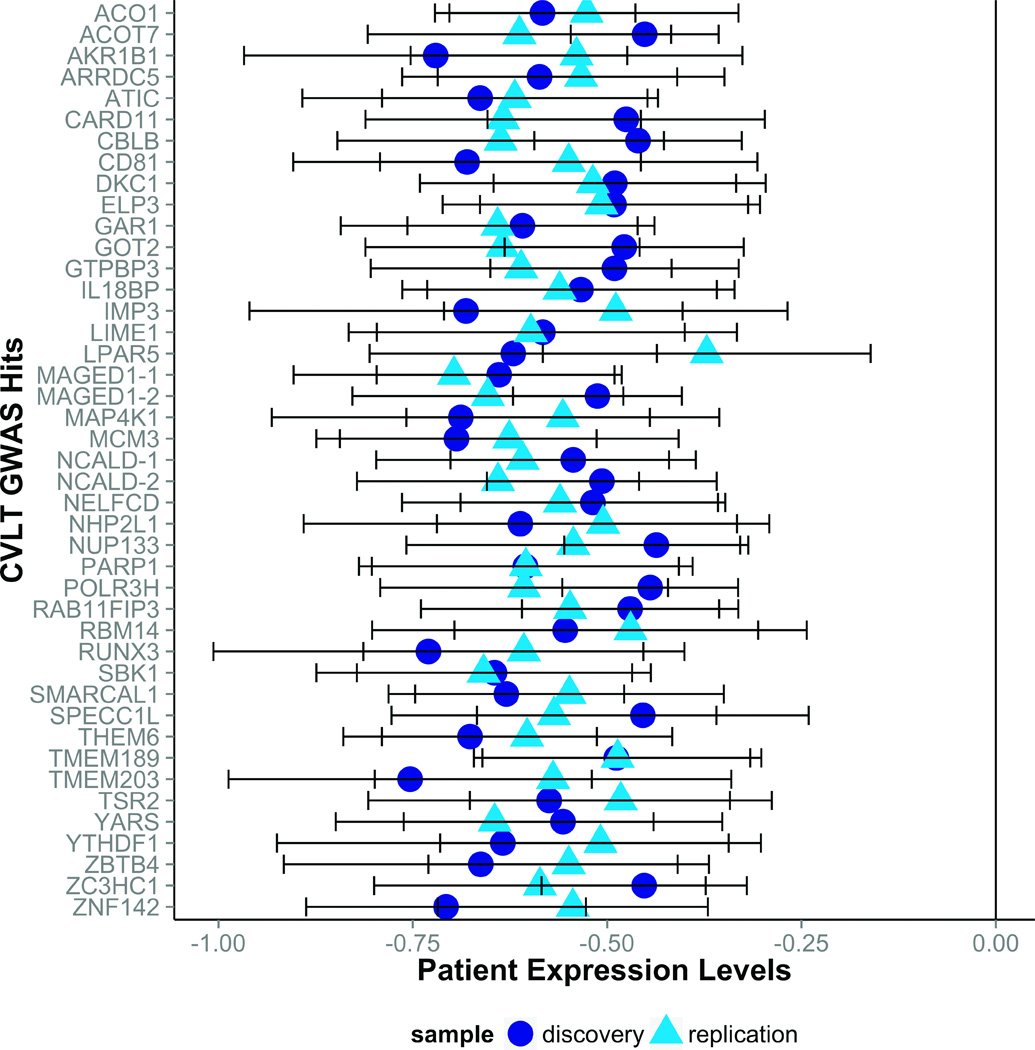
Forty-three memory-related genes were under-expressed in patients with schizophrenia as compared to controls. Gene expression values for schizophrenia patients in the discovery (Swedish) and replication (Finnish) samples are displayed as z-scores normed within sample to the control mean and standard deviation for each gene. For some genes, two corresponding probes were identified; in these cases, probes were arbitrarily denoted gene-1 or gene-2. Error bars represent +/− standard error.
Fourteen of the 43 probes related to diagnosis in the discovery sample (Swedish twins) were confirmed as showing significantly lower expression in patients with schizophrenia compared to controls in the replication sample (Finnish twins), p < 0.05, one-tailed (S4). However, the effect sizes for all 43 genes in the replication sample (N = 73) were similar in magnitude and in the same direction (i.e., reduced expression in patients) as those observed in the larger Swedish sample (N = 151). A power analysis revealed that, given the observed effect sizes for these 43 probes in the Finnish sample, if the replication sample had been comparable in size to the discovery sample (i.e., N = 151 instead of 73), the diagnostic effects would have been statistically significant for all 43 probes in the replication sample (Figure 1) (Faul et al., 2007). Patients with schizophrenia also had significantly lower expression than their unaffected co-twins for 13 probes, p < 0.05, one-tailed, but again effect sizes were similar in magnitude and in the same direction as in the discovery sample across all of the genes. Unaffected co-twins and controls did not differ significantly in expression levels. Mean expression levels for each group in the replication sample are reported in Supplementary Materials (S4).
Heritability Analyses of Gene Expression
Heritability was assessed for the 43 probes related to both memory performance and schizophrenia. For 41 of the 43 probes, the best fitting model included a genetic component, ranging from 24% to 58% (M = 44%, 95% CIs [42%, 47%]), indicating moderate heritability of expression levels of the genes relating to both memory and schizophrenia. All of these models included a genetic term (A, D) and a unique environment term (E), but no shared environment term (C). As such, the genetic term was reported in Table 2 and the E term was simply the remaining variance. Heritability models are considered to fit the data well if there is no significant difference, as tested using chi-square, between the model and the measured data (Plomin et al., 2013). 40 of the 43 best-fitting models had non-significant chi-square values, p > .05 (Table 2). For each model, we also explicitly tested whether the genetic and common environmental terms accounted for significant proportions of variance by performing a chi-square difference test with the genetic (A or D) or comment environmental (C) terms in and out of the model. The C term was not significant for any of the 43 probes or CVLT performance, p > .05. Conversely, exclusion of the genetic term resulted in a significant decrement in model fit for CVLT and 37 of the 41 heritable markers, p < .05 (Table 2), confirming that the observed data are best explained by a model including a genetic factor.
Gene Classification
The genes associated with both CVLT and diagnostic status were input into DAVID for functional annotation clustering. Twelve clusters were identified, two of which exceeded the significance threshold for enrichment, p < .05; these two clusters contained genes related to RNA processing and DNA replication (S5). Cluster assignments for each of the genes are reported in Table 3. All clusters, included annotation terms, number of genes assigned to each term, cluster enrichment values, and cluster significance values are listed in the Supplementary Materials (S5).
Table 3.
Biological Relevance of CVLT-Associated Genes Expressed Differentially by Diagnostic Status
| Illumina Probe | Chr | Gene Symbol | Gene Description | Functional Annotation Clusters | Previous Associations |
|---|---|---|---|---|---|
| ILMN_1740265 | 1 | ACOT7 | acyl-CoA thloesterase 7 | SZ-exome | |
| ILMN_1751338 | 1 | NUP133 | nucleoporin 133kDa | RNA processing; Protein localization and transport; Membrane |
SZ-exome |
| ILMN_1686871 | 1 | PARP1 | poly (ADP-ribose) polymerase 1 | RNA processing; DNA replication; Nucleotide and ATP binding; Regulation of transcriptionIon binding |
SZ-exome |
| ILMN_1787461 | 1 | RUNX3 | runt-related transcription factor 3 | RNA processing; Regulation of cell proliferation and death; Nucleotide and ATP binding; Regulation of transcription; Ion binding |
SZ-exome |
| ILMN_1766010 | 1 | YARS | tyrosyl-tRNA synthetase | RNA processing; Nucleotide and ATP binding |
SZ-exome |
| ILMN_1673991 | 2 | ATIC | 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase / IMP cyclohydrolase |
SZ-exome; SZ- linkage |
|
| ILMN_1752111 | 2 | SMARCAL1 | SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a-like 1 |
RNA processing; DNA replication; Nucleotide and ATP binding; Regulation of transcription; Ion binding |
SZ-exome; SZ- linkage |
| ILMN_1676745 | 2 | ZNF142 | zinc finger protein 142 | RNA processing; Regulation of transcription; Ion binding |
SZ-exome; SZ-linkage |
| ILMN_1685580 | 3 | CBLB | Cas-Br-M (murine) ecotropic retroviral transforming sequence B |
Regulation of cell proliferation and death; Calcium ion binding; Ion binding; Catabolic processes; Protein localization and transport |
SZ-exome; SZ- linkage |
| ILMN_2412549 | 4 | GAR1 | GAR1 ribonucleoprotein homolog | RNA processing; DNA replication | SZ-exome |
| ILMN_2224143 | 6 | MCM3 | minichromosome maintenance complex component 3 |
RNA processing; DNA replication; Nucleotide and ATP binding; Regulation of transcription; Ion binding |
SZ-exome; SZ- linkage |
| ILMN_1701731 | 7 | AKR1B1 | aldo-keto reductase family 1, member B1 (aldose reductase) |
SZ-exome | |
| ILMN_1721978 | 7 | CARD11 | caspase recruitment domain family, member 11 |
Regulation of cell proliferation and death; Regulation of transcription; Ion binding; Cell membrane; Membrane |
BP; SZ-exome |
| ILMN_1761479 | 7 | ZC3HC1 | zinc finger, C3HC-type containing 1 | RNA processing; Regulation of cell proliferation and death; Ion binding; Catabolic processes |
SZ-exome |
| ILMN_1744068 | 8 | ELP3 | elongation protein 3 homolog | RNA processing; DNA replication; Regulation of transcription; Ion binding |
SZ-exome; SZ- linkage |
| ILMN_1788538 | 8 | NCALD | neurocalcin delta | Calcium ion binding; Ion binding | COG; SZ- exome |
| ILMN_2343097 | 8 | NCALD | neurocalcin delta | Calcium ion binding; Ion binding | COG; SZ-exome |
| ILMN_1777740 | 8 | THEM6 | thioesterase superfamily member 6 | ||
| ILMN_1750800 | 9 | ACO1 | aconitase 1 | RNA processing; Ion binding | SZ-exome |
| ILMN_2073012 | 9 | TMEM203 | transmembrane protein 203 | Cell membran; eMembrane | SZ-exome |
| ILMN_1689953 | 11 | CD81 | CD81 molecule | Regulation of cell proliferation and death; Protein localization and transport; Phosphorylation; Cell membrane; Membrane |
SZ-exome |
| ILMN_2334296 | 11 | IL18BP | interleukin 18 binding protein | SZ-exome | |
| ILMN_1700604 | 11 | RBM14 | RNA binding motif protein 14 | RNA processing; DNA replication; Nucleotide and ATP binding; Regulation of transcription; Ion binding |
BP; SZ-exome |
| ILMN_2044453 | 12 | LPAR5 | lysophosphatidic acid receptor 5 | Cell membrane; Membrane | SZ-exome |
| ILMN_1733696 | 15 | IMP3 | IMP3, U3 small nucelolar ribonucleoprotein homolog |
RNA processing | SZ-exome |
| ILMN_1801348 | 16 | GOT2 | glutamic-oxaloacetic transminase 2, mitochondrial (aspartate aminotransferase 2) |
RNA processing; Cell membrane; Membrane | SZ-exome |
| ILMN_1708328 | 16 | RAB11FIP3 | RAB11 family interacting protein 3 | Calcium ion binding; Ion binding; Protein localization and transport; Cell membrane; Membrane |
SZ-exome |
| ILMN_1728298 | 16 | SBK1 | SH3-binding domain kinase 1 | Nucleotide and ATP binding; Phosphorylation |
SZ-exome; SZ- linkage |
| ILMN_2097793 | 17 | ZBTB4 | zinc finger and BTB domain containing 4 |
RNA processing; Regulation of transcription; Ion binding |
SZ-exome |
| ILMN_2153787 | 19 | ARRDC5 | arrestin domain containing 5 | SZ-exome | |
| ILMN_1748894 | 19 | GTPBP3 | GTP binding protein 3 (mitochondrial) | RNA processingNucleotide and ATP binding | SZ-exome |
| ILMN_1665943 | 19 | MAP4K1 | mitogen-activated protein kinase kinase kinase kinase 1 |
Nucleotide and ATP binding; Phosphorylation |
SZ-exome |
| ILMN_2183687 | 20 | LIME1 | Lck interacting transmembrane adapter 1 |
Cell membrane; Membrane | SZ-exome |
| ILMN_1663954 | 20 | NELFCD | negative elongation factor complex member C/D |
||
| ILMN_2162989 | 20 | TMEM189 | transmembrane protein 189 | RNA processing; Regulation of transcriptionIon binding; Catabolic processes; Cell membrane; Membrane |
SZ-exome |
| ILMN_1753885 | 20 | YTHDF1 | YTH domain family, member 1 | SZ-exome | |
| ILMN_1763460 | 22 | NHP2L1 | NHP2 non-histone chromosome protein 2-like 1 |
RNA processing | |
| ILMN_1786024 | 22 | POLR3H | polymerase (RNA) III (DNA directed) polypeptide H (22.9kD) |
RNA processing; DNA replication; Ion binding |
SZ-exome |
| ILMN_1779185 | 22 | SPECC1L | sperm antigen with calponin homology and coiled-coil domains 1-like |
SZ-exome | |
| ILMN_1671257 | X | DKC1 | dyskeratosis congenita 1, dyskerin | RNA processing; DNA replication | SZ-exome |
| ILMN_1775522 | X | MAGED1 | melanoma antigen family D1 | Regulation of cell proliferation and death; Regulation of transcription; Ion binding; Cell membrane; Membrane |
SZ-exome |
| ILMN_2306189 | X | MAGED1 | melanoma antigen family D1 | Regulation of cell proliferation and death; Regulation of transcription; Ion binding; Cell membrane; Membrane |
SZ-exome |
| ILMN_1670948 | X | TSR2 | TSR2, 20S rRNA accumulation, homolog |
RNA processing |
Note. SZ-exome = previously associated with schizophrenia in an exome sequencing study; SZ-linkage = previously associated with schizophrenia in a linkage study; BP = previously associated with bipolar disorder; COG = previously associated with a cognitive phenotype.
Discussion
In this study we have found evidence of heritable gene expression contributing to both memory and schizophrenia, suggesting that some gene expression changes may increase risk for schizophrenia via effects on cognitive endophenotypes, such as memory impairment. Specifically, after correction for multiple testing at the whole genome level, we identified 76 RNA probes whose expression varied significantly in relation to memory function, 43 of which – reflecting 41 independent genes – also showed differential expression in schizophrenia patients compared with their unaffected co-twins and control twins. For all of these genes, higher expression correlated with better memory performance in the overall sample, and expression was reduced in patients with schizophrenia as compared to co-twins and controls. While many genes negatively correlated with both CVLT and diagnostic status (S2), none of these survived correction for multiple comparisons. As the samples were run in duplicate and averaged, it is unlikely that the lack of significant negative correlations were due to technical artifact. A smaller, independent sample of schizophrenia patients, co-twins, and control twins confirmed diagnostic differences for 14 of the probes, which represented nearly all functional clusters present in the full set of 43 genes. However, for all 43 probes, effect sizes in the smaller replication sample were consistent with those of the larger discovery sample both in direction and magnitude (Figure 1); power analyses confirmed that all 43 effects would be significant with a replication sample of comparable size to the discovery sample. Heritability analyses utilizing the twin design revealed a genetic contribution for 41 genes within this subset indicating that these effects are at least partially related to inherited variations. We did not find any genes significantly expressed in relation to general ability after correction. This is likely because our sample included a high proportion of patients with schizophrenia, who, consistent with previous work (Cannon et al., 2000; Schaefer et al., 2013), had a much more severe deficit in verbal memory than in general cognitive ability (i.e., 1.8 SD vs. .49 SD decrement compared with controls), conferring relatively higher power to detect schizophrenia-related gene expression impacting memory as opposed to general ability. In a general population sample, in which schizophrenia would occur at the population base rate of ~1%, there would likely be no increased sensitivity to detect memory-related genes than those impacting general ability.
The specific genes identified as differentially expressed in relation to both memory and diagnostic status in this study are largely overlapping with genes for which there is prior evidence of association with schizophrenia and related disorders and/or with neurocognition from linkage, exome sequencing, and genome-wide association studies (Table 2). Specifically, 37 of the 41 genes have been previously related to schizophrenia (Fromer et al., 2014; Gulsuner et al., 2013; Purcell et al., 2014), one was recently identified in a GWAS of cognitive phenotypes (Ibrahim-Verbaas et al., 2015), and two have been identified in studies of bipolar disorder (Group, 2011; Sprooten et al., 2011), which is estimated to share 40% of its genetic etiology with schizophrenia (Lichtenstein et al., 2009).
Seven genes from the current study (SMARCAL1, MCM3, SBK1, ZNF142, ATIC, CBLB, ELP3) are in regions significantly associated with schizophrenia in a meta-analysis of 32 European ancestry linkage studies (Jia, Sun, Guo, & Zhao, 2010; Ng et al., 2009). An exome sequencing study comparing schizophrenia patients to controls identified de novo mutations in patients in 37 of the 41 genes identified here (Purcell et al., 2014). De novo mutations in four of these genes (ACO1, GTPBP3, YARS, and RUNX3) were also found in exome sequencing studies comparing patients to parents (Fromer et al., 2014), and unaffected siblings (Gulsuner et al., 2013).
A SNP in NCALD, a gene producing a calcium binding protein associated with regulation of G-protein coupled signal transduction, which has also been associated with schizophrenia (Purcell et al., 2014), has recently been associated with processing speed in a large genome-wide association study of control individuals (Ibrahim-Verbaas et al., 2015). Here, we report a strong association between gene expression of NCALD and memory performance, a related cognitive process, as well as reduced expression in patients with schizophrenia. Schizophrenia has consistently been associated with substantial deficits in memory and processing speed (Schaefer et al., 2013), as well as mutations in calcium-associated genes (Hertzberg et al., 2015; Ripke et al., 2013). This is promising evidence that at least some schizophrenia-related genes confer risk via impacting cognition.
Schizophrenia and bipolar disorder share substantial genetic overlap, many endophenotypes, and several symptom-level similarities, including cognitive impairment and psychosis (Lichtenstein et al., 2009). Two memory-related, schizophrenia-associated genes identified in the current study, which have both previously been associated with schizophrenia (Purcell et al., 2014), have also previously been associated with bipolar disorder (Group, 2011; Sprooten et al., 2011). A SNP in BMP14, a gene that regulates gene transcription splicing, was identified in a genome-wide association study of bipolar disorder (Group, 2011). Changes in gene transcription may underlie some of the shared etiology between bipolar disorder and schizophrenia, potentially mediated by effects on cognition. Additionally, a SNP in CARD11 has previously been associated with decreased white matter in siblings of bipolar patients as compared to controls (Sprooten et al., 2011). Decreased white matter integrity is a shared endophenotype between bipolar disorder and schizophrenia, which suggests that CARD11 could plausibly confer risk for these disorders via impacts on white matter integrity (Camchong, Lim, Sponheim, & Macdonald, 2009; Sprooten et al., 2011).
Four genes implicated in our study are novel genes not previously associated with schizophrenia, bipolar disorder, or cognition: NELFCD, NHP2L1, THEM6, and TSR2. NELFCD, an essential component of the NELF complex, which suppresses the elongation of transcription by RNA polymerase II, NHP2L1, a highly conserved nuclear protein associated with RNA binding, and TSR2, a pre-rRNA processing protein with a known ASD-related de novo mutation (O’Roak et al., 2012), are all related to transcription and translation (Safran et al., 2002). Furthermore, a cluster analysis using DAVID, a bioinformatics platform that accesses many gene ontology databases to group lists of genes, identified a significant enrichment of DNA- and RNA-regulating genes within the memory- and schizophrenia-related genes (S5) (Huang da et al., 2009). Twenty-one of the 41 identified genes fell into this category, suggesting a significant role of transcriptional and translational regulation in cognitive impairment in schizophrenia. This lends further evidence to the hypothesis that many of the genetic effects relevant in schizophrenia reflect regulatory processes rather than coding changes per se (Kim et al., 2014; Maurano et al., 2012).
We also identified six genes related to T and B cell signaling pathways, consistent with previous work implicating immune-related genes (Schizophrenia Working Group of the Psychiatric Genomics, 2014), altered pro-inflammatory cytokine expression and protein levels (Dimitrov et al., 2013; Na, Jung, & Kim, 2014; Pandey, Ren, Rizavi, & Zhang, 2015; Song et al., 2014), and inflammation during pregnancy (Meyer, Feldon, & Dammann, 2011; Miller, Culpepper, Rapaport, & Buckley, 2013) in risk for schizophrenia (for review see: Fineberg & Ellman, 2013).
The primary competing explanation of the findings in this study are that the gene expression changes associated with memory and schizophrenia may be secondary to illness expression (e.g., symptoms) or treatment. While we cannot fully rule out that such factors contribute, they are unlikely to completely account for the present results, for several reasons. First, in considering the sources of the relationships between gene expression and CVLT scores, if the effects were driven purely by such secondary factors, they would disappear when probands with schizophrenia or bipolar disorder are excluded. However, the average effect size for the 76 probes significantly associated with CVLT (partial R2 = .131, 95% CIs [.128, .134]) was reduced only moderately when excluding schizophrenia and bipolar patients (partial R2 = .095, 95% CIs [.090, .099]) (S1). A power analysis to determine the sample size required for detecting the average effect size found among the non-patient participants (partial R2 = .095) at genome-wide significance revealed that only 100 additional subjects would be necessary (Faul et al., 2007). Furthermore, expression levels of the genes associated with memory were found to be moderately heritable. Taken together, these results are consistent with an inherited basis for the relationships between expression levels of these genes and memory in the general population. Future work may discern the specificity of these genotype-phenotype relationships, as genes can both impinge on a variety of cognitive processes (e.g., BDNF; Ahmed, Mantini, Fridberg, & Buckley, 2015), or only certain components of a single process (Kremen et al., 2014; Panizzon et al., 2011; Papassotiropoulos & de Quervain, 2011). Furthermore, as memory impairment is a feature of many psychiatric illnesses, schizophrenia only one among them, it is possible that some of these genes confer risk for psychopathology more broadly.
Given that probands with schizophrenia and bipolar disorder are enriched (relative to non-affected individuals) for genetic variations conferring cognitive impairment, larger effect sizes for the associations between gene expression and memory performance would be expected when probands are included compared to those when probands are excluded, as was observed. Alternatively, the higher effect sizes when probands are included could reflect the impacts of clinical state and/or medications on both gene expression and memory performance. However, the assessments of memory performance and gene expression were done when probands were clinically stable, and expression levels of the genes associated with both memory and schizophrenia were not related to positive symptom severity or current antipsychotic medication status. These findings, combined with the fact that previous work has shown associations of genotypic variations in nearly all of the identified genes with schizophrenia, indicate that a shared inherited substrate is likely to represent a major source of the overlap between gene expression patterns associated with reduced memory performance and schizophrenia. That there were no significant expression differences between the non-affected co-twins of probands and controls somewhat complicates this interpretation; however, as would be expected from a heritable source, co-twins’ expression levels were intermediate between probands and controls on most of the markers, and power was limited for this contrast. In any case, because these are correlational data, they do not provide direct evidence for causal relationships between expression of these genes and either memory performance or schizophrenia. Animal model studies employing genetic manipulations could more directly test whether decreased expression of these genes cause decrements in memory performance or changes on other behavioral assays related to schizophrenia.
Other methodological limitations of this study include the assay of gene expression from peripheral tissue and the relatively small sample size. We examined gene expression in PBMCs, which has been shown to correlate with central nervous system expression and is heritable in our sample, but is nonetheless not as direct a read-out of expression levels in the brain as would be true of samples in neurons or glia (Cheung et al., 2003). Peripheral gene expression is a pragmatic approach – it is relatively low cost and less invasive to assay than other tissues, and biomarkers of disease ascertainable at this level are advantageous for future research and clinical application (Cattane et al., 2015). Further, to protect against identifying gene expression changes irrelevant to brain function, we first screened the genome for genes associating with a cognitive phenotype. We also used a microarray assay to measure gene expression, which only captures expression of primary transcripts and cannot quantify absolute levels of expression. Future work using RNA-Seq could be useful in this regard.
Additionally, our sample size is relatively modest. Because we used Bonferroni correction for multiple testing on the whole genome level at the discovery stage and found evidence of replication of effect sizes and direction of schizophrenia-related variations in an independent sample, the small sample size is of concern primarily in regards to Type II (false negative) rather than Type I (false positive) error. That is, there are likely many hundreds of additional genes relating to both memory and schizophrenia that were not detected in this study. Even in the case that some or all of the genes not meeting statistical significance in the replication sample are false positives, the interpretation of the findings would remain the same as the 14 genes meeting statistical significance represent nearly all the functional clusters identified in the full set of 43 probes. Nonetheless, individual RNA transcripts within these functional groups that did not show statistical replication may not be genes of high effect.
In summary, consistent with substantial previous work linking schizophrenia-related genotypic variation to memory (Greenwood et al., 2012; Greenwood et al., 2013; Heck et al., 2014; Karlsgodt et al., 2011; Kauppi et al., 2015; Nicodemus et al., 2014; Toulopoulou et al., 2007; van Erp et al., 2008; Walton et al., 2013), we have provided evidence that a subset of genes expressed in relation to memory performance are under-expressed in schizophrenia patients relative to unaffected co-twins of patients and controls and that this expression is moderately heritable. Thus, genes increasing risk for schizophrenia may do so via effects on memory-related processes. Further, enrichment of translation and transcription related genes identified in the current study suggests this may be a functional mechanism by which genes promoting memory impairment in schizophrenia may specifically confer risk. This study represents only a first step in uncovering gene expression changes that promote memory impairment in schizophrenia. Investigations of other endophenotypes for schizophrenia may uncover relevant genetic overlap or dissociation between cognitive processes, revealing important functional connections between risk-conferring genes and specific neurobiological systems.
Supplementary Material
References
- Ahmed AO, Mantini AM, Fridberg DJ, Buckley PF. Brain-derived neurotrophic factor (BDNF) and neurocognitive deficits in people with schizophrenia: a meta-analysis. Psychiatry Res. 2015;226(1):1–13. doi: 10.1016/j.psychres.2014.12.069. [DOI] [PubMed] [Google Scholar]
- Akaike H. New Look at Statistical-Model Identification. Ieee Transactions on Automatic Control, Ac19. 1974;(6):716–723. [Google Scholar]
- Association AP. Diagnostic and statistical manual of mental disorders : DSM-IV. 4th ed. Washington, DC: American Psychiatric Association; 1994. [Google Scholar]
- Bates D, Maechler M, Bolker B, Walker S. lme4: Linear mixed-effects models using Eigen and S4. 2014 Retrieved from http://CRAN.R-project.org/package=lme4.
- Camchong J, Lim KO, Sponheim SR, Macdonald AW. Frontal white matter integrity as an endophenotype for schizophrenia: diffusion tensor imaging in monozygotic twins and patients’ nonpsychotic relatives. Front Hum Neurosci. 2009;3:35. doi: 10.3389/neuro.09.035.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon TD, Huttunen MO, Lonnqvist J, Tuulio-Henriksson A, Pirkola T, Glahn D, Koskenvuo M. The inheritance of neuropsychological dysfunction in twins discordant for schizophrenia. Am J Hum Genet. 2000;67(2):369–382. doi: 10.1086/303006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon TD, Kaprio J, Lonnqvist J, Huttunen M, Koskenvuo M. The genetic epidemiology of schizophrenia in a Finnish twin cohort. A population-based modeling study. Arch Gen Psychiatry. 1998;55(1):67–74. doi: 10.1001/archpsyc.55.1.67. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/9435762. [DOI] [PubMed] [Google Scholar]
- Cannon TD, Keller MC. Endophenotypes in the genetic analyses of mental disorders. Annu Rev Clin Psychol. 2006;2:267–290. doi: 10.1146/annurev.clinpsy.2.022305.095232. [DOI] [PubMed] [Google Scholar]
- Carmelli D, Swan GE, DeCarli C, Reed T. Quantitative genetic modeling of regional brain volumes and cognitive performance in older male twins. Biol Psychol. 2002;61(1–2):139–155. doi: 10.1016/s0301-0511(02)00056-x. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/12385673. [DOI] [PubMed] [Google Scholar]
- Cattane N, Minelli A, Milanesi E, Maj C, Bignotti S, Bortolomasi M, Gennarelli M. Altered gene expression in schizophrenia: findings from transcriptional signatures in fibroblasts and blood. Plos One. 2015;10(2):e0116686. doi: 10.1371/journal.pone.0116686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung VG, Conlin LK, Weber TM, Arcaro M, Jen KY, Morley M, Spielman RS. Natural variation in human gene expression assessed in lymphoblastoid cells. Nat Genet. 2003;33(3):422–425. doi: 10.1038/ng1094. [DOI] [PubMed] [Google Scholar]
- Dimitrov DH, Lee S, Yantis J, Valdez C, Paredes RM, Braida N, Walss-Bass C. Differential correlations between inflammatory cytokines and psychopathology in veterans with schizophrenia: potential role for IL-17 pathway. Schizophr Res. 2013;151(1–3):29–35. doi: 10.1016/j.schres.2013.10.019. [DOI] [PubMed] [Google Scholar]
- Dunning M, Lynch A, Eldridge M. illuminaHumanv4.db: Illumina HumanHT12v4 annotation data (chip illuminaHumanv4) Retrieved from http://www.bioconductor.org/packages/release/data/annotation/html/illuminaHumanv4.db.html.
- Faul F, Erdfelder E, Lang AG, Buchner A. G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods. 2007;39(2):175–191. doi: 10.3758/bf03193146. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/17695343. [DOI] [PubMed] [Google Scholar]
- Fineberg AM, Ellman LM. Inflammatory cytokines and neurological and neurocognitive alterations in the course of schizophrenia. Biol Psychiatry. 2013;73(10):951–966. doi: 10.1016/j.biopsych.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, O’Donovan MC. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506(7487):179–184. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner EJ, Cairns MJ, Liu B, Beveridge NJ, Carr V, Kelly B, Tooney PA. Gene expression analysis reveals schizophrenia-associated dysregulation of immune pathways in peripheral blood mononuclear cells. J Psychiatr Res. 2013;47(4):425–437. doi: 10.1016/j.jpsychires.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glahn DC, Almasy L, Blangero J, Burk GM, Estrada J, Peralta JM, Escamilla MA. Adjudicating neurocognitive endophenotypes for schizophrenia. Am J Med Genet B Neuropsychiatr Genet. 2007;144B(2):242–249. doi: 10.1002/ajmg.b.30446. [DOI] [PubMed] [Google Scholar]
- Greenwood TA, Braff DL, Light GA, Cadenhead KS, Calkins ME, Dobie DJ, Schork NJ. Initial heritability analyses of endophenotypic measures for schizophrenia: the consortium on the genetics of schizophrenia. Arch Gen Psychiatry. 2007;64(11):1242–1250. doi: 10.1001/archpsyc.64.11.1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood TA, Light GA, Swerdlow NR, Radant AD, Braff DL. Association Analysis of 94 Candidate Genes and Schizophrenia-Related Endophenotypes. Plos One. 2012;7(1) doi: 10.1371/journal.pone.0029630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood TA, Swerdlow NR, Gur RE, Cadenhead KS, Calkins ME, Dobie DJ, Braff DL. Genome-Wide Linkage Analyses of 12 Endophenotypes for Schizophrenia From the Consortium on the Genetics of Schizophrenia. American Journal of Psychiatry. 2013;170(5):521–532. doi: 10.1176/appi.ajp.2012.12020186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Group PGCBDW. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet. 2011;43(10):977–983. doi: 10.1038/ng.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillozet-Bongaarts AL, Hyde TM, Dalley RA, Hawrylycz MJ, Henry A, Hof PR, Kleinman JE. Altered gene expression in the dorsolateral prefrontal cortex of individuals with schizophrenia. Mol Psychiatry. 2014;19(4):478–485. doi: 10.1038/mp.2013.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulsuner S, Walsh T, Watts AC, Lee MK, Thornton AM, Casadei S, McClellan JM. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell. 2013;154(3):518–529. doi: 10.1016/j.cell.2013.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halekoh U, Hojsgaard S. A Kenward-Roger Approximation and Parametic Bootstrap Methods for Tests in Linear Mixed Models - The R Package pbkrtest. Journal of Statistical Software. 2014;59(9):1–30. Retrieved from http://www.jstatsoft.org/v59/i09/ [Google Scholar]
- Haut K, Karlsgodt KH, Bilder RM, Congdon E, Freimer N, London ED, Sabb FW, Ventura J, Cannon TD. Phenomics of memory dysfunction in schizophrenia, bipolar disorder and ADHD. in prep [Google Scholar]
- Heck A, Fastenrath M, Ackermann S, Auschra B, Bickel H, Coynel D, Papassotiropoulos A. Converging genetic and functional brain imaging evidence links neuronal excitability to working memory, psychiatric disease, and brain activity. Neuron. 2014;81(5):1203–1213. doi: 10.1016/j.neuron.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertzberg L, Katsel P, Roussos P, Haroutunian V, Domany E. Integration of gene expression and GWAS results supports involvement of calcium signaling in Schizophrenia. Schizophr Res. 2015 doi: 10.1016/j.schres.2015.02.001. [DOI] [PubMed] [Google Scholar]
- Higier RG, Jimenez AM, Hultman CM, Borg J, Roman C, Kizling I, Cannon TD. Enhanced neurocognitive functioning and positive temperament in twins discordant for bipolar disorder. Am J Psychiatry. 2014;171(11):1191–1198. doi: 10.1176/appi.ajp.2014.13121683. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Ibrahim-Verbaas CA, Bressler J, Debette S, Schuur M, Smith AV, Bis JC, Mosley TH. GWAS for executive function and processing speed suggests involvement of the CADM2 gene. Mol Psychiatry. 2015 doi: 10.1038/mp.2015.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia P, Sun J, Guo AY, Zhao Z. SZGR: a comprehensive schizophrenia gene resource. Mol Psychiatry. 2010;15(5):453–462. doi: 10.1038/mp.2009.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsgodt KH, Robleto K, Trantham-Davidson H, Jairl C, Cannon TD, Lavin A, Jentsch JD. Reduced dysbindin expression mediates N-methyl-D-aspartate receptor hypofunction and impaired working memory performance. Biol Psychiatry. 2011;69(1):28–34. doi: 10.1016/j.biopsych.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppi K, Westlye LT, Tesli M, Bettella F, Brandt CL, Mattingsdal M, Andreassen OA. Polygenic Risk for Schizophrenia Associated With Working Memory-related Prefrontal Brain Activation in Patients With Schizophrenia and Healthy Controls. Schizophr Bull. 2015;41(3):736–743. doi: 10.1093/schbul/sbu152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Xia K, Tao R, Giusti-Rodriguez P, Vladimirov V, van den Oord E, Sullivan PF. A meta-analysis of gene expression quantitative trait loci in brain. Transl Psychiatry. 2014;4:e459. doi: 10.1038/tp.2014.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline RB. Principles and practice of structural equation modeling. 3rd Ed. New York, NY: The Guilford Press; 2011. [Google Scholar]
- Kremen WS, Panizzon MS, Franz CE, Spoon KM, Vuoksimaa E, Jacobson KC, Lyons MJ. Genetic complexity of episodic memory: a twin approach to studies of aging. Psychol Aging. 2014;29(2):404–417. doi: 10.1037/a0035962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lencz T, Knowles E, Davies G, Guha S, Liewald DC, Starr JM, Malhotra AK. Molecular genetic evidence for overlap between general cognitive ability and risk for schizophrenia: a report from the Cognitive Genomics consorTium (COGENT) Mol Psychiatry. 2014;19(2):168–174. doi: 10.1038/mp.2013.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenstein P, De Faire U, Floderus B, Svartengren M, Svedberg P, Pedersen NL. The Swedish Twin Registry: a unique resource for clinical, epidemiological and genetic studies. J Intern Med. 2002;252(3):184–205. doi: 10.1046/j.1365-2796.2002.01032.x. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/12270000. [DOI] [PubMed] [Google Scholar]
- Lichtenstein P, Yip BH, Bjork C, Pawitan Y, Cannon TD, Sullivan PF, Hultman CM. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population-based study. Lancet. 2009;373(9659):234–239. doi: 10.1016/S0140-6736(09)60072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo XJ, Mattheisen M, Li M, Huang L, Rietschel M, Borglum AD, Yao YG. Systematic Integration of Brain eQTL and GWAS Identifies ZNF323 as a Novel Schizophrenia Risk Gene and Suggests Recent Positive Selection Based on Compensatory Advantage on Pulmonary Function. Schizophr Bull. 2015 doi: 10.1093/schbul/sbv017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Stamatoyannopoulos JA. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337(6099):1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U, Feldon J, Dammann O. Schizophrenia and autism: both shared and disorder-specific pathogenesis via perinatal inflammation? Pediatr Res. 2011;69(5 Pt 2):26R–33R. doi: 10.1203/PDR.0b013e318212c196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BJ, Culpepper N, Rapaport MH, Buckley P. Prenatal inflammation and neurodevelopment in schizophrenia: a review of human studies. Prog Neuropsychopharmacol Biol Psychiatry. 2013;42:92–100. doi: 10.1016/j.pnpbp.2012.03.010. [DOI] [PubMed] [Google Scholar]
- Na KS, Jung HY, Kim YK. The role of pro-inflammatory cytokines in the neuroinflammation and neurogenesis of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2014;48:277–286. doi: 10.1016/j.pnpbp.2012.10.022. [DOI] [PubMed] [Google Scholar]
- Neale M, Boker S, Xie G, Maes H. Mx: Statistical modeling. Richmond, VA: Department of Psychiatry, Virginia Institute for Psychiatric and Behavior Genetics, Virginia Commonwealth University; 2003. [Google Scholar]
- Ng MY, Levinson DF, Faraone SV, Suarez BK, DeLisi LE, Arinami T, Lewis CM. Meta-analysis of 32 genome-wide linkage studies of schizophrenia. Mol Psychiatry. 2009;14(8):774–785. doi: 10.1038/mp.2008.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicodemus KK, Hargreaves A, Morris D, Anney R, Gill M, Corvin A Wellcome Trust Case Control, C. Variability in working memory performance explained by epistasis vs polygenic scores in the ZNF804A pathway. JAMA Psychiatry. 2014;71(7):778–785. doi: 10.1001/jamapsychiatry.2014.528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Eichler EE. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485(7397):246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oresic M, Seppanen-Laakso T, Sun D, Tang J, Therman S, Viehman R, Cannon TD. Phospholipids and insulin resistance in psychosis: a lipidomics study of twin pairs discordant for schizophrenia. Genome Med. 2012;4(1):1. doi: 10.1186/gm300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey GN, Ren X, Rizavi HS, Zhang H. Proinflammatory cytokines and their membrane-bound receptors are altered in the lymphocytes of schizophrenia patients. Schizophr Res. 2015;164(1–3):193–198. doi: 10.1016/j.schres.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panizzon MS, Lyons MJ, Jacobson KC, Franz CE, Grant MD, Eisen SA, Kremen WS. Genetic architecture of learning and delayed recall: a twin study of episodic memory. Neuropsychology. 2011;25(4):488–498. doi: 10.1037/a0022569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papassotiropoulos A, de Quervain DJ. Genetics of human episodic memory: dealing with complexity. Trends Cogn Sci. 2011;15(9):381–387. doi: 10.1016/j.tics.2011.07.005. [DOI] [PubMed] [Google Scholar]
- Pinheiro J, Bates D, DebRoy S, Sarkar D, Team RC. nlme: Linear and Nonlinear Mixed Effects Models. 2015 Retrieved from http://CRAN.R-project.org/package=nlme.
- Plomin R, DeFries JC, Knopik V, Neiderhiser J. Behavioral genetics. 6th ed. New York: Worth Publishers; 2013. [Google Scholar]
- Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, Sklar P. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506(7487):185–190. doi: 10.1038/nature12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, Sullivan PF, Scolnick EM. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460(7256):748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke S, O’Dushlaine C, Chambert K, Moran JL, Kahler AK, Akterin S, Sullivan PF. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet. 2013;45(10):1150–1159. doi: 10.1038/ng.2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safran M, Solomon I, Shmueli O, Lapidot M, Shen-Orr S, Adato A, Lancet D. GeneCards 2002: towards a complete, object-oriented, human gene compendium. Bioinformatics. 2002;18(11):1542–1543. doi: 10.1093/bioinformatics/18.11.1542. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/12424129. [DOI] [PubMed] [Google Scholar]
- Schaefer J, Giangrande E, Weinberger DR, Dickinson D. The global cognitive impairment in schizophrenia: consistent over decades and around the world. Schizophr Res. 2013;150(1):42–50. doi: 10.1016/j.schres.2013.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics, C. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511(7510):421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Fan X, Li X, Zhang W, Gao J, Zhao J, Lv L. Changes in pro-inflammatory cytokines and body weight during 6-month risperidone treatment in drug naive, first-episode schizophrenia. Psychopharmacology (Berl) 2014;231(2):319–325. doi: 10.1007/s00213-013-3382-4. [DOI] [PubMed] [Google Scholar]
- Spitzer RL, Williams JB, Gibbon M, First MB. The Structured Clinical Interview for DSM-III-R (SCID). I: History, rationale, and description. Arch Gen Psychiatry. 1992;49(8):624–629. doi: 10.1001/archpsyc.1992.01820080032005. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/1637252. [DOI] [PubMed] [Google Scholar]
- Sprooten E, Sussmann JE, Clugston A, Peel A, McKirdy J, Moorhead TW, McIntosh AM. White matter integrity in individuals at high genetic risk of bipolar disorder. Biol Psychiatry. 2011;70(4):350–356. doi: 10.1016/j.biopsych.2011.01.021. [DOI] [PubMed] [Google Scholar]
- Stone WS, Giuliano AJ, Tsuang MT, Braff DL, Cadenhead KS, Calkins ME, Seidman LJ. Group and site differences on the California Verbal Learning Test in persons with schizophrenia and their first-degree relatives: findings from the Consortium on the Genetics of Schizophrenia (COGS) Schizophr Res. 2011;128(1–3):102–110. doi: 10.1016/j.schres.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone WS, Mesholam-Gately RI, Braff DL, Calkins ME, Freedman R, Green MF, Seidman LJ. California Verbal Learning Test-II performance in schizophrenia as a function of ascertainment strategy: comparing the first and second phases of the Consortium on the Genetics of Schizophrenia (COGS) Schizophr Res. 2015;163(1–3):32–37. doi: 10.1016/j.schres.2014.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan HY, Callicott JH, Weinberger DR. Intermediate phenotypes in schizophrenia genetics redux: is it a no brainer? Mol Psychiatry. 2008;13(3):233–238. doi: 10.1038/sj.mp.4002145. [DOI] [PubMed] [Google Scholar]
- Toulopoulou T, Picchioni M, Rijsdijk F, Hua-Hall M, Ettinger U, Sham P, Murray R. Substantial genetic overlap between neurocognition and schizophrenia - Genetic modeling in twin samples. Arch Gen Psychiatry. 2007;64(12):1348–1355. doi: 10.1001/archpsyc.64.12.1348. [DOI] [PubMed] [Google Scholar]
- van Eijk KR, de Jong S, Strengman E, Buizer-Voskamp JE, Kahn RS, Boks MP, Ophoff RA. Identification of schizophrenia-associated loci by combining DNA methylation and gene expression data from whole blood. Eur J Hum Genet. 2014 doi: 10.1038/ejhg.2014.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Erp TG, Therman S, Pirkola T, Tuulio-Henriksson A, Glahn DC, Bachman P, Cannon TD. Verbal recall and recognition in twins discordant for schizophrenia. Psychiatry Res. 2008;159(3):271–280. doi: 10.1016/j.psychres.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton E, Turner J, Gollub RL, Manoach DS, Yendiki A, Ho BC, Ehrlich S. Cumulative genetic risk and prefrontal activity in patients with schizophrenia. Schizophr Bull. 2013;39(3):703–711. doi: 10.1093/schbul/sbr190. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.