

Abstract
Objective
The lysosomal storage disease alpha‐mannosidosis is caused by the deficiency of the lysosomal acid hydrolase alpha‐mannosidase (LAMAN) leading to lysosomal accumulation of neutral mannose‐linked oligosaccharides throughout the body, including the brain. Clinical findings in alpha‐mannosidosis include skeletal malformations, intellectual disabilities and hearing impairment. To date, no curative treatment is available. We previously developed a beneficial enzyme replacement therapy (ERT) regimen for alpha‐mannosidase knockout mice, a valid mouse model for the human disease. However, humoral immune responses against the injected recombinant human alpha‐mannosidase (rhLAMAN) precluded long‐term studies and chronic treatment.
Methods
Here, we describe the generation of an immune‐tolerant alpha‐mannosidosis mouse model that allowed chronic injection of rhLAMAN by transgenic expression of a catalytically inactive variant of human LAMAN in the knockout background.
Results
Chronic ERT of rhLAMAN revealed pronounced effects on primary substrate storage throughout the brain, normalization of lysosomal enzyme activities and morphology as well as a decrease in microglia activation. The positive effect of long‐term ERT on neuronal lysosomal function was reflected by an improvement of cognitive deficits and exploratory activity. in vivo and in vitro uptake measurements indicate rapid clearance of rhLAMAN from circulation and a broad uptake into different cell types of the nervous system.
Interpretation
Our data contribute to the understanding of neurological disorders treatment by demonstrating that lysosomal enzymes such as rhLAMAN can penetrate into the brain and is able to ameliorate neuropathology.
Introduction
Alpha‐mannosidosis is a devastating inherited autosomal recessive lysosomal storage disease (LSD) caused by deficiency of the lysosomal acid alpha‐mannosidase (LAMAN) encoded by MAN2B1.1, 2 To date, 125 different disease‐causing mutations were identified affecting enzymatic activity, stability and intracellular transport of LAMAN.3, 4, 5, 6 Lysosomal deficiency of LAMAN leads to the accumulation of free mannose‐containing oligosaccharides of asparagine (N)‐linked glycans derived from proteolysis of glycoproteins within the lysosomal compartment7 as well as impaired trimming of N‐linked oligosaccharides on native lysosomal proteins.8 Cells with storage pathology are found throughout the body, including the peripheral and central nervous system (CNS). Alpha‐mannosidosis presents with a wide spectrum of symptoms typically, including recurrent infections, impaired hearing, dysostosis multiplex, and progressive mental retardation.9, 10 To date, no curative therapy for alpha‐mannosidosis is in clinical practice. Bone marrow transplantation was efficient in alpha‐mannosidosis cats11 but with variable outcome for patients.12
A promising therapeutic approach for an effective treatment of alpha‐mannosidosis is the application of recombinant human LAMAN (rhLAMAN) into the bloodstream, designated as enzyme replacement therapy (ERT). This concept was based on findings on cross‐correction in cultured patient's fibroblasts from Hurler Syndrome with healthy cells.13 So far, several LSDs including Morbus Gaucher (Type I), mucopolysaccharidoses (I, II, VI), Morbus Fabry, and Morbus Pompe have been shown to benefit from ERT.14 We have previously demonstrated the value of high‐dose short‐term ERT on clearance of primary brain substrate storage in alpha‐mannosidase‐deficient (knockout) mice,15 a valid mouse model for the human disease, using rhLAMAN of a laboratory scale batch.16, 17, 18 However, immunological responses to the injected enzyme associated with high mortality precluded long‐term ERT of this mouse strain. To evaluate the chronic curative effect of enzyme application, we have generated an immune‐tolerant mouse model and studied the effect of high‐dose long‐term ERT on CNS pathology, using industrial produced rhLAMAN which is used to treat alpha‐mannosidosis patients in a phase I‐II clinical ERT trial.19 Strikingly, despite the low number of patients, a significant improvement of motor and to some extent also of cognitive function was observed. Due to the limited accessibility of human brain material, no correlation with a decline of primary substrate storage and other neuropathological parameters could be drawn.
We report a substantial clearance of primary stored oligosaccharides in brain of immune‐tolerant alpha‐mannosidosis mice after long‐term ERT accompanied by a normalization of lysosomal enzyme activity, protein expression and morphology. Furthermore, a deterioration of secondary pathological alterations such as neuroinflammation was prevented and an improvement of behavioral anomalies achieved. Overall, our data suggest that chronic ERT with rhLAMAN is a valuable tool to ameliorate the observed CNS pathology in alpha‐mannosidosis.
Material and Methods
Materials
A detailed description of the material, reagents and antibodies used can be found under Data S1.
Experimental animals
Alpha‐mannosidase knockout mice were described previously15 and are here referred to alpha‐mannosidosis mice (‐/‐). A detailed description of the generation of the transgene can be found under Data S1. Animals were maintained in a conventional animal facility. All procedures performed in this study involving animals were in accordance with the ethical standards set by the National Animal Care Committee of Germany.
Purification of rhLAMAN and intravenous injection
Purification of Chinese hamster ovary‐derived rhLAMAN from cell culture supernatants was described previously.16, 19 RhLAMAN (64 U/mL; 2 mg/mL) was injected into the tail vein at doses of 25, 125, 500, or 1000 U/kg per body weight (bw) (corresponding to 0.9, 4.4, 15.6, and 31.2 mg/kg per bw, respectively). Mock‐injected mice received the same volume of phosphate‐buffered saline (PBS). Prior to dissecting organs for biochemical and histological analysis, if not stated otherwise, mice were perfused at least 24 h after the last enzyme injection with 0.1 mol/L phosphate buffer in order to clear the circulation from residual enzyme. All animal experiments were approved by local authorities (Tierversuchsantragsnummer: V 312‐72241.121‐3 (32‐3/10)).
Behavioral experiments
Prior to behavioral experiments, mice received biweekly injections during 12 weeks of either 500 U/kg rhLAMAN (ERT‐treated ‐/‐ tg+ mice) or saline solution (mock‐treated ‐/‐ tg+ and +/‐ tg+ control mice) (all n = 13‐14). Behavioral experiments were executed during the following 2 weeks while treatment continued. Mice were 3‐4 months at the start of treatment. Injections were given after daily testing to avoid interference with behavioral experiments. The behavioral test battery included an open field (exploration and emotion), water maze (learning and memory), and treadmill (motor performance). A detailed description of the behavioral analyses can be found under Data S1.
Immunohistology and immunofluorescence
Immunohistological and immunofluorescent staining were performed on formalin‐fixed free‐floating tissue sections or cultured cells according to standard laboratory protocols. A detailed description can be found in Data S1.
Preparation of primary mixed neuronal and glial cultures
Primary mixed neurons and glial cells were prepared as described previously20 from the cortices of gestational day 15 embryos. Cells were fixed in 4 % paraformaldehyde diluted in PBS after 14 days in vitro.
Enzyme‐linked immunosorbent assay (ELISA) for detection of LAMAN‐specific IgG in plasma
A detailed description can be found under Data S1.
Preparation of tissue lysates, SDS‐PAGE, immunoblotting, and enzyme activity measurements
Tissues were homogenized in PBS‐containing Triton X‐100 and cleared by centrifugation at 16.000 × g for 20 min at 4°C. Lysates were separated on 12.5 % sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE) gels, blotted on nitrocellulose membrane and detected with appropriate antibodies according to standard procedures, using Horse radish peroxidase (HRP)‐coupled secondary antibodies and chemiluminescence. The activity of standard lysosomal enzymes was determined colorimetrically in tissue homogenates under acidic conditions as described.16
Oligosaccharide extraction, TLC and HPLC
Oligosaccharide extraction and thin layer chromatography (TLC) analysis of neutral oligosaccharides was performed essentially as described previously.16 A detailed description of oligosaccharide extraction, TLC and high‐performance liquid chromatography (HPLC) analyses can be found under Data S1.
Statistical analyses
For statistical analysis, all samples were normalized to controls in each experiment, and quantification was represented as the mean ± SEM. Data were plotted with GraphPad Prism. Statistical significance, using unpaired Student's t‐test was calculated with GraphPad Prism (Version 5.04) (GraphPad Software, La Jolla CA, USA) and statistical significance was set at: ns, nonsignificant, P > 0.05, *P < 0.05, **P < 0.01, and ***P < 0.001.
Results
Generation of immune‐tolerant alpha‐mannosidosis mice that permit chronic injections of rhLAMAN
Repeated injections every other week of low and high doses (25 and 500 U/kg, corresponding to 0.9 and 15.6 mg/kg per bw) rhLAMAN in alpha‐mannosidosis mice (‐/‐) led to increased mortality (Fig. 1A) during 16 weeks of treatment due to a humoral immune response revealed by an increase in LAMAN‐specific IgG in the plasma of ERT‐treated mice (Fig. S1A). Treatment with 500 U/kg caused an earlier onset of the immune response compared to the low dose, whereas the survival rate was dosage‐independent and all mice died during treatment. To enable chronic ERT, we developed immune‐tolerant alpha‐mannosidosis mice by transgenic expression of an active site mutant of human LAMAN under the control of the β‐actin promotor in the alpha‐mannosidase knockout background. The insertion of a clinical mutation (c.215: A>T) within the human LAMAN cDNA leads to an exchange of a histidine to leucine at position 72 (H72L) in the active site resulting in an inactive form of the enzyme, which still localizes to lysosomes4, 5 (Fig. S1B–D). Finally, homozygote alpha‐mannosidase knockout mice that were heterozygote for the LAMAN‐Mut‐H72L transgene (‐/‐ +tg) were obtained. Transgene expression was validated by immunoblot analysis, which revealed a transgenic‐driven expression of LAMAN in all tissues investigated (brain, spleen, liver, heart, kidney, and lung) as depicted for brain and spleen in Figure 1B. Brain sections that were immunologically stained for LAMAN revealed broad expression of the transgene throughout the CNS, including cerebellum, hippocampus, and cortex (Fig. 1C). The specificity of the antibody was verified in nontransgenic knockout mice (Fig. S1E). Alpha‐mannosidase activity was detectable in the wild‐type (+/+) but not in lysates derived from the CNS (brain) and visceral tissues (spleen) of both knockout strains (‐/‐ +tg and ‐/‐) (Fig. 1D), indicating expression of transgenic inactive LAMAN. Finally, we determined LAMAN‐specific IgG in the plasma of transgenic and nontransgenic knockout mice which received seven weekly injections of 500 U/kg (Fig. 1E). Nontransgenic mice developed high antibody levels immediately after the first injection and died upon further treatment, whereas transgenic knockout animals developed only negligible plasma levels of LAMAN‐specific IgG and tolerated the injected enzyme. Overall, our data demonstrate successful induction of immune‐tolerance to recombinant LAMAN due to transgenic expression of an inactive variant of the enzyme.
Figure 1.
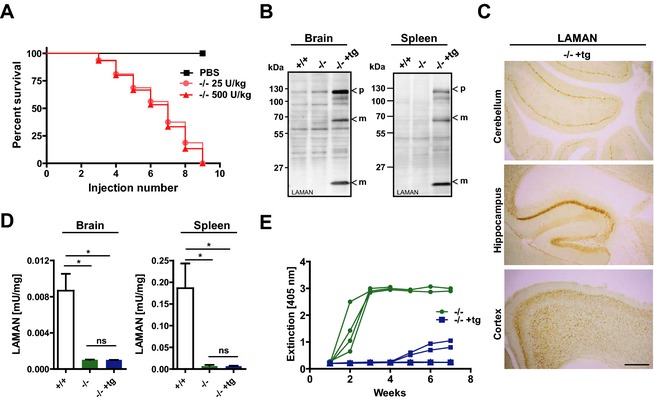
Transgenic alpha‐mannosidase knockout mice express inactive human LAMAN and are immune‐tolerant. (A) Repeated injections every other week of recombinant human LAMAN with doses of 25 and 500 U/kg into the tail vein of nontransgenic alpha‐mannosidase knockout mice (‐/‐) leads to premature death during 16 weeks of treatment whereas mock‐injected mice (‐/‐ PBS) were not affected. (B) Immunoblot analyses of tissue lysates revealed transgene driven expression of inactive human LAMAN in brain and spleen of transgenic 14 knockout mice (‐/‐ +tg) but not in nontransgenic wild‐type (+/+) and knockout (‐/‐) tissue. LAMAN‐specific bands detected at about 130, 70 and 14 kDa correspond to the precursor (p) and lysosomal processed forms (m) of LAMAN. (C) Transgenic expression of LAMAN throughout the central nervous system including cerebellum, hippocampus and cortex as determined by 3,3'‐Diaminobenzidine (DAB)‐immunohistochemistry, using a LAMAN‐specific antibody (Scale bars = 500 μm). (D) alpha‐mannosidase activity of brain and spleen homogenates indicates lack of enzymatic activity in both knockout mouse strains. (E) The antibody (LAMAN‐specific IgG) response in plasma of transgenic knockout mice compared to nontransgenic animals is reduced as determined by an ELISA after seven weekly injections of 500 U/kg (*P < 0.05). LAMAN, lysosomal acid alpha‐mannosidase; PBS, phosphate‐buffered saline; ELISA, enzyme‐linked immunosorbent assay.
Neuropathological characterization of immune‐tolerant alpha‐mannosidosis mice
We then assessed whether transgenic knockout mice reflect the human pathology comparable to the conventional alpha‐mannosidase knockout mice15 and are suitable to study the impact of chronic ERT on neuropathology. Therefore, transgenic and nontransgenic knockout mice as well as wild‐type mice were analyzed for storage and secondary pathological changes.8, 16 TLC analyses revealed comparable accumulation of oligosaccharides linked to 2‐9 mannose residues (Man2‐Man9) between both genotypes in brain (Fig. 2A) and spleen (Fig. S2A) which was confirmed by HPLC‐based quantification of Man2‐Man4 (Fig. 2B, Fig. S2B). In brain, both genotypes displayed a similar increase in the specific activity of the lysosomal enzymes β‐glucuronidase (GUS), β‐hexosaminidase and β‐galactosidase (Fig. 2C). The presence of storage vacuoles is a prominent feature of alpha‐mannosidosis in mice15, 16 and men.21, 22 Ultrastructural analyses revealed comparable presence of storage vacuoles in neurons of the ganglion trigeminale, hippocampus, and Purkinje cells of the cerebellum (Fig. 2D). Visceral tissues such as the pancreas also displayed a comparable storage phenotype between both genotypes (Fig. S2C). These storage vacuoles are strongly positive for the Lysosome‐associated membrane glycoprotein 1 (LAMP‐1).17 Morphological examination of brain sections immunologically stained for LAMP‐1 displayed a similar increase in vesicular labeling of LAMP‐1 within neurons of the hippocampus, cerebellum, and cortex (Fig. 2E) of both genotypes. Finally, we compared neuropathological alterations such as microglia activation which appear secondary to primary substrate storage.17 A similar degree of microglia activation as indicated by increased labeling with CD68 throughout the cortex was evident (Fig. 2F). We conclude that immune‐tolerant alpha‐mannosidosis mice are a valuable disease model to study the impact of preclinical long‐term ERT on neuropathology, using recombinant human enzyme.
Figure 2.
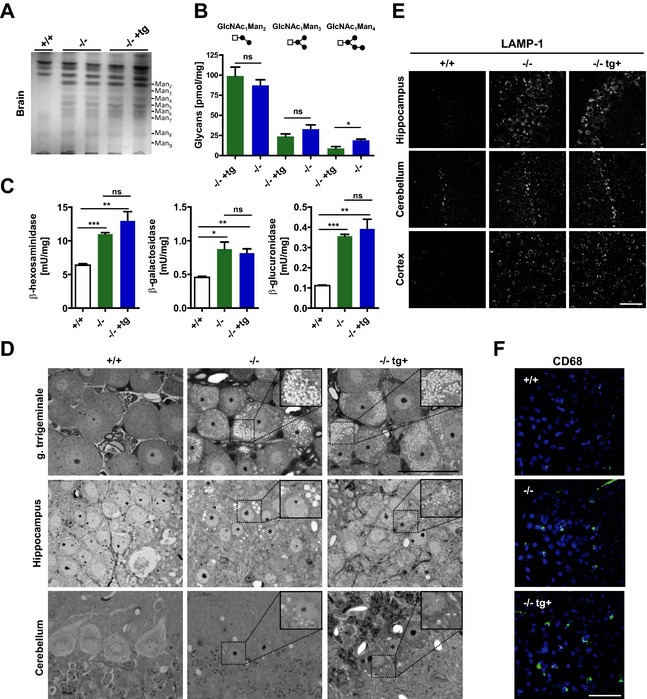
Immune‐tolerant transgenic alpha‐mannosidosis mice are phenotypically indistinguishable from nontransgenic animals. (A) Separation of neutral oligosaccharides extracted from brains of 3‐4 months old wild‐type (+/+), nontransgenic (‐/‐) and transgenic (‐/‐ +tg) mice by TLC shows comparable amounts of all glycan species (Man2–Man9). (B) Quantitative analysis of oligosaccharides by high pressure liquid chromatography indicate equal levels of the major glycan species Man2 and Man3 (n = 3‐4, per genotype). (C) α‐glucuronidase, β‐hexosaminidase and α‐galactosidase show a similar increase in their specific activity in brain extracts of nontransgenic and transgenic knockout mice compared to wild‐type animals (n = 3, per genotype). (D) Light microscopy of Toluidine‐stained semithin sections from the ganglion (G.) trigeminale, hippocampus and cerebellum reveals similar amount of storage vacuoles in both knockout strains. Scale bar: 50 μm, insets show zoomed images of the outlined area; stars (*) highlight vacuolated storage cells. (E) Immunofluorescence labeling of LAMP‐1 revealed a similar increase in LAMP‐1 levels in ‐/‐ and ‐/‐ tg+ mice compared to +/+ animals in the hippocampus, cerebellum and the cerebral cortex (Scale bar: 50 μm). (F) Reactive microglia (green) are present to a similar degree in the molecular layer of the cerebellum of ‐/‐ and ‐/‐ tg+ mice (4’,6‐Diamidin‐2‐phenylindol (DAPI), blue; Scale bar: 50 μm) (*P < 0.05, **P < 0.01, ***P < 0.001). TLC, thin layer chromatography; LAMP‐1, Lysosome‐associated membrane glycoprotein 1.
Effect of ERT on primary substrate storage in immune‐tolerant alpha‐mannosidosis mice
We next evaluated the efficacy of a 12‐week ERT protocol in reducing primary substrate storage of the CNS and peripheral tissues. In this experimental set‐up either nontransgenic wild‐type or nontransgenic heterozygote animals served as controls.15 In previous studies, we demonstrated that four biweekly injections of 500 U/kg rhLAMAN were necessary to significantly reduce CNS storage when applied short term.16 In order to investigate the chronic impact of this ERT regimen and whether chronic treatment can compensate for a lower dose, 4‐6 weeks old mice with an established blood‐brain barrier23, 24 were biweekly injected with 125 and 500 U/kg rhLAMAN (corresponding to 4.4 and 15.6 mg/kg per bw), respectively. ERT was completed at an age of 16‐18 weeks, a time point where alpha‐mannosidosis mice display a prominent storage phenotype.16, 17 Of note, for this and the following experiments prior to harvesting brain and other organs, mice were perfused with phosphate buffer to avoid contamination with plasma‐derived rhLAMAN. Enzyme injections led to a dose‐dependent increase of alpha‐mannosidase activity in brain reaching 72 % of the endogenous wild‐type level at 500 U/kg (Fig. 3A). Accordingly, TLC analysis of brain oligosaccharides revealed a dose‐dependent decrease of all glycan species in mice treated with 125 and 500 U/kg (Fig. 3B). Similar results were obtained by quantitative HPLC analysis which revealed a 37 % (125 U/kg) and 68 % (500 U/kg) decrease of the most abundant oligosaccharide species Man2 in rhLAMAN‐treated mice when compared to control mice (Fig. 3C). In wild‐type brains, mannosylated‐glycans were below detection levels. Histological examination of brain sections revealed less storage vacuoles in hippocampal neurons of 125 U/kg treated animals in comparison to control mice, whereas use of 500 U/kg led to an almost complete disappearance of storage vacuoles (Fig. 3D). The normalization of lysosomal function in response to the applied ERT regimen was verified by a significant decline in GUS and β‐hexosaminidase activity (Fig. 3E). Furthermore, a decrease in LAMP‐1 immunoreactivity in brain sections of ERT‐treated mice was also evident (Fig. 3F). Among all brain regions investigated, hippocampal and cortical neurons responded best to ERT as indicated by a gradually decrease in LAMP‐1 immunoreactivity after treatment with either 125 or 500 U/kg, respectively. The cerebellum apparently benefited least from the applied ERT regimen (Fig. 3F, lower panel). In contrast to the CNS, peripheral tissues such as kidney (Fig. S3A and B) and spleen (Fig. S3C and D) showed an efficient clearance of oligosaccharides as determined by TLC and HPLC analyses at either dose. This was also reflected by a complete absence of storage vacuoles within all histologically examined peripheral tissues, including pancreas, kidney, and ganglion trigeminale (Fig. S3E). Our data suggest an efficient and beneficial uptake of chronically injected rhLAMAN into cells of visceral tissues and the CNS with a clear dose‐ and regional‐dependent effect in the brain.
Figure 3.
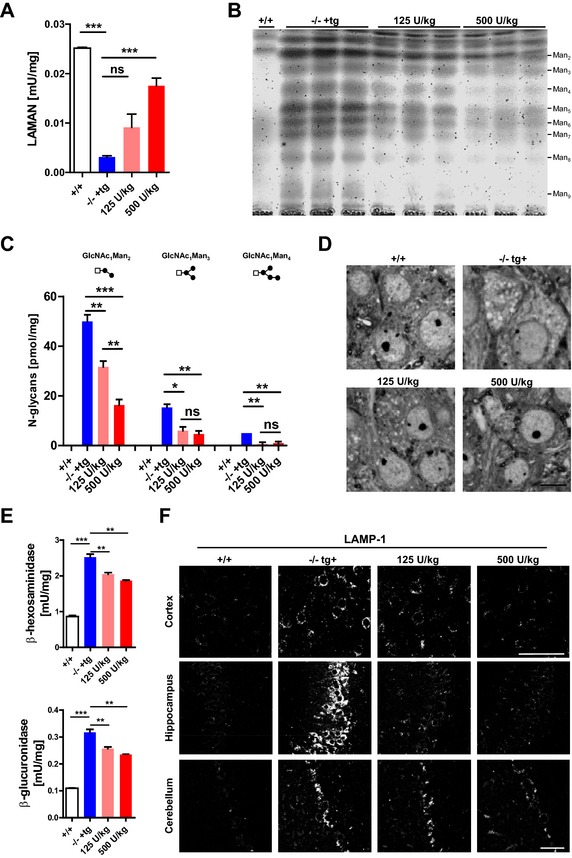
Long‐term high‐dose ERT efficiently reduces oligosaccharide storage within brain. (A) Alpha‐mannosidase activity increases dose‐dependently in brain homogenates of transgenic (‐/‐ +tg) mice after biweekly injections of 125 or 500 U/kg over 12 weeks of treatment. (B) TLC analysis of oligosaccharide brain extracts of wild‐type (+/+) and both knockout strains shows decreased levels of all major oligosaccharide species in ‐/‐ +tg mice after ERT with both indicated doses. (C) HPLC‐based quantification reveals a significant dose‐dependent decrease of Man2 and Man3 in treated ‐/‐ tg+ mice when compared to mock treated animals. (D) Toluidine blue‐stained semithin sections display less storage vacuoles in high dose (500 U/kg) treated ‐/‐ +tg mice when compared to non‐ and 125 U/kg treated mice. (E) ERT leads to a significant dose‐dependent decrease of lysosomal enzyme activities such as β‐hexosaminidase and α‐galactosidase and (F) a normalization of LAMP‐1 immunoreactivity in cerebral cortex (upper panel) and hippocampus (middle panel) but not in Purkinje cells of the cerebellum (*P < 0.05, **P < 0.01, ***P < 0.001). ERT, enzyme replacement therapy; TLC, thin layer chromatography; HPLC, high‐performance liquid chromatography; LAMP‐1, Lysosome‐associated membrane glycoprotein 1.
Effect of ERT on secondary neuropathological alterations in immune‐tolerant alpha‐mannosidosis mice
After evaluating the long‐term effect of ERT on neuronal oligosaccharide accumulation and lysosomal morphology we determined the effect of ERT on secondary neuropathological alterations in elder mice in which neuropathology, including microgliosis is apparent. The ERT regimen was started at 4 months of age by injecting (biweekly) 500 U/kg rhLAMAN (corresponding to 15.6 mg/kg per bw) during 14 weeks. The efficacy of ERT was verified by a decline in primary oligosaccharide storage and LAMP‐1 levels as well as a normalization of lysosomal enzyme activities (Fig. S4). Chronic treatment of immune‐tolerant mice significantly attenuated microglia activation in the hippocampus, cerebral cortex, and thalamus as determined by immunofluorescence staining for CD68. However, no positive effect of ERT treatment was observed in the cerebellum, where phagocytic microglia (Fig. 4A) and reactive Bergmann glia persisted even after long‐term ERT (Fig. 4B).
Figure 4.
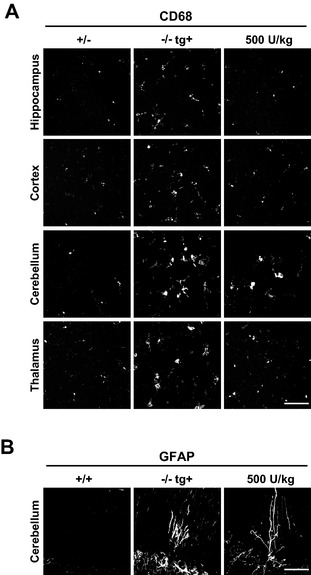
Long‐term high‐dose enzyme replacement therapy (ERT) attenuates microgliosis in most parts of the brain. (A) Immunofluorescence of brain sections using the glial marker CD68 and GFAP reveals less activation of CD68 positive microglia after long‐term ERT (14 weeks) with biweekly injections of 500 U/kg in the hippocampus, cerebral cortex and thalamus. In the cerebellum, no obvious change in the presence of activated microglia (A, lower panel) or GFAP positive Bergman glia (B) is evident (Scale bar: A, 50 μm; B, 100 μm). In (A) representative z stack images with maximum projection are shown.
Effect of ERT on behavioral anomalies observed in immune‐tolerant alpha‐mannosidosis mice
We evaluated the functional benefit in the same cohort of long‐term ERT‐treated mice described above, using a behavioral test battery including open field (exploration and emotion), water maze (learning and memory), and treadmill (motor performance). Exploratory locomotion was video‐tracked during 10 min in a brightly illuminated open field arena. Different zones were identified in the analysis arena (corners, center) as well as time intervals selected to evaluate spatial and temporal components of exploration. Spatial analysis of the tracks revealed significant differences between the groups for time in the center (Fig. 5A; P < 0.05) and % path length in the center (Fig. 5B; P < 0.05), and a trend for the number of center entries (P = 0.06; not shown). In general, ‐/‐ tg+ mice displayed reduced exploration of the center of the arena showing no improvement after ERT. However, temporal analysis of exploration showed different relationships between the experimental groups. Analysis of cumulative path length (Fig. 5C) indicated an early onset decline in mock‐treated ‐/‐ tg+ mice, while enzyme‐treated animals progressively fell behind control mice. Indeed, when dividing the 10 min exploration time into three equal intervals (3 min 20 sec; Fig. 5D), there was a significant Group × Interval interaction on total path length (P < 0.05). Control mice (P < 0.05) showed increased exploratory activity in the first interval in comparison with mock‐treated ‐/‐ tg+ mice. Of note, mice receiving enzyme treatment were indistinguishable from control mice. Moreover, control mice and enzyme‐treated ‐/‐ tg+ mice showed similar distribution of exploratory activity, indicated by an initial exploratory peak and significant decline of activity over time. There was a significant difference between interval I and III for control mice (P < 0.01) and enzyme‐treated ‐/‐ tg+ mice (P < 0.001). On the contrary, mock‐treated ‐/‐ tg+ mice showed no changes in exploration over time. In conclusion, ERT did not improve central exploratory measures in ‐/‐ tg+ mice but normalized the temporal aspect of exploration.
Figure 5.
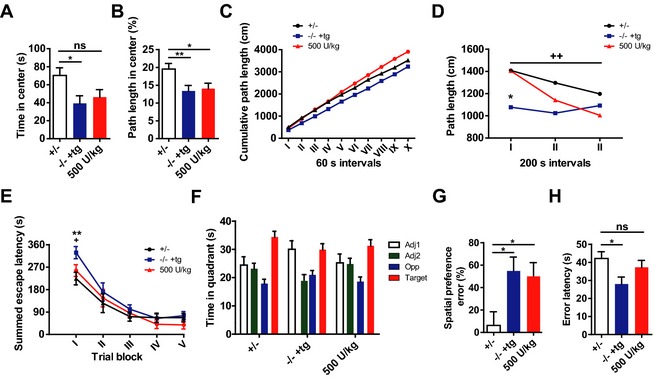
Long‐term high‐dose enzyme replacement therapy (ERT) partially rescues behavioral deficits. After 12–14 weeks of biweekly ERT with 500 U/kg, decreased exploratory activity of ‐/‐ tg+ mice is partially rescued (A–D). No improvement was found in spatial parameters, as reduced exploration of the center was unaltered (A; time in center and [B] % path length in center; main effects of Group P < 0.05). Cumulative path length indicated changes in temporal distribution of exploratory activity however (C). ERT‐treated ‐/‐ tg+ mice showed an initial exploratory peak in the beginning of the test with a progressive decline similar to control mice ([D]; interaction effect of Group × Time P < 0.05). Improvements after long‐term ERT were also found in the water maze task (E–G). Learning impairment in the acquisition phase was corrected after long‐term ERT (E; day I main effect of Group P < 0.05). However, no benefit was observed for long‐term memory in the probe trial. Control mice showed consistent preference for the target quadrant, while this was less pronounced for both ‐/‐ tg+ groups (F). Erroneous preference for other quadrants was indeed more frequent in both ‐/‐ tg+ groups, independent of treatment (G; main effect of Group P < 0.05). Treadmill performance was improved as well, as reflected in an increase of error latencies (H; main effect of Group P < 0.05). Asterisks indicate significant difference versus the control group: *P < 0.05; **P < 0.01. Plus‐signs indicate significance of difference versus mock‐treated ‐/‐ tg+ mice: + P < 0.05; ++ P < 0.01.
During the acquisition phase of the water maze experiment, mice were trained for 5 days (trial blocks I‐V) to learn the location of a hidden escape platform (Fig. 5E). Mock‐treated ‐/‐ tg+ mice were slower to locate the platform during the first trial block than control and ERT‐treated ‐/‐ tg+ mice (P < 0.05 and <0.01), but eventually reached a similar level of performance. After 2 days of rest, a 100 sec probe trial is performed during which the platform is removed. Preference for the target quadrant is analyzed as an index of long‐term reference memory. Control mice showed a preference for the target quadrant, spending significantly more time there in comparison with the three other quadrants (Fig. 5F). ERT‐treated ‐/‐ tg+ mice spent most time in the target quadrant, but displayed less spatial accuracy (no significant difference vs. the adjacent quadrants). Mock‐treated ‐/‐ tg+ mice preferred the target quadrant, but also quadrant Adj1 over Adj2 and the opposite quadrant. Individual analysis of spatial preference (Fig. 5G) revealed that 55 % of the mock‐treated ‐/‐ tg+ mice (P < 0.05) and 50 % of the ERT‐treated ‐/‐ tg+ mice (P < 0.05) showed an erroneous preference for another quadrant, while this was only the case for 6 % of control mice (Group effect P < 0.05). In conclusion, The ERT treatment did not improve the impaired long‐term reference memory, but led to a correction of the learning deficit in the first acquisition trial block, which more likely reflects improvements in short‐term working memory.
During the treadmill experiment (Fig. 5H), motor performance is analyzed by evaluating the ability of mice to keep pace with a motorized treadmill belt. Mock‐treated ‐/‐ tg+ mice showed significantly less error latencies in comparison with the control group (P < 0.05), while enzyme treatment revealed a trend of ‐/‐ tg+ mice for increased error latencies. This confirms previous findings of improved motor performance after short‐term ERT.
Uptake studies of rhLAMAN in brain and primary cultured neuronal and glial cells
We next investigated the clearance of rhLAMAN from circulation as well as the accessibility of the CNS to the injected enzyme by injecting one‐single high enzyme dose (1000 U/kg, corresponding to 31.2 mg/kg per bw) to obtain measurable enzyme activity in brain.16 At different time points, after administration (5 min, 1 h, 5 h, and 16 h), blood was taken and LAMAN activity determined. We included both wild‐type and nontransgenic knockout mice in our analysis to exclude genotype‐specific effects such as impairment of the blood‐brain barrier or the presence of high levels of mannose‐rich oligosaccharides in the serum of alpha‐mannosidosis mice that might influence enzyme uptake by competing with the recombinant glycoprotein via carbohydrate‐specific receptors. As shown in Figure 6A, in both genotypes, the injected enzyme was cleared from circulation rapidly and after 16 h only about a tenth of the initial enzyme activity in plasma was detectable. LAMAN activity measurements 16 h after injection indicate a similar uptake of enzyme into the brain of wild‐type and knockout mice. Furthermore, a single injection of 1000 U/kg rhLAMAN is sufficient to fully restore endogenous levels of LAMAN activity in brain lacking alpha‐mannosidase activity (Fig. 6B).
Figure 6.
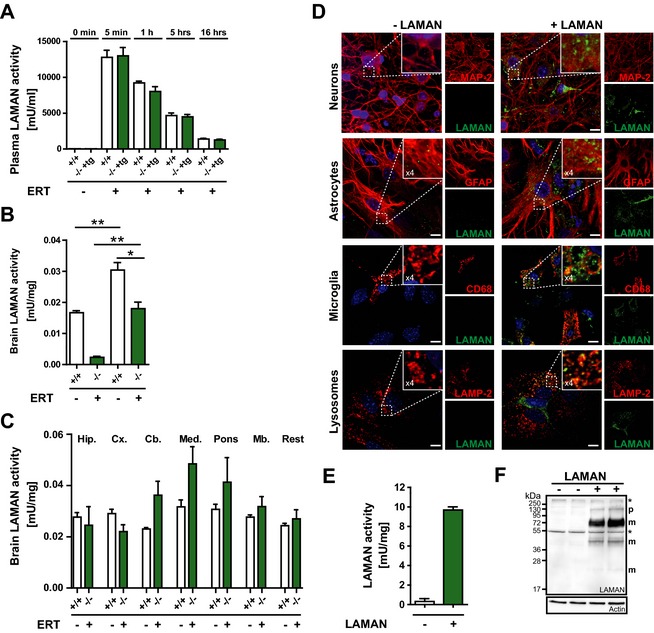
Broad uptake of rhLAMAN into brain fully restores endogenous LAMAN activity. (A) Wild‐type (+/+) and transgenic knockout mice (‐/‐ tg+) received one single injection of 1000 U/kg and LAMAN plasma activity measured at different time (t) points (before injection [0], 5 min, 1 h, 5h, and 16 h). 16 h after injection mice were perfused with phosphate buffer to eliminate remaining enzyme from circulation and LAMAN activity determined in (B) whole brain and (C) isolated brain regions, respectively (*P < 0.05, **P < 0.01). (D) Mixed culture of neurons, astrocytes and microglial cells incubated with rhLAMAN (green) for 16 h. Neurons were visualized, using MAP‐2 as a marker (red), whereas astrocytes and microglia are highlighted by staining with markers against GFAP (red) and CD68 (red), respectively. Lysosomal localization of rhLAMAN is reflected by co‐staining with LAMP‐2 (red). (E) Enhanced LAMAN activity was observed in the same mixed cultures after treatment with the enzyme. (F) Similarly, uptake of the enzyme was verified via immunoblotting. (Scale bar: 10 μm). rhLAMAN, recombinant human lysosomal acid alpha‐mannosidase; LAMP‐2, lysosomal‐associated membrane protein 2.
Our morphological analyses suggest a differential response of various brain regions to high‐dose ERT. To determine whether a different accessibility of rhLAMAN to various brain regions accounts for this effect, we have used the same injection protocol as described above and measured LAMAN activity within different brain regions, including hippocampus, cortex, cerebellum, medulla oblongata, pons and midbrain (Fig. 6C). In all brain regions investigated, the applied ERT regimen was sufficient to reach endogenous LAMAN activity with no significant differences between LAMAN activities in various brain regions.
To evaluate the ability of rhLAMAN to target different cell types of the CNS, we have performed in vitro uptake experiments using mixed primary neuronal and glial primary cultures followed by immunofluorescence staining, using an rhLAMAN‐specific antibody. Co‐staining with different cellular markers such as the Microtubule‐associated protein type 2 (MAP‐2), the Glial fibrillary acidic protein (GFAP) and macrosialin/CD68 were used to identify neurons, astroglia, and microglia cells, respectively (Fig. 6D). RhLAMAN was preferentially taken up by CD68‐labeled microglia cells whereas only a few neurons and astrocytes stained positive for the recombinant enzyme. Lysosomal localization of rhLAMAN was verified by co‐staining with LAMP‐2. Enhanced LAMAN activity was observed in the same mixed cultures after treatment with the enzyme (Fig. 6E). Similarly, uptake of the enzyme was verified via immunoblotting (Fig. 6F). Overall, our data indicate rapid clearance of rhLAMAN from circulation and a preferential uptake into microglial cells in vitro. Importantly, a single injection of a high dose of rhLAMAN is sufficient to restore endogenous alpha‐mannosidase activity in the brain.
Discussion
Lysosomal storage disorders (LSDs) are devastating, mostly neurodegenerative diseases with limited therapeutic options. However, despite the long‐standing dogma that lysosomal enzymes cannot cross the blood‐brain barrier, within recent years, preclinical research has demonstrated the power of specific ERT regimens to even ameliorate CNS pathology. In this paper, we describe the generation of a novel immune‐tolerant mouse model for alpha‐mannosidosis showing histopathological, biochemical and behavioral characteristics comparable to the conventional alpha‐mannosidase knockout mice.15 A similar approach, describing the successful induction of immune tolerance by transgenic expression of an active site mutant in various LSD mouse models has been used by others.25, 26, 27 Immune‐tolerant mice were instrumental in investigating the effect of long‐term ERT in other LSDs such as Pompe disease,28 mucopolysaccharidosis‐type VII (MPSVII),26 and metachromatic leukodystrophy.25 Beside its value for systemic high‐dose ERT, our immune‐tolerant mouse model is also useful in order to test other therapeutic approaches such as intrathecal delivery of (un)modified rh LAMAN preparations or gene therapy. In contrast to visceral tissues, the long‐term benefit of ERT on neuronal function in alpha‐mannosidosis mice, represented by a substantial decline in primary substrate storage and a normalization of lysosomal function as well as an improvement of behavioral deficits, was only apparent after high‐dose enzyme injections (500 U/kg). This is in agreement with our previous studies16 showing that high enzyme doses are necessary to get sufficient amounts of enzyme into the CNS leading to a substantial reduction of brain storage when applied short term, suggesting a dose‐dependent uptake of the recombinant enzyme into the brain. Our studies are in agreement with other preclinical ERT studies in LSD animal models showing mostly beneficial effects of high‐dose injections of recombinant enzymes on primary lysosomal storage reduction and improvement of secondary pathological alterations such as neuroinflammation and behavioral deficits.29, 30, 31, 32, 33 Of note, in our preclinical study, the beneficial effect of ERT was most prominent for the 500 U/kg dosage but still significant at 125 U/kg for most of the parameters analyzed, suggesting that chronic treatment can at least to some extent compensate for lower enzyme activity. The precise mechanism as to how the systemically applied recombinant enzymes can cross the blood‐brain barrier remains to be established. According to Sly and coworkers using recombinant GUS in preclinical ERT studies with MPSVII mice,24, 34 mannose‐6 phosphate receptors (MPRs) that are responsible for intracellular lysosomal targeting of most lysosomes enzymes and their cellular uptake35, 36 cannot account for the uptake of recombinant lysosomal enzymes into adult brain due to their restricted expression in brain endothelial cells within the perinatal period (2 weeks after birth). In adult animals which lack expression of endothelial MPRs, administration of recombinant enzymes bearing mannose 6‐phosphate (M6P) residues might even diminish their uptake into brain as demonstrated for GUS.24, 30, 34 Of note, recombinant rhLAMAN which, in comparison with other lysosomal enzymes, is highly efficient in clearance of brain primary substrate storage is only marginally phosphorylated, suggesting primarily M6P‐independent uptake.16 Low phosphorylation content was shown to correlate with an increased half‐life of recombinant enzymes such as GUS34, 37 and alpha‐mannosidase.38 A prolonged circulation was suggested to enable the transfer of higher amounts of lysosomal enzymes into the brain. Therefore, deglycosylation or chemical modification was used to increase the circulatory half‐life of lysosomal enzymes such as Tripeptidyl peptidase type 1 (TTP1)39 and GUS.30, 40 However, in contrast to GUS, glycan modification of TTP1 did not improve its targeting to brain. Further uptake studies, using different preparations of recombinant enzymes that are commercially available are therefore necessary to elucidate whether modification of the glycan structure and/or a low phosphorylation content in general, or only in the case of specific enzymes can improve brain uptake. Furthermore, it should be investigated if an earlier onset of treatment might result in improved efficacy of ERT, as described for other preclinical ERT in murine LSD models.41, 42, 43, 44 Impairment of the blood–brain barrier integrity in LSDs represents another pathway of how an enzyme can reach the brain.14 However, in our preclinical studies, the uptake of rhLAMAN into the brain of wild‐type and alpha‐mannosidosis mice was comparable, suggesting that the specific nature of the enzyme rather than permeable tight‐junctions account for the observed brain uptake. The application of high enzyme doses is not a guarantee for sufficient brain uptake since several other ERT approaches did not show beneficial effects on the CNS even upon use of high doses.45, 46, 47
Most ERT studies showing beneficial effects on the CNS reached only the sub‐endogenous enzyme activity level.32, 46, 48 In contrast, with the applied ERT regimen, we fully restored endogenous LAMAN activity levels in all CNS regions investigated. Of note, in comparison with other lysosomal enzymes such as β‐hexosaminidase and GUS, the endogenous activity of LAMAN in brain is extremely low and 20‐fold less as compared to the liver. Therefore, it is tempting to speculate that due to the low endogenous activity of LAMAN in the CNS, only minor fractions are needed to restore endogenous activities.
It should be highlighted that by including a factor of 10 that relates surface area to body weight between mouse and men,49 the applied 500 U/kg dosage used in our preclinical studies exactly matches the dose of 50 U/g applied weekly to alpha‐mannosidosis patients in a first phase I–II clinical trial.19 Remarkably, despite the low number of patients (n = 10), chronic ERT of 12 months led to a significant decline of oligosaccharide levels in urine, serum, and cerebrospinal fluid (CSF) as well as decreased levels of the CSF‐biomarkers Tau‐protein and GFAP. Moreover, a significant improvement of motor abilities and most notably of cognitive functions was achieved, indicating that our findings in the mouse model can be well assigned to the clinical studies, at least for patients with enzymatically inactive LAMAN variants. Patients lacking expression of LAMAN1, 3, 6 may develop antibodies against the recombinant enzyme affecting ERT efficacy. However, in this first clinical study for alpha‐mannosidosis the frequency of infusion‐related reactions as well as the production of antibodies against the recombinant enzyme was low19 in comparison with other ERT studies.50, 51, 52, 53, 54 Furthermore, these rhLAMAN‐specific antibodies were shown not to be neutralizing, suggesting only mild immunological complications with regard to future ERT. Our results implicate that a further increase in enzyme dosing could be beneficial to the treatment of the disease and that alpha‐mannosidosis might be the first LSD for which systemic ERT leads to significantly improved CNS pathology.
Conflict of Interest
None declared.
Supporting information
Figure S1. Development of rhLAMAN‐specific IgG in alpha‐mannosidase knockout mice and generation of immune‐tolerant alpha‐mannosidosis mice. (A) Conventional nontransgenic alpha‐mannosidase knockout mice (‐/‐) develop significant antibody responses after the second injection of recombinant rhLAMAN as determined by ELISA measuring LAMAN‐specific IgG in plasma of both low (25 U/kg) and high (500 U/kg) dose‐treated animals. (B) Schematic drawing of the pTVC vector containing the mutated cDNA of human LAMAN carrying the clinical H72L mutation under the control of the beta‐actin promotor. The SalI/NotI‐excised expression cassette of pTVC‐LAMAN(MutH72L was injected into the pronucleus of fertilized oocytes. (C) Genotyping strategy with exon‐specific primers flanking the inserted H72L mutation (hLAMAN‐H72L‐F: 5′‐CACACACTTGATGACGTCGGC‐3′ and hLAMAN‐670R: 5′‐GATAATCAAGGCGCC CAAAGAAG‐3′) identified three transgene‐positive founder lines (lines 4, 8 and 11). (D) Breeding scheme for the generation of immune‐tolerant transgenic knockout mice (‐/‐ +tg): transgene positive founder mice (Founder line #4) were crossed with alpha‐mannosidase knockout mice (‐/‐) in the F0 generation. Transgene positive LAMAN heterozygote animals were further crossed in the F1 generation with homozygous alpha‐mannosidase knockout mice (‐/‐) in order to obtain transgene‐positive transgenic knockout mice (‐/‐ +tg) in the homozygote LAMAN background in the F2 generation. (E) Antibody specificity for the rhLAMAN staining shown in Figure 1 was proved by absence of signals in DAB‐stained brain sections of nontransgenic alpha‐mannosidase knockout mice (‐/‐) using a rhLAMAN‐specific antibody. The images presented in Figure 1C and E were acquired under the same experimental and microscopical settings.”
Figure S2. Immune‐tolerant alpha‐mannosidase knockout mice are phenotypically indistinguishable from nontransgenic animals. (A) Separation of neutral oligosaccharides extracted from spleens of 3–4 months old wild‐type (+/+), nontransgenic (‐/‐) and transgenic (‐/‐ +tg) mice by thin layer chromatography (TLC) shows comparable amounts of all glycan species (Man2–Man9). (B) Quantitative analysis of oligosaccharides by high‐performance liquid chromatography (HPLC) indicate equal levels of the major glycan species Man2 and Man3 (n = 3–4, per genotype). Light microscopy of Toluidine‐stained semi‐thin sections from the pancreas reveals similar amounts of storage vacuoles in both knockout strains (Scale bar: 50 μm).
Figure S3. Long‐term high‐dose ERT efficiently reduces oligosaccharide storage within peripheral tissues. (A) TLC analysis and (B) HPLC of oligosaccharides extracted from kidney of wild‐type (+/+) and both knockout strains shows complete clearance of all major oligosaccharide species in ‐/‐ +tg mice after ERT with both indicated doses. Similar results were obtained for the spleen as determined by both (C) TLC and (D) HPLC analyses. (E) Light microscopy of Toluidine‐stained ultrathin sections from the pancreas, the kidney and trigeminal ganglion of untreated and 125 U/kg treated animals shows complete disappearance of vacuoles in all tissues after ERT.
Figure S4. Long‐term high‐dose ERT efficiently reduces oligosaccharide storage in the brain of mice with established neuropathology. (A) The specific activity of α‐glucuronidase and β‐hexosaminidase show a normalization to control (+/‐) levels after rhLAMAN treatment with 500 U/kg (n = 4–6, per genotype). (B) TLC analysis of oligosaccharide brain extracts (upper panel) of both knockout strains shows decreased levels of all major oligosaccharide species in ‐/‐ +tg mice after ERT with 500 U/kg. Quantitative clearance of oligosaccharides is also observed in the spleen (lower panel). (C) HPLC‐based quantification revealed a clear decrease of Man2–Man4 oligosaccharides in treated ‐/‐ tg+ mice when compared to untreated ‐/‐ +tg mice animals in the brain (upper panel) and a nearly complete clearance in the spleen (lower panel). (D) Immunofluorescence labeling of LAMP‐1 revealed a normalization of LAMP‐1 levels in ‐/‐ and ‐/‐ tg+ mice compared to control mice (+/) in the hippocampus, cerebellum, and the cerebral cortex (Scale bar: 50 μm).
Data S1. Supplemental methods.
Acknowledgment
This work was supported by the Research Training Group (GRK1459), funded by the Deutsche Forschungsgemeinschaft to J. B., and an EU grant to P. S. and J. B. (EU/ALPHA‐MAN 261331). S. S. received support from the MM Delacroix foundation. We thank Meryem Senkara, Inez Götting, and Dagmar Niemeier for excellent technical assistance. The authors thank William Sly (St. Louis, MO, USA) for providing the plasmid pTVC.
References
- 1. Malm D, Nilssen O. Alpha‐mannosidosis. Orphanet J Rare Dis 2008;3:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ockerman PA. Deficiency of beta‐galactosidase and alpha‐mannosidase–primary enzyme defects in gargoylism and a new generalized disease? Acta Paediatr Scand 1967:suppl 177:135–176. [PubMed] [Google Scholar]
- 3. Berg T, Riise HM, Hansen GM, et al. Spectrum of mutations in alpha‐mannosidosis. Am J Hum Genet 1999;64:77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gotoda Y, Wakamatsu N, Kawai H, et al. Missense and nonsense mutations in the lysosomal alpha‐mannosidase gene (MANB) in severe and mild forms of alpha‐mannosidosis. Am J Hum Genet 1998;63:1015–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hansen G, Berg T, Riise Stensland HM, et al. Intracellular transport of human lysosomal alpha‐mannosidase and alpha‐mannosidosis‐related mutants. Biochem J 2004;381:537–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Riise Stensland HM, Klenow HB, Van Nguyen L, et al. Identification of 83 novel alpha‐mannosidosis‐associated sequence variants: functional analysis of MAN2B1 missense mutations. Hum Mutat 2012;33:511–520. [DOI] [PubMed] [Google Scholar]
- 7. Michalski JC, Klein A. Glycoprotein lysosomal storage disorders: alpha‐ and beta‐mannosidosis, fucosidosis and alpha‐N‐acetylgalactosaminidase deficiency. Biochim Biophys Acta 1999;1455:69–84. [DOI] [PubMed] [Google Scholar]
- 8. Damme M, Morelle W, Schmidt B, et al. Impaired lysosomal trimming of N‐linked oligosaccharides leads to hyperglycosylation of native lysosomal proteins in mice with alpha‐mannosidosis. Mol Cell Biol 2010;30:273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Beck M, Olsen KJ, Wraith JE, et al. Natural history of alpha mannosidosis a longitudinal study. Orphanet J Rare Dis 2013;8:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Malm D, Riise Stensland HM, Edvardsen O, Nilssen O. The natural course and complications of alpha‐mannosidosis–a retrospective and descriptive study. J Inherit Metab Dis 2014;37:79–82. [DOI] [PubMed] [Google Scholar]
- 11. Walkley SU, Thrall MA, Dobrenis K, et al. Bone marrow transplantation corrects the enzyme defect in neurons of the central nervous system in a lysosomal storage disease. Proc Natl Acad Sci USA 1994;91:2970–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mynarek M, Tolar J, Albert MH, et al. Allogeneic hematopoietic SCT for alpha‐mannosidosis: an analysis of 17 patients. Bone Marrow Transplant 2012;47:352–359. [DOI] [PubMed] [Google Scholar]
- 13. Migeon BR, Sprenkle JA, Liebaers I, et al. X‐linked Hunter syndrome: the heterozygous phenotype in cell culture. Am J Hum Genet 1977;29:448–454. [PMC free article] [PubMed] [Google Scholar]
- 14. Lachmann RH. Enzyme replacement therapy for lysosomal storage diseases. Curr Opin Pediatr 2011;23:588–593. [DOI] [PubMed] [Google Scholar]
- 15. Stinchi S, Lullmann‐Rauch R, Hartmann D, et al. Targeted disruption of the lysosomal alpha‐mannosidase gene results in mice resembling a mild form of human alpha‐mannosidosis. Hum Mol Genet 1999;8:1365–1372. [DOI] [PubMed] [Google Scholar]
- 16. Blanz J, Stroobants S, Lullmann‐Rauch R, et al. Reversal of peripheral and central neural storage and ataxia after recombinant enzyme replacement therapy in alpha‐mannosidosis mice. Hum Mol Genet 2008;17:3437–3445. [DOI] [PubMed] [Google Scholar]
- 17. Damme M, Stroobants S, Walkley S, et al. Cerebellar alterations and gait defects as therapeutic outcome measures for enzyme replacement therapy in α‐mannosidosis. J Neuropathol Exp Neurol 2011;70:83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Roces DP, Lullmann‐Rauch R, Peng J, et al. Efficacy of enzyme replacement therapy in alpha‐mannosidosis mice: a preclinical animal study. Hum Mol Genet 2004;13:1979–1988. [DOI] [PubMed] [Google Scholar]
- 19. Borgwardt L, Dali CI, Fogh J, et al. Enzyme replacement therapy for alpha‐mannosidosis: 12 months follow‐up of a single centre, randomised, multiple dose study. J Inherit Metab Dis 2013;36:1015–1024. [DOI] [PubMed] [Google Scholar]
- 20. Dobrenis K, Chang HY, Pina‐Benabou MH, et al. Human and mouse microglia express connexin36, and functional gap junctions are formed between rodent microglia and neurons. J Neurosci Res 2005;82:306–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kjellman B, Gamstorp I, Brun A, et al. Mannosidosis: a clinical and histopathologic study. J Pediatr 1969;75:366–373. [DOI] [PubMed] [Google Scholar]
- 22. Sung JH, Hayano M, Desnick RJ. Mannosidosis: pathology of the nervous system. J Neuropathol Exp Neurol 1977;36:807–820. [DOI] [PubMed] [Google Scholar]
- 23. Bar T. The vascular system of the cerebral cortex. Adv Anat Embryol Cell Biol 1980;59:I–VI, 1–62. [DOI] [PubMed] [Google Scholar]
- 24. Urayama A, Grubb JH, Sly WS, Banks WA. Mannose 6‐phosphate receptor‐mediated transport of sulfamidase across the blood‐brain barrier in the newborn mouse. Mol Ther 2008;16:1261–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Matzner U, Matthes F, Herbst E, et al. Induction of tolerance to human arylsulfatase A in a mouse model of metachromatic leukodystrophy. Mol Med 2007;13:471–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sly WS, Vogler C, Grubb JH, et al. Active site mutant transgene confers tolerance to human beta‐glucuronidase without affecting the phenotype of MPS VII mice. Proc Natl Acad Sci USA 2001;98:2205–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tomatsu S, Orii KO, Vogler C, et al. Production of MPS VII mouse (Gus(tm(hE540A x mE536A)Sly)) doubly tolerant to human and mouse beta‐glucuronidase. Hum Mol Genet 2003;12:961–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Raben N, Nagaraju K, Lee A, et al. Induction of tolerance to a recombinant human enzyme, acid alpha‐glucosidase, in enzyme deficient knockout mice. Transgenic Res 2003;12:171–178. [DOI] [PubMed] [Google Scholar]
- 29. Ahn SY, Chang YS, Sung DK, et al. High‐dose enzyme replacement therapy attenuates cerebroventriculomegaly in a mouse model of mucopolysaccharidosis type II. J Hum Genet 2013;58:728–733. [DOI] [PubMed] [Google Scholar]
- 30. Grubb JH, Vogler C, Levy B, et al. Chemically modified beta‐glucuronidase crosses blood‐brain barrier and clears neuronal storage in murine mucopolysaccharidosis VII. Proc Natl Acad Sci USA 2008;105:2616–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Matzner U, Lullmann‐Rauch R, Stroobants S, et al. Enzyme replacement improves ataxic gait and central nervous system histopathology in a mouse model of metachromatic leukodystrophy. Mol Ther 2009;17:600–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ou L, Herzog T, Koniar BL, et al. High‐dose enzyme replacement therapy in murine Hurler syndrome. Mol Genet Metab 2014;111:116–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vogler C, Levy B, Grubb JH, et al. Overcoming the blood‐brain barrier with high‐dose enzyme replacement therapy in murine mucopolysaccharidosis VII. Proc Natl Acad Sci USA 2005;102:14777–14782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Urayama A, Grubb JH, Sly WS, Banks WA. Developmentally regulated mannose 6‐phosphate receptor‐mediated transport of a lysosomal enzyme across the blood‐brain barrier. Proc Natl Acad Sci USA 2004;101:12658–12663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Braulke T, Bonifacino JS. Sorting of lysosomal proteins. Biochim Biophys Acta 2009;1793:605–614. [DOI] [PubMed] [Google Scholar]
- 36. Kornfeld S. Trafficking of lysosomal enzymes. FASEB J 1987;1:462–468. [DOI] [PubMed] [Google Scholar]
- 37. Urayama A, Grubb JH, Banks WA, Sly WS. Epinephrine enhances lysosomal enzyme delivery across the blood brain barrier by up‐regulation of the mannose 6‐phosphate receptor. Proc Natl Acad Sci USA 2007;104:12873–12878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Crawley AC, King B, Berg T, et al. Enzyme replacement therapy in alpha‐mannosidosis guinea‐pigs. Mol Genet Metab 2006;89:48–57. [DOI] [PubMed] [Google Scholar]
- 39. Meng Y, Sohar I, Wang L, et al. Systemic administration of tripeptidyl peptidase I in a mouse model of late infantile neuronal ceroid lipofuscinosis: effect of glycan modification. PLoS One 2012;7:e40509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Huynh HT, Grubb JH, Vogler C, Sly WS. Biochemical evidence for superior correction of neuronal storage by chemically modified enzyme in murine mucopolysaccharidosis VII. Proc Natl Acad Sci USA 2012;109:17022–17027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dunder U, Valtonen P, Kelo E, Mononen I. Early initiation of enzyme replacement therapy improves metabolic correction in the brain tissue of aspartylglycosaminuria mice. J Inherit Metab Dis 2010;33:611–617. [DOI] [PubMed] [Google Scholar]
- 42. Gliddon BL, Hopwood JJ. Enzyme‐replacement therapy from birth delays the development of behavior and learning problems in mucopolysaccharidosis type IIIA mice. Pediatr Res 2004;56:65–72. [DOI] [PubMed] [Google Scholar]
- 43. Matthes F, Stroobants S, Gerlach D, et al. Efficacy of enzyme replacement therapy in an aggravated mouse model of metachromatic leukodystrophy declines with age. Hum Mol Genet 2012;21:2599–2609. [DOI] [PubMed] [Google Scholar]
- 44. Vogler C, Levy B, Galvin NJ, et al. Enzyme replacement in murine mucopolysaccharidosis type VII: neuronal and glial response to beta‐glucuronidase requires early initiation of enzyme replacement therapy. Pediatr Res 1999;45:838–844. [DOI] [PubMed] [Google Scholar]
- 45. Hu J, Lu JY, Wong AM, et al. Intravenous high‐dose enzyme replacement therapy with recombinant palmitoyl‐protein thioesterase reduces visceral lysosomal storage and modestly prolongs survival in a preclinical mouse model of infantile neuronal ceroid lipofuscinosis. Mol Genet Metab 2012;107:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rozaklis T, Beard H, Hassiotis S, et al. Impact of high‐dose, chemically modified sulfamidase on pathology in a murine model of MPS IIIA. Exp Neurol 2011;230:123–130. [DOI] [PubMed] [Google Scholar]
- 47. Wang D, Bonten EJ, Yogalingam G, et al. Short‐term, high dose enzyme replacement therapy in sialidosis mice. Mol Genet Metab 2005;85:181–189. [DOI] [PubMed] [Google Scholar]
- 48. Dunder U, Kaartinen V, Valtonen P, et al. Enzyme replacement therapy in a mouse model of aspartylglycosaminuria. FASEB J 2000;14:361–367. [DOI] [PubMed] [Google Scholar]
- 49. Reagan‐Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J 2008;22:659–661. [DOI] [PubMed] [Google Scholar]
- 50. Burton BK, Whiteman DA. Incidence and timing of infusion‐related reactions in patients with mucopolysaccharidosis type II (Hunter syndrome) on idursulfase therapy in the real‐world setting: a perspective from the Hunter Outcome Survey (HOS). Mol Genet Metab 2011;103:113–120. [DOI] [PubMed] [Google Scholar]
- 51. Eng CM, Banikazemi M, Gordon RE, et al. A phase 1/2 clinical trial of enzyme replacement in fabry disease: pharmacokinetic, substrate clearance, and safety studies. Am J Hum Genet 2001;68:711–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Eng CM, Guffon N, Wilcox WR, et al. Safety and efficacy of recombinant human alpha‐galactosidase A–replacement therapy in Fabry's disease. N Engl J Med 2001;345:9–16. [DOI] [PubMed] [Google Scholar]
- 53. Harmatz P, Giugliani R, Schwartz IV, et al. Long‐term follow‐up of endurance and safety outcomes during enzyme replacement therapy for mucopolysaccharidosis VI: final results of three clinical studies of recombinant human N‐acetylgalactosamine 4‐sulfatase. Mol Genet Metab 2008;94:469–475. [DOI] [PubMed] [Google Scholar]
- 54. Pastores GM. Laronidase (Aldurazyme): enzyme replacement therapy for mucopolysaccharidosis type I. Expert Opin Biol Ther 2008;8:1003–1009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Development of rhLAMAN‐specific IgG in alpha‐mannosidase knockout mice and generation of immune‐tolerant alpha‐mannosidosis mice. (A) Conventional nontransgenic alpha‐mannosidase knockout mice (‐/‐) develop significant antibody responses after the second injection of recombinant rhLAMAN as determined by ELISA measuring LAMAN‐specific IgG in plasma of both low (25 U/kg) and high (500 U/kg) dose‐treated animals. (B) Schematic drawing of the pTVC vector containing the mutated cDNA of human LAMAN carrying the clinical H72L mutation under the control of the beta‐actin promotor. The SalI/NotI‐excised expression cassette of pTVC‐LAMAN(MutH72L was injected into the pronucleus of fertilized oocytes. (C) Genotyping strategy with exon‐specific primers flanking the inserted H72L mutation (hLAMAN‐H72L‐F: 5′‐CACACACTTGATGACGTCGGC‐3′ and hLAMAN‐670R: 5′‐GATAATCAAGGCGCC CAAAGAAG‐3′) identified three transgene‐positive founder lines (lines 4, 8 and 11). (D) Breeding scheme for the generation of immune‐tolerant transgenic knockout mice (‐/‐ +tg): transgene positive founder mice (Founder line #4) were crossed with alpha‐mannosidase knockout mice (‐/‐) in the F0 generation. Transgene positive LAMAN heterozygote animals were further crossed in the F1 generation with homozygous alpha‐mannosidase knockout mice (‐/‐) in order to obtain transgene‐positive transgenic knockout mice (‐/‐ +tg) in the homozygote LAMAN background in the F2 generation. (E) Antibody specificity for the rhLAMAN staining shown in Figure 1 was proved by absence of signals in DAB‐stained brain sections of nontransgenic alpha‐mannosidase knockout mice (‐/‐) using a rhLAMAN‐specific antibody. The images presented in Figure 1C and E were acquired under the same experimental and microscopical settings.”
Figure S2. Immune‐tolerant alpha‐mannosidase knockout mice are phenotypically indistinguishable from nontransgenic animals. (A) Separation of neutral oligosaccharides extracted from spleens of 3–4 months old wild‐type (+/+), nontransgenic (‐/‐) and transgenic (‐/‐ +tg) mice by thin layer chromatography (TLC) shows comparable amounts of all glycan species (Man2–Man9). (B) Quantitative analysis of oligosaccharides by high‐performance liquid chromatography (HPLC) indicate equal levels of the major glycan species Man2 and Man3 (n = 3–4, per genotype). Light microscopy of Toluidine‐stained semi‐thin sections from the pancreas reveals similar amounts of storage vacuoles in both knockout strains (Scale bar: 50 μm).
Figure S3. Long‐term high‐dose ERT efficiently reduces oligosaccharide storage within peripheral tissues. (A) TLC analysis and (B) HPLC of oligosaccharides extracted from kidney of wild‐type (+/+) and both knockout strains shows complete clearance of all major oligosaccharide species in ‐/‐ +tg mice after ERT with both indicated doses. Similar results were obtained for the spleen as determined by both (C) TLC and (D) HPLC analyses. (E) Light microscopy of Toluidine‐stained ultrathin sections from the pancreas, the kidney and trigeminal ganglion of untreated and 125 U/kg treated animals shows complete disappearance of vacuoles in all tissues after ERT.
Figure S4. Long‐term high‐dose ERT efficiently reduces oligosaccharide storage in the brain of mice with established neuropathology. (A) The specific activity of α‐glucuronidase and β‐hexosaminidase show a normalization to control (+/‐) levels after rhLAMAN treatment with 500 U/kg (n = 4–6, per genotype). (B) TLC analysis of oligosaccharide brain extracts (upper panel) of both knockout strains shows decreased levels of all major oligosaccharide species in ‐/‐ +tg mice after ERT with 500 U/kg. Quantitative clearance of oligosaccharides is also observed in the spleen (lower panel). (C) HPLC‐based quantification revealed a clear decrease of Man2–Man4 oligosaccharides in treated ‐/‐ tg+ mice when compared to untreated ‐/‐ +tg mice animals in the brain (upper panel) and a nearly complete clearance in the spleen (lower panel). (D) Immunofluorescence labeling of LAMP‐1 revealed a normalization of LAMP‐1 levels in ‐/‐ and ‐/‐ tg+ mice compared to control mice (+/) in the hippocampus, cerebellum, and the cerebral cortex (Scale bar: 50 μm).
Data S1. Supplemental methods.