

Key Clinical Message
Neurofibromatosis type 1 is a common cancer predisposing condition. Tumors, particularly gastrointestinal tumors, are commonly associated with NF1 but are not widely known. In addition, the relationship between lung cancer and neurofibromatosis has been controversial until recently with the discovery of oncogenes such as p53.
Keywords: Neuroendocrine tumor, neurofibromatosis type 1, non‐small cell lung cancer, tumors in neurofibromatosis
Background
Neurofibromatosis type 1 (NF1) is an autosomal dominant disorder that classically features café au lait spots, cutaneous neurofibromas, axillary and inguinal freckling, and iris Lisch nodules; however, the manifestations are extremely variable, even within families. NF1 is also one of the most common cancer predisposing disorders with a prevalence of 1 in 3000 1. The development of malignancies is a major cause of morbidity and mortality in NF1. Therefore, it is important to be familiar with the different malignancies associated with NF1 to provide early detection and intervention.
Case Presentation
We present a 45‐year‐old male with past medical history significant for NF1 and active smoking who presents with a 1‐month history of painless jaundice with associated pruritus. The patient initially presented to an outside hospital where he underwent an ERCP. During that ERCP, he was found to have an esophageal mass at the gastroesophageal junction as well as a bulging duodenum. As a result, the ERCP was aborted and he was transferred to a tertiary center for further evaluation.
Investigations
Laboratory work revealed: a total bilirubin of 7.9 mg/dL; alkaline phosphatase (ALP) 1260 IU/L; aspartate aminotransferase (AST) 94 IU/L; and alanine transaminase (ALT) 73 IU/L. An ERCP revealed an ulcerated submucosal lesion at the distal esophagus (Fig. 1). Furthermore, the main bile duct was severely dilated due to choledocholithiasis. Removal of the stones was accomplished with a biliary sphincterotomy and placement of stents in the common bile duct.
Figure 1.
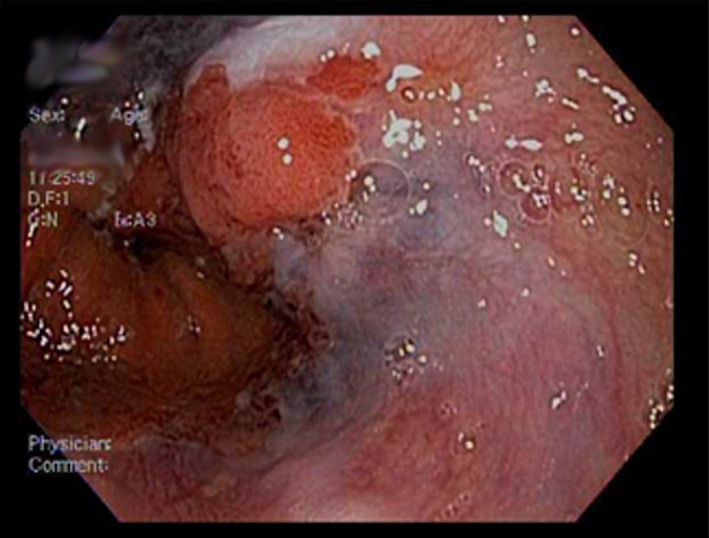
Ulcerated submucosal lesion at the distal esophagus.
Three days following his procedure, his liver function tests (LFTs) improved: total bilirubin 3.6 mg/dL; ALP 784 IU/L; AST 41 IU/L; and ALT 35 IU/L. The patient subsequently underwent an endoscopic ultrasound (EUS) for further evaluation of the esophageal mass. The EUS revealed: esophagitis with nodularity; an irregular mediastinal mass adjacent to the middle third of the esophagus (right hilar region); a subepithelial lesion in the ampulla (Fig. 2); and malignant‐appearing lymph nodes in the periduodenal region. All four of these suspicious areas were biopsied. The preliminary biopsy results were suggestive of malignancy – the ampullary lesion was well differentiated while the mediastinal and periduodenal lymph nodes were poorly differentiated. Laboratory studies revealed: carbohydrate antigen 19‐9 (CA 19‐9) 151 U/mL and carcinoembryonic antigen (CEA) 4.3 ng/mL.
Figure 2.
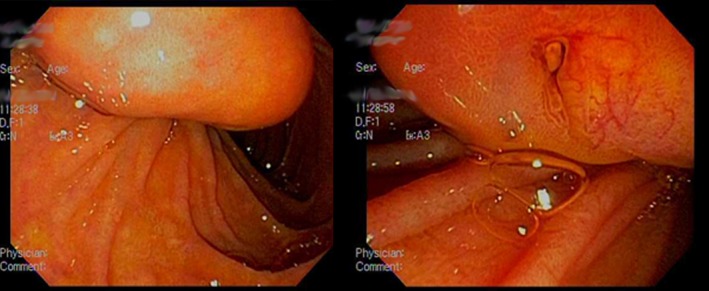
Periampullary neuroendocrine tumor discovered during endoscopy.
Differential Diagnosis
Cholangiocarcinoma
Given that the ampullary lesion was low grade and well differentiated compared to the mediastinal and periduodenal specimens, it was theorized that the ampullary lesion was the primary site and may have lost its differentiation in the process of metastasizing to the lymph nodes and right hilar region. Common clinical features of cholangiocarcinoma that were present upon presentation include painless jaundice and pruritus. The diagnosis of cholangiocarcinoma is often made based upon the clinical scenario and radiographic findings. Tumor markers such as CA 19‐9 and CEA, neither have the sensitivity nor specificity to make the diagnosis. In fact, some tumors have been found to produce little to no CA 19‐9 2. The clinical diagnosis is then confirmed with cytology and/or pathology.
Lung cancer
Given the single, right hilar mass, the differential included a lung primary with metastasis to the periduodenal lymph nodes and ampulla. Risk factors for the development of lung cancer in the patient included a 10‐pack year smoking history and the history of NF1. However, the discrepancy in the level of differentiation between the mediastinal, periduodenal, and ampullary specimens argues against a lung primary with metastasis to the periduodenal lymph nodes and ampulla, though it did not exclude the possibility of multiple primaries.
Malignant peripheral nerve sheath tumor (MPNST)
One of the characteristic features of neurofibromatosis is the presence of neurofibromas. These neurofibromas are divided into two categories: cutaneous and plexiform neurofibromas 3. Cutaneous neurofibromas are present in the majority of adult patients with NF1 4. Plexiform neurofibromas (PNFs) are present in 30–50% of patients with NF1 and tend to be associated with large nerves and present as larger, more diffuse tumors 3, 5. It is estimated that 5–10% of PNFs will undergo malignant transformation to MPNSTs, which are highly aggressive and invasive soft tissues sarcomas. MPNSTs represent a major cause of morbidity and mortality in NF1 patients 3.
Carcinoid Tumor
Patients with NF1 have an increased risk of developing certain gastrointestinal tumors including: gastrointestinal stromal tumors (GISTs); hyperplasia of the submucosal and intestinal myenteric nerve plexuses; and periampullary carcinoid tumors, which are most commonly identified as somatostatinomas 6, 7, 8. The majority of these carcinoid tumors are nonfunctional and symptoms typically occur secondary to a mass effect 8. While nonfunctional, these carcinoid tumors have potential to metastasize, particularly to the lymph nodes and liver, though this phenomenon is more rare when compared to their pancreatic counterparts 9.
Follow‐up and outcome
The patient re‐presented 4 days after initial discharge with a cough and fevers. A chest X‐ray revealed a right upper lobe consolidation and a right hilar mass that was previously seen on EUS. These findings were confirmed with a CT scan of the chest, which also noted multiple small, round lung cysts, paraseptal, and centrilobular emphysema along with scattered bullae with an apical predominance (Fig. 3). A CT abdomen revealed: abdominal lymphadenopathy and a lobular mesenteric lesion inferior to the gastric antrum. The patient was started on intravenous antibiotics for a postobstructive pneumonia. In the interim, the results of the biopsies from his previous admission became available.
Figure 3.

A. Chest X‐ray demonstrating right hilar mass. B. Coronal view demonstrating right hilar mass. C. Sagittal view demonstrating multiple small, round lung cysts, and apical bullae. D. Cross‐sectional view demonstrating right hilar mass with paraseptal and centrilobular emphysema.
Results from the esophagus revealed focal interstitial metaplasia with no dysplasia identified, which was suggestive of Barrett's esophagus. Biopsy of the ampullary mass was consistent with a well‐differentiated neuroendocrine tumor (Fig. 4). The cells were positive for cytokeratin (OSCAR), chromogranin A, and synaptophysin. Ki67 stain showed a low proliferative index of 2%. The biopsy results from the mediastinal mass (Fig. 5) and periduodenal lymph nodes (Fig. 6) were suggestive of a poorly differentiated non‐small‐cell carcinoma, given the diffuse positive staining with cytokeratin AE1/AE3. Furthermore, the samples were positive for TTF1 and CK7, which was strongly suggestive of a lung primary. A malignant germ cell neoplasm was unlikely given the lack of reactivity with OCT3/4, PLAP, and CD117. Angiosarcoma was excluded due to a negative CD31 and CD34 stain. Ki67 stain showed a proliferative index of >70%. Ultimately, this extensive immunostaining pattern excluded melanoma, mesothelioma, colorectal, and neuroendocrine primaries.
Figure 4.
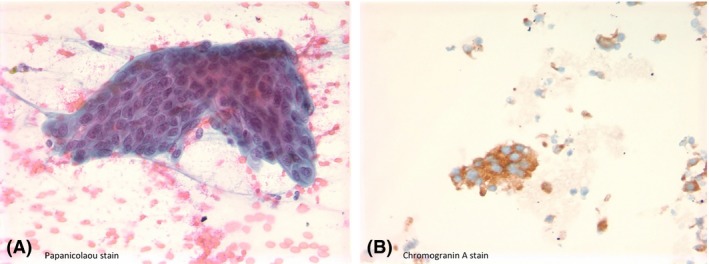
A. Well‐differentiated neuroendocrine neoplasm; a cluster of cohesive monomorphic population of tumor cells with mildly overlapping, salt and pepper appearing nuclei, Papanicolaou stain. B. Tumor cells were positive for Chromogranin A.
Figure 5.
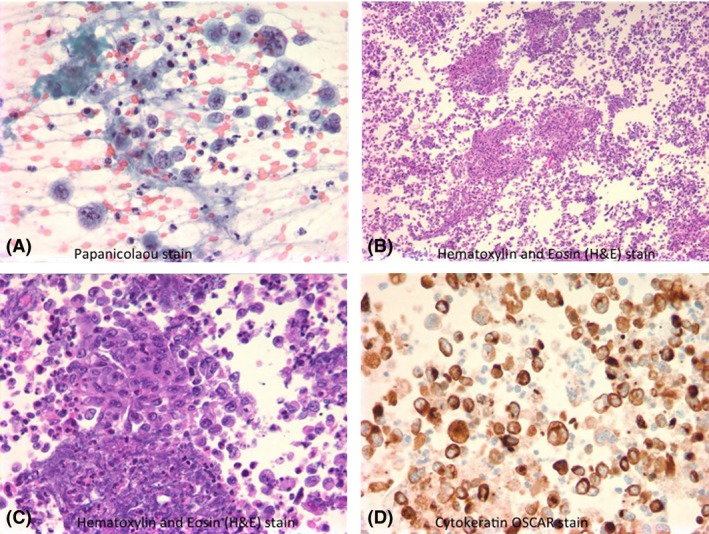
Mediastinal mass FNA – Poorly differentiated non‐small‐cell malignant epithelioid neoplasm. A. Demonstrates large pleomorphic atypical cells with irregular nuclear contour and scattered prominent nucleoli, Papanicolaou stain. B. Cell block preparation of the same lesion, showing abundant necrotic debris mixed with tumor cells, Hematoxylin and Eosin (H&E) stain. C. Clusters and single‐cell population of tumor cells with highly atypical features, H&E stain. D. Cytokeratin (OSCAR) immunostain highlights the atypical tumor cells.
With continued antibiotic therapy, the patient clinically improved and was discharged. Radiation oncology and medical oncology had been consulted during the hospitalization and recommended further outpatient work‐up, including a PET scan and CT scan of the head, to complete his staging. Of note, the patient refused to undergo a MRI of the head due to claustrophobia. PET scan revealed FDG uptake in the porta hepatis, left adrenal gland, and cervical lymph nodes in addition to the right hilar mass and mesenteric nodes.
The patient was seen at the interdisciplinary oncology clinic and was to be treated for a stage IV non‐small‐cell carcinoma of the lung. Prior to completion of his radiation and prior to initiation of chemotherapy, the patient re‐presented with right upper quadrant abdominal pain. A CT abdomen revealed: new hepatic lesions with an increased left adrenal metastatic lesion and increased upper abdominal lymphadenopathy. His acute‐onset abdominal pain was attributed to the new metastatic lesions in his liver. The patient was initiated on chemotherapy with carboplatin and paclitaxel during his hospitalization. After achieving adequate pain control, the patient was discharged with instruction to continue the outpatient chemotherapy and radiation therapy. However, during his subsequent follow‐up with oncology, the patient declined further treatment with chemotherapeutics and was transitioned to hospice care. The patient expired 4 months after his initial presentation.
Discussion
Neurofibromatosis type 1 (NF1) is a cancer predisposing disorder that results from mutations in the NF1 gene on chromosome 17q11.2 10. NF1 encodes the tumor suppressor, neurofibromin, which regulates cell growth by inactivating the p21 RAS and MAP kinase pathway 11. NF1 is classically associated with café au lait spots, cutaneous neurofibromas, axillary and inguinal freckling, and iris Lisch nodules. In addition, patients with NF1 have an increased risk of developing certain gastrointestinal tumors. The most common and characteristic gastrointestinal tumors are somatostatinomas 6, 7, 8 of the periampullary region and ampulla of Vater; which occurs in 43% of NF1 patients 12. The majority of these carcinoid tumors are nonfunctional and symptoms only occur due to a mass effect, 8 which was the case in our patient who presented with painless jaundice. While duodenal carcinoid tumors are commonly associated with NF1, there has been controversy over the pathogenesis of lung cancer in NF1.
Studies have reported an association between the development of lung cancer and NF1 10, 13. Two predominate theories have been postulated in an attempt to explain this association. One of which includes the development of tumors from previous scar tissue or bullae secondary to interstitial fibrosis 14. However, the association between interstitial fibrosis and NF1 has been controversial 15. Several retrospective studies involving smokers and nonsmokers with NF1 attempted to examine this association and found that patients with NF1 demonstrated ground‐glass opacities, bibasilar reticular opacities, bullae, cysts, and emphysema regardless of smoking status 16, 17, 18. The radiographic findings most commonly reported small, round lung cysts and asymmetrical, thin‐walled, bullae with an apical predominance. These findings are similar to, but distinct from those found in smoking‐related emphysema – the borders of the bullae and cysts are thicker and more sharply defined in NF1‐related disease 16. The CT chest in our patient noted multiple small, round cysts, paraseptal, and centrilobular emphysema along with scattered bullae with an apical predominance. Furthermore, the patient's tumor originated in the right hilar region. This area was associated with the paraseptal and centrilobular emphysematous changes. However, given our patient's smoking history, it remains unclear whether these changes were secondary to tobacco use, underlying NF1‐related lung disease, or a combination of both. Studies have suggested that NF1 could lead to early development of emphysematous change when exposed to cigarette smoke 15, 19. This predisposition in conjunction with his smoking could have led to early changes in our patient's lung parenchyma making them more susceptible in the development of his lung cancer. The second theory that could explain the development of lung cancer in NF1 involves deletions on chromosome 17p.
NF1 is an autosomal dominant disease that results from mutations of the NF1 gene on chromosome 17. A study examining the characteristics of benign and malignant neurofibrosarcomas in NF1 discovered that deletions in 17p were associated with tumor progression and malignant transformation to MPNSTs. The study noted that the p53 tumor suppressor gene was located in the 17p region and its inactivation was a likely mechanism for tumorgenesis in NF1 20. Two subsequent studies revealed that the loss of heterozygosity on chromosome 17p was critical in the development and progression of small‐cell lung cancer 21, 22. One of these studies specifically examined patients with NF1 and also implicated the inactivation of p53 in the development of small‐cell lung cancer in NF1 21. Multiple mutations have been associated with the development of non‐small‐cell lung cancer – mutations in p53 have been found in about 50% of non‐small‐cell lung cancers 23, 24, 25. Given these findings, one can theorize that patients with NF1 are at higher risk of developing lung cancers secondary to deletions in 17p, specifically the p53 gene. This increased risk can be further compounded with tobacco use – studies have demonstrated a higher frequency of p53 mutations in smokers when compared to never smokers 26, 27, 28.
In our patient, it is difficult to isolate one single process in the development of his lung cancer because his history is complicated with a 10‐pack year smoking history. However, studies have suggested that cigarette smoke could lead to the early development of emphysematous change in patients with NF1. This scarring of his lung parenchyma could have been the foundation for the development of his lung cancer. In addition, mutations in p53 have been implicated the development of lung cancer. Studies have isolated p53 on chromosome 17p, a region where deletions have been associated with tumor progression in NF1. The patient's smoking history further contributes to the genesis of his non‐small‐cell lung cancer as it also leads to increased frequency of p53 mutations – putting him at an even higher risk for developing lung cancer.
Conclusion
NF1 is classically associated with café au lait spots, cutaneous neurofibromas, axillary and inguinal freckling, and iris Lisch nodules. However, this case demonstrates that gastrointestinal manifestations such as periampullary carcinoid tumors should also be thought of in association with NF1. Furthermore, while pulmonary involvement is not commonly associated with NF1, this case exhibits the need to consider NF1 as an underlying risk factor for development of lung cancer. It is important to be familiar with the different malignancies associated with NF1 to allow for a meticulous investigation leading to early detection and intervention.
Conflict of Interest
None declared.
Clinical Case Reports 2015; 3(12): 990–996
References
- 1. Jett, K. , and Friedman J. M.. 2010. Clinical and genetic aspects of neurofibromatosis 1. Genet. Med. 12:1–11. [DOI] [PubMed] [Google Scholar]
- 2. Shah, N. , Cardinal J.. 2014. The Bethesda Handbook of Clinical Oncology. 4th ed. Biliary Tract Cancer. Lippincott Williams & Wilkins, Philadelphia, PA: Pp. 87–92. [Google Scholar]
- 3. Laycock‐van Spyk, S. , Thomas N., Cooper D. N., and Upadhyaya M.. 2011. Neurofibromatosis type 1‐associated tumours: their somatic mutational spectrum and pathogenesis. Hum. Genomics 5:623–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Upadhyaya, M. , Huson S., Davies M., and Thomas N.. 2007. An absence of cutaneous neurofibromas associated with a 3‐bp inframe deletion in exon 17 of the NF1 gene (c.2970‐2972 delAAT): Evidence of a clinically significant NF1 genotype‐phenotype correlation. Am. J. Hum. Genet. 80:140–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Upadhyaya, M. 2011. Genetic basis of tumorigenesis in NF1 malignant peripheral nerve sheath tumors. Front Biosci. 16:937–951. [DOI] [PubMed] [Google Scholar]
- 6. Bhandari, R. , Riddiough G., Lokan J., Weinberg L., Efthymiou M., and Nikfarjam M.. 2015. Somatostatinoma of the minor papilla treated by local excision in a patient with neurofibromatosis type 1. JOP 16:81–84. [DOI] [PubMed] [Google Scholar]
- 7. Tewari, N. , Rollins K., Gandhi N., Kaye P., and Lobo D. N.. 2014. Mixed periampullary adenocarcinoma and somatostatinoma with small bowel gastrointestinal stromal tumour in neurofibromatosis type 1. JOP 15:600–603. [DOI] [PubMed] [Google Scholar]
- 8. Kistler, C. A. , Johnson J. M., Winter J. M., Baliff J. P., Siddiqui A. A., and Sama A. R.. 2014. A case of periampullary adenocarcinoma in neurofibromatosis type 1. J. Gastrointest. Oncol. 5:E96–E99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mao, C. , Shah A., Hanson D. J., and Howard J. M.. 1995. Von Recklinghausen's disease associated with duodenal Somatostatinoma: contrast of duodenal versus pancreatic Somatostatinomas. J. Surg. Oncol. 59:67. [DOI] [PubMed] [Google Scholar]
- 10. Walker, L. , Thompson D., Easton D., Ponder B., Ponder M., Frayling I., et al. 2006. A prospective study of neurofibromatosis type 1 cancer incidence in the UK. Br. J. Cancer 95:233–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lodish, M. B. , and Stratakis C. A.. 2010. Endocrine tumours in neurofibromatosis type 1, tuberous sclerosis and related syndromes. Best Pract. Res. Clin. Endocrinol. Metab. 24:439–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tanaka, S. , Yamasaki S., Matsushita H., Ozawa Y., Kurosaki A., Takeuchi K., et al. 2000. Duodenal somatostatinoma: a case report and review of 31 cases with special reference to the relationship between tumor size and metastasis. Pathol. Int. 50:146–152. [DOI] [PubMed] [Google Scholar]
- 13. Kim, E. T. , Namgung H., Shin H. D., Lee S. I., Kwon J. E., Chang M. C., et al. 2012. Oncologic manifestations of neurofibromatosis type 1 in Korea. J. Korean Surg. Soc. 82:205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shimizu, Y. , Tsuchiya S., Watanabe S., and Saitoh R.. 1994. von Recklinghausen's disease with lung cancer derived from the wall of emphysematous bullae. Intern. Med. 33:167–171. [DOI] [PubMed] [Google Scholar]
- 15. Ryu, J. H. , Parambil J. G., McGrann P. S., and Aughenbaugh G. L.. 2005. Lack of evidence for an association between neurofibromatosis and pulmonary fibrosis. Chest 128:2381–2386. [DOI] [PubMed] [Google Scholar]
- 16. Zamora, A. C. , Collard H. R., Wolters P. J., Webb W. R., and King T. E.. 2007. Neurofibromatosis‐associated lung disease: a case series and literature review. Eur. Respir. J. 29:210–214. [DOI] [PubMed] [Google Scholar]
- 17. Porterfield, J. K. , Pyeritz R. E., and Traill T. A.. 1986. Pulmonary hypertension and interstitial fibrosis in von Recklinghausen neurofibromatosis. Am. J. Med. Genet. 25:531–535. [DOI] [PubMed] [Google Scholar]
- 18. McCaffrey, J. T. 1982. Interstitial pulmonary fibrosis in von Recklinghausen's neurofibromatosis: report of case. J. Am. Osteopath. Assoc. 81:688–691. [PubMed] [Google Scholar]
- 19. Yokoyama, A. , Kohno N., Sakai K., Kondo K., Hirasawa Y., and Hiwada K.. 1997. Distal acinar emphysema and interstitial pneumonia in a patient with von Recklinghausen's disease: five‐year observation following quitting smoking. Intern. Med. 36:413–416. [DOI] [PubMed] [Google Scholar]
- 20. Menon, A. G. , Anderson K. M., Riccardi V. M., Chung R. Y., Whaley J. M., Yandell D. W., et al. 1990. Chromosome 17p deletions and p53 gene mutations associated with the formation of malignant neurofibrosarcomas in von Recklinghausen neurofibromatosis. Proc. Natl Acad. Sci. USA 87:5435–5439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shimizu, E. , Shinohara T., Mor N., Yokota J., Tani K., Izumi K., et al. 1993. Loss of heterozygosity on chromosome arm 17p in small cell lung carcinomas, but not in neurofibromas, in a patient with von Recklinghausen neurofibromatoiss. Cancer 71:725–728. [DOI] [PubMed] [Google Scholar]
- 22. Yokota, J. , Wada M., Shimosato Y., Terada M., and Sugimura T.. 1987. Loss of heterozygosity on chromosomes 3, 13, and 17 in small‐cell carcinoma and on chromosome 3 in adenocarcinoma of the lung. Proc. Natl Acad. Sci. USA 84:9252–9256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Thomas, A. , Brzezniak C., and Giaccone G.. 2014. The Bethesda Handbook of Clinical Oncology. 4th ed. Non‐Small Cell Lung Cancer. Lippincott Williams & Wilkins, Philadelphia, PA: Pp. 31–41. [Google Scholar]
- 24. Takahashi, T. , Nau M. M., Chiba I., Birrer M. J., Rosenberg R. K., Vinocour M., et al. 1989. p53: a frequent target for genetic abnormalities in lung cancer. Science 246:491–494. [DOI] [PubMed] [Google Scholar]
- 25. Campling, B. G. , and el‐Deiry W. S.. 2003. Clinical implications of p53 mutations in lung cancer. Methods Mol. Med. 75:53–77. [DOI] [PubMed] [Google Scholar]
- 26. Husgafvel‐Pursiainen, K. , Boffetta P., Kannio A., Nyberg F., Pershagen G., Mukeria A., et al. 2000. p53 mutations and exposure to environmental tobacco smoke in a multicenter study on lung cancer. Cancer Res. 60:2906–2911. [PubMed] [Google Scholar]
- 27. Takagi, Y. , Osada H., Kuroishi T., Mitsudomi T., Kondo M., Niimi T., et al. 1998. p53 mutations in non‐small‐cell lung cancers occurring in individuals without a past history of active smoking. Br. J. Cancer 77:1568–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vähäkangas, K. H. , Bennett W. P., Castrén K., Welsh J. A., Khan M. A., Blömeke B., et al. 2001. p53 and K‐ras mutations in lung cancers from former and never‐smoking women. Cancer Res. 61:4350–4356. [PubMed] [Google Scholar]