

Abstract
Somatic sensitizing mutations in the epidermal growth factor receptor (EGFR) are associated with response to EGFR tyrosine kinase inhibitors (TKIs), however acquired resistance and subsequent progression of disease inevitably occurs. One such mechanism of acquired resistance (AR), a second site mutation in the EGFR, T790M, has been shown to wax and wane in the presence and absence of selection pressure in the form of EGFR TKI therapy. Another less common mechanism of AR is development of a secondary driver pathway via MET amplification. Here we describe waxing and waning MET amplification in to the presence and absence of erlotinib supporting the idea that mechanisms of AR other than T790M also respond to EGFR TKI selection pressure, with implications for standard practice and clinical trial design.
Keywords: EGFR mutation, MET
Introduction
Somatic sensitizing mutations in the epidermal growth factor receptor (EGFR) gene are associated with marked responses to first and second generation EGFR tyrosine kinase inhibitors (TKI) such as gefitinib, erlotinib or afatinib.1,2,3 However, acquired resistance to therapy inevitably occurs. In 50-60% of cases, the mechanism of resistance involves a second EGFR mutation, T790M, occurring in cis with the original mutation.4,5 In the remainder of cases, multiple other mechanisms have been described, including the development of second driver pathways, for example through MET gene amplification which occurs in 5-20% of cases.4,6,7
Each resistance mechanism is assumed to be selected out along Darwinian evolutionary lines due to their conferring specific growth advantages in the presence of the EGFR TKI. In addition, the lack of predominance of these genotypes at baseline, suggests that the growth advantage is specific to the EGFR TKI environment. Consistent with this, while co-existence of T790M with a standard sensitizing mutation allows kinase signaling in the presence of EGFR TKIs, this activity is less than the initial EGFR mutation in the absence of the TKI.8 In addition, T790M+ clones wax and in the presence and absence, respectively, of a specific EGFR TKI selection pressure.9 This phenomenon is felt to underlie the basis for re-challenge responses which can occur in EGFR mutant patients who have progressed on a TKI, but then had a period of time on other non-EGFR directed therapies and are then re-exposed to the original TKI at a later date.10 Here we report a similar phenomenon of waxing and waning MET amplification in a patient with a sensitizing EGFR mutation in the presence and absence of EGFR TKI selection pressure.
Case
A previously healthy 55 year old female never smoker presented with persistent cough in 4/2006. Staging revealed two lesions within the right upper lobe (RUL) of the lung, right hilar lymphadenopathy and multiple bone metastases. A CT guided bone biopsy demonstrated TTF-1 and CK7 positive adenocarcinoma. The patient was initially started on carboplatin and gemcitabine with a good partial response, before stopping after seven cycles. The patient then received consolidation stereotactic body radiation therapy to her RUL lesion. In 7/2007 the patient had progression in multiple pulmonary nodules and erlotinib 150mg commenced, initially in the absence of molecular information. In 3/2009 after an initial minor response PET/CT showed a new hypermetabolic paratracheal lymphnode, new right pleural effusion, new right apical pleural thickening (Figure 1A). Thoracentesis and preparation of a cell pellet from the pleural fluid allowed assessment as formalin-fixed paraffin embedded material. Mutational analysis of the pleural fluid showed EGFR exon 21 L858R mutation, but no T790M. FISH analysis, conducted as previously described can also be applied to pleural pellets and showed widespread MET amplification (mean MET per cell 13.07, ratio MET/CEP7 = 4.13) (Figure 2A).11,12 Erlotinib was stopped and treatment started in a Phase I trial with sunitinib/pemetrexed/cisplatin. Cisplatin was stopped after one cycle due to intolerance. She continued on sunitinib/pemetrexed. After 4 cycles (cycle length 21 days) she had no metabolic evidence of malignancy on PET/CT (Figure 1B). She was taken off pemetrexed secondary to myelosupression after cycle 5 and was continued on sunitinib alone through cycle 12.
Figure 1. Re-emergence of partial EGFR TKI sensitivity during time off EGFR TKI therapy (relaxation of specific selection pressure).
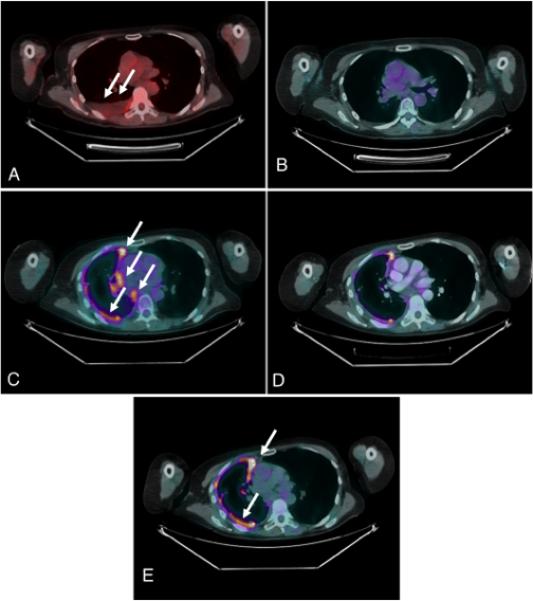
PET/CT chest imaging. A, Progression of disease after 20 months on erlotinib therapy (right pleural effusion) (arrows). B, No metabolic evidence of malignancy after 5 cycles of chemotherapy (sunitinib +/− pemetrexed trial). C, Progression of disease after 16 cycles of sunitinib (arrows). D, Minor response to erlotinib rechallenge after 5 weeks on erlotinib. E, Progression of disease after 14 weeks of erlotinib rechallenge (arrows).
Figure 2. Waning MET expression in the absence of erlotinib selection pressure.
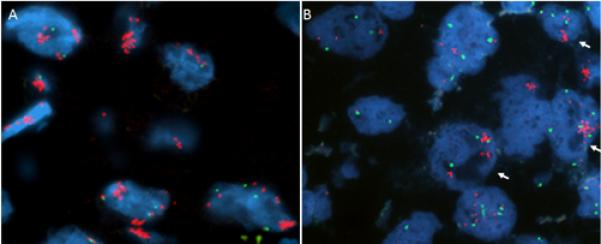
FISH analysis of malignant cells from pleural fluid. A, Widespread MET copy number gain (MET/CEP7 = 4.13) in the setting of progression of disease on erlotinib in March 2009. B, Waning of MET copies per cell after 10 months off erlotinib therapy in Jan 2010. Only tumor nuclei on the right exhibit MET gene clusters (arrows). Red = MET probe, green = CEP7 probe.
After cycle 12 (now approximately 10 months off erlotinib) the patient developed increased dyspnea and was found to have a new right pleural effusion on CT. Repeat sampling of the fluid revealed the same EGFR exon 21 mutation (L858R) but FISH analysis now showed that the MET amplification was confined to small tumor foci representing less than <10% of the tumor cells in the specimen tested. The tumor cells with MET amplification showed similar results compared with the prior specimen tested (mean MET per cell 13.47, MET/CEP7 ratio 4.16) but the tumor cells without MET amplification, which made up the bulk of the specimen, had much lower MET copy numbers (mean MET per cell 4.23, MET/CEP7 ratio 1.09) (Figure 2B). Pemetrexed was added back with the sunitinib through cycle 16. In 4/2010 PET/CT showed increased prominence and metabolic activity of right pleural nodular thickening as well as a newly involved right paratracheal lymphnode (Figure 1C) and increased involvement of retroperitoneal lymphnodes and nodes around the right psoas. Given these findings the patient was taken off sunitinib/pemetrexed and restarted on erlotinib at 150mg daily in 4/2010, 14 months after previous EGFR TKI exposure. PET/CT in 6/2010 showed a minor response (Figure 1D). However, in 8/2010 PET/CT indicated worsening pleural disease (Figure 1E) retroperitoneal and hepatic metastases. Erlotinib was stopped at this time and the patient was started on docetaxel, then gemcitabine, then a clinical trial of a second generation EGFR inhibitor combination all with limited success before succumbing to CNS progression in 4/2011.
Discussion
Mutations of T790M have been shown to wax and wane in response to selection pressure in the form of EGFR TKI therapy.4 Here, we describe a case in which MET amplification, as a separate mechanism of acquired resistance to EGFR TKIs in EGFR mutant patients, also waxed and waned in the presence and absence of erlotinib therapy. Unlike the presence or absence of T790M, MET copy number (either mean MET/cell or the MET:CEP7 ratio) represents a continuous variable. While there is now data on the predictive value of different cut-points in the MET:CEP7 ratio with regard to sensitivity to crizotinib (a MET inhibitor) in NSCLC cases in which MET is acting as a primary oncogenic driver,13 similar data to define the optimal cut-point in MET as a secondary driver in the setting of EGFR mutant NSCLC continues to be generated. Combinations of EGFR and MET inhibition in patients with EGFR mutations and MET positivity in the acquired resistance setting recently demonstrated an objective partial response rate of 24%, where MET positivity was defined as either a MET copy number ≥5 or ≥50% of tumor cells with 2+ or 3+ MET staining by immunohistochemistry. When only cases with MET copy number ≥5 were included, the response rate increased to 40%.14 In the case presented here of MET amplification detected in the presence of erlotinib, levels of MET (mean MET per cell 13.07, ratio MET/CEP7 = 4.13) were significantly greater than the levels reported in the above EGFR/MET inhibitor combination study. MET copy number was also significantly lower after 10 months off erlotinib, with just a subset of tumor cells carrying gene amplification. While the higher copy number gain was associated with progression on erlotinib after a PFS of 20 months, the much lower copy number was associated with a further minor response and a second PFS of 14 weeks. These data support the idea that MET, as a mechanism of acquired resistance, as with T790M, must have some growth disadvantage in the absence of exposure to an EGFR TKI and will wane, both biologically and in its clinical impact, without this specific selection pressure. As selection is manifested by clones increasing in proportion among the bulk of the tumor due to an inherent growth advantage, deselection manifests as a reducing proportion of amplified clones. Of note, one key advantage of the pleural fluid based analyses conducted in this patient was the ability of this medium to more clearly include representation of different clones present in the tumor at the same time. With deselection the fraction of cells with MET amplification fell dramatically in the pleural cell pellet of this patient. These data have implications for both standard practice (rechallenge responses to EGFR TKIs can occur even in non-T790M driven cases after significant time without a specific EGFR selection pressure) and clinical trial design and interpretation (to avoid the confounding effect of re-challenge responses, MET directed trials in the EGFR mutant acquired resistance setting should control the period of time from prior EGFR therapy). In addition, tumoral heterogeneity with regard to MET amplification can clearly exist leading to potentially misleading biopsy results, just as with T790M.10
Acknowledgements
Probe development and validation was supported through both the Colorado Lung Cancer SPORE (P50CA058187) and CU Cancer Center Support Grant (NCI CCSG P30CA046934) (Molecular Pathology Shared Resource).
REFERENCES
- 1.Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009 Sep 3;361(10):947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 2.Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012 Mar;13(3):239–246. doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 3.Sequist LV, Yang JC, Yamamoto N, et al. Phase III Study of Afatinib or Cisplatin Plus Pemetrexed in Patients With Metastatic Lung Adenocarcinoma With EGFR Mutations. J Clin Oncol. 2013 Jul 1; doi: 10.1200/JCO.2012.44.2806. [DOI] [PubMed] [Google Scholar]
- 4.Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011 Mar 23;3(75):75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu HA, Arcila ME, Rekhtman N, et al. Analysis of Tumor Specimens at the Time of Acquired Resistance to EGFR-TKI Therapy in 155 Patients with EGFR-Mutant Lung Cancers. Clin Cancer Res. 2013 Apr 15;19(8):2240–2247. doi: 10.1158/1078-0432.CCR-12-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bean J, Brennan C, Shih JY, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007;104:20932–20937. doi: 10.1073/pnas.0710370104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007 May 18;316(5827):1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 8.Chmielecki J, Foo J, Oxnard GR, et al. Optimization of dosing for EGFR-mutant non-small cell lung cancer with evolutionary cancer modeling. Sci Transl Med. 2011 Jul 6;3(90):90ra59. doi: 10.1126/scitranslmed.3002356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maheswaran S, Sequist LV, Nagrath S, et al. Detection of mutations in EGFR in circulating lung-cancer cells. N Engl J Med. 2008;359:366–77. doi: 10.1056/NEJMoa0800668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Camidge DR, Pao W, Sequist LV. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat Rev Clin Oncol. 2014 Aug;11(8):473–81. doi: 10.1038/nrclinonc.2014.104. [DOI] [PubMed] [Google Scholar]
- 11.Cappuzzo F, Marchetti A, Skokan M, et al. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J Clin Oncol. 2009 Apr 1;27(10):1667–74. doi: 10.1200/JCO.2008.19.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bubendorf L. ALK Analysis in Cytology. In: Tsao MS, Hirsch FR, Yatabe Y, editors. IALSC Atlas of ALK Testing in Lung Cancer. IASLC Press; Aurora, CO: 2013. pp. 53–57. [Google Scholar]
- 13.Camidge DR, Ou SI, Shapiro G, et al. Efficacy and safety of crizotinib in patients with advanced c-MET-amplified non-small cell lung cancer (NSCLC). J Clin Oncol. 2014;32:5s. (suppl; abstr 8001) [Google Scholar]
- 14.Wu YL, Yang JC, Kim DW, et al. Safety and efficacy of INC280 in combination with gefitinib (gef) in patients with EGFR-mutated (mut), MET-positive NSCLC: A single-arm phase lb/ll study. J Clin Oncol. 2014;32:5s. (suppl; abstr 8017) [Google Scholar]