

Abstract
Clinical and experimental evidence indicate that polymorphisms within the interleukin 4 (IL‐4) receptor (IL‐4R) chain are sufficient for altered strength of IL‐4/IL‐13 signaling, leading to an exaggerated allergic inflammatory response and increase susceptibility to allergic phenotypes. In the present study, we show that ablation of IL‐4Rα–induced phosphatidylinositol 3‐kinase (PI3K) activating signal by germline point mutation within the IL‐4Rα motif (Y500F) did not alter susceptibility to IgE‐mediated, food‐induced experimental anaphylaxis. Moreover, diarrhea occurrence, antigen‐specific IgE and intestinal mastocytosis were comparable between WT and IL‐4RαY500F mice. However, mice unable to stimulate IL‐4Rα–mediated PI3K signaling had accelerated disease progression. Notably, the accelerated anaphylactic response was associated with more rapid histamine‐induced hypovolemia. Mechanistic in vitro and in vivo analyses revealed that endothelial IL‐4Rα PI3K signaling negatively regulates the histamine‐induced endothelial leak response. These results define an unanticipated role for IL‐4Rα–mediated PI3K signaling in negative regulation of IgE‐mediated anaphylactic reactions.
Keywords: food‐induced anaphylaxis, IgE and mast cells, interleukin 4 (IL‐4) receptor (IL‐4R) chain
Introduction
Food allergy is currently on the rise in the Western world: the prevalence of pediatric peanut allergy has doubled from 1997 to 2002 1, 2, 3, 4, and the Centers for Disease Control and Prevention has recently documented an 18% increase in the prevalence of reported food allergy in children from 1997 to 2007 5. Severe food allergy‐related reactions are most often caused by peanuts (50–62%) and tree nuts (15–30%) 6, placing 2.7–5.4 million people at risk for food‐induced anaphylaxis.
A food‐induced anaphylactic reaction encompasses a variety of symptoms that may affect one or more target organs including gastrointestinal, cutaneous, respiratory, and cardiovascular systems 7, 8. Clinical and experimental analyses have identified a central role for IgE/FcϵR/mast cells and mast cell‐derived mediators, including histamine, platelet‐activating factor (PAF), serotonin, proteases (tryptase and chymase), lipid‐derived mediators (prostaglandins [PGD2] and leukotrienes [LTC4, LTD4, and LTE4]), in promoting the clinical manifestations associated with food‐triggered anaphylaxis 9, 10, 11, 12, 13, 14, 15. The interleukin (IL)‐4 /IL‐13 signaling pathway is integral to the food allergic reaction via regulating CD4+ Th2 responses, IgE synthesis and mast cell and vascular endothelial cell function 16, 17. Indeed, targeted ablation of IL‐4/IL‐13 signaling alleviates IgE‐mediated, food‐induced allergic reactions 16.
The biological activity of IL‐4 and IL‐13 is regulated via receptor (R) binding: IL‐4 can bind the type I (IL‐4Rα chain and γc chain) and type II (IL‐4Rα chain and IL‐13Rα1 chain) IL‐4R, and IL‐13 can bind the type II IL‐4R and type II IL‐13R (IL‐13Rα1 and IL‐13Rα2 chains). Ligand (IL‐4 and/or IL‐13) interaction with the type I IL‐4R and type II IL‐4R induces downstream signaling including the signal transducer and activator of transcription (STAT) 6 and phosphatidylinositol 3‐kinase (PI3K) pathways. Phosphorylation of Y575, Y603 and Y633 of human IL‐4Rα mobilizes the transcription factor STAT‐6, which induces IL‐4‐ and IL‐13‐responsive genes 18, 19, 20. Phosphorylation of Y497 of IL‐4Rα, which is part of the IL‐4R motif necessary for recruiting insulin receptor substrate (IRS) 1 and IRS‐2, activates the PI3K and mitogen‐activated protein kinase (MAPK) pathways and mediates IL‐4 proliferative activity 20. Y713 of IL‐4Rα is part of an immunoreceptor tyrosine‐based inhibition motif (ITIM) that binds Src homology 2 (SH2) domain‐containing phosphatases, including SH2 domain‐containing tyrosine phosphatase (SHP) 1 and SHP‐2, and inositol phosphatases and thereby negatively regulates IL‐4/IL‐13 responses 21, 22, 23.
Clinical studies have identified a number of atopic susceptibility genes linking polymorphisms in the IL‐4R/IL‐13 axis with atopic diseases including food allergy and asthma 24, 25. This has been supported by corroborative evidence provided by studies employing mice deficient in components of the IL‐4/IL‐13 signaling pathway and knock‐in murine models demonstrating that disruption of individual signaling domains within the IL‐4Rα in mice can amplify IgE responses and elicits enhanced allergic responses 26, 27, 28. One such mutation is within the part of the IL‐4R motif (Y497 of IL‐4Rα in humans and Y500 of IL‐4Rα in mouse) that regulates PI3K signaling. IL‐4RαY500F mice possess a germline mutation in the Il4ra gene resulting in a loss of IL‐4Rα‐induced PI3K signaling and leading to impaired IL‐4‐induced CD4+ T‐cell proliferation, increased allergen‐induced IgE production and an allergic asthma phenotype 29. In this study, we examined the effects of the IL‐4RαY500F mutation on susceptibility of mice to food‐induced anaphylaxis. Unexpectedly, we show that loss of IL‐4Rα‐induced PI3K signaling did not alter susceptibility to IgE‐mediated food‐induced reactions but rather increased histamine‐induced endothelial leak response and accelerated disease progression.
Materials and Methods
Animals
Wild‐type (WT) BALB/c and IL‐4RαY500F (BALB/c) were originally provided by The Jackson Laboratory, Bar Harbor, ME, USA 29. The mice were crossed to generate heterozygotes (F1 IL‐4RαY500F/WT) and subsequently backcrossed to generate age‐, sex‐, and litter‐matched IL‐4Rα WT and IL‐4RαY500F/Y500F mice as described 29. The mice were maintained in a barrier facility, and animals were handled under Institutional Animal Care & Use Committee‐approved protocols from Cincinnati Children's Hospital Medical Center.
Oral antigen‐induced anaphylaxis
Six‐ to 8‐week‐old mice were sensitized subcutaneously with 50 µg of ovalbumin (OVA) (Sigma–Aldrich, St. Louis, MO, USA) in the presence of 2 mg of aluminum potassium sulfate adjuvant (alum: AIK(SO4)2‐12H2O) (Sigma–Aldrich) in sterile saline. Two weeks later, mice were deprived of food for 5 h and received repeated intragastric (i.g) challenge of OVA (50 mg/250 µL saline) via i.g. feeding needles (Fisher Scientific Co., Pittsburgh, PA, USA). Rectal temperature was monitored at 0, 10, 15, 30, 45, and 60 min following the sixth or seventh OVA challenge with a rectal probe (Physitemp Model BAT‐12) as previously described 30. In some experiments, mice were administered an i.v. (final volume 200 μL) injection with the histamine receptor antagonists Triprolidine (200 μg) and Cimetidine (200 μg) 30 min prior to OVA challenge.
IL‐4‐ and histamine‐induced hypothermia
Histamine biphosphate monohydrate (Sigma–Aldrich) (25 µg/1 mL saline per mouse) and/or IL‐4C (recombinant, IL‐4‐neutralizing, anti–IL‐4 monoclonal antibody [mAb] complex produced by mixing recombinant mouse IL‐4 with an anti‐IL‐4 mAb [BVD4‐1D11] at a 2:1 molar [1:5 weight] ratio, which saturates the mAb with IL‐4. We have previously demonstrated that these complexes have an in vivo half‐life of approximately 1 day and slowly dissociate, releasing biologically active IL‐4 31. IL‐4C or histamine was i.v. injected, and body temperature was monitored by rectal thermometry every 10 min for 60 min, as we have previously described 30.
Hematocrit
Blood was drawn from incised mouse tail veins into heparinized capillary tubes and centrifuged for 5 min at 10,000 rpm. Hematocrit (percentage of packed red blood cell [RBC] volume) was calculated as the length of packed RBCs divided by the total length of serum and red cells in the capillary tube and multiplied by 100%, as previously described 15.
Mast cell quantification
Jejunum (7–10 cm distal to the stomach) were collected and fixed in 10% formalin and processed by standard histologic techniques. Longitudinal sections (5 µm) were stained for mast cells with chloroacetate esterase (CAE) activity, as described previously 30. At least four random sections per mouse per area examined were analyzed. Quantification of stained cells was performed by counting the number of CAE‐positive cells in 5 fields for tongue, 10 fields for ear, and 20 fields for intestine (magnification 400×).
Enzyme‐Linked Immunosorbent Assay measurements
Mast cell protease 1 (MCPT‐1) serum levels were measured by the mouse MCPT‐1 ELISA Ready‐SET‐Go!, according to the manufacturer's instructions (ebioscience, San Diego, CA, USA). Serum total IgE levels were determined using the ELISA MAX Deluxe SET Mouse IgE Kit (Biolegend, San Diego, CA, USA). Serum OVA‐specific IgE levels were determined by means of ELISA. Plates were coated with anti‐IgE antibody (EM‐95; 10 µg/mL; BD PharMingen, San Jose, CA, USA) and blocked with 200 µL of 10% fetal bovine serum (FBS) before adding serial dilutions of plasma samples (100 µL per well). After overnight incubation, plates were washed and incubated with biotinylated OVA (2.5 mg/mL, 100 µL per well). After 1 h of incubation, streptavidin–horseradish peroxidase (1 mg/mL; Biosource, Camarillo, CA, USA) was added and the assay developed with 100 µL of substrate (TMB substrate reagent set; BD OptEIA, San Diego, CA, USA). Colorimetric reaction was stopped with 1 mol/L H2SO4 and was quantified by measuring optical density with an ELISA plate reader at 450 nm.
In vitro permeability
The human vascular endothelial cell line EA.hy926 (ATCC, Manassas, VA, USA) was maintained in DMEM supplemented with 10% FCS, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, 10 mM HEPES and 1X penicillin/streptomycin (Invitrogen, Grand Island, NY, USA) in a humidified incubator (5% CO2, 37°C). On snap wells (12‐mm diameter, 0.4‐µm pore; Corning Glass, Corning, NY, USA), 5 × 105 cells were seeded and cultured for 18–21 days under maintenance media conditions as described above. Transendothelial resistance (TER) was monitored with an EVOM/Endohm (WPI Inc, Sarasota, FL, USA), and endothelial monolayers with TER >100 ohms · cm2 were used for all experiments. Monolayers were mounted between the hemi‐chambers of an Ussing apparatus (U2500 dual Ussing chamber, Warner Instruments, Hamden, CT, USA), and 0.112 cm2 of tissue was exposed to 10 mL of Krebs buffer at 37°C. The transendothelial potential difference was detected with two paired electrodes that contain 4% agar in 3 M KCl. The electrodes were connected to a voltage clamp amplifier (EC‐800, epithelial voltage clamp, Warner Instruments, Hamden, CT, USA). The electrode potential difference and fluid resistance were compensated before mounting tissue segments into the chamber. To establish equilibrium, potential difference was continuously monitored under open‐circuit conditions for 15 min. Thereafter, the tissues were voltage‐clamped at 0 mV while continuously measuring the short circuit current (I sc). Voltage pulses (3‐mV square waves sustained for 5 s) were delivered every 50 s to yield a current response for calculation of the resistance across a mucosa from Ohm's law. IL‐4 (10 ng/mL)‐, histamine (100 μM)‐ and vehicle‐stimulated endothelial monolayers were placed in Ussing chambers in the presence and absence of DHEA (100 nM) and allowed to equilibrate for 15 min; basal I sc and TER were measured as described previously 21.
Western Blot analysis
EA.hy926 cell lysates (30 µg) were loaded in 4–12% BisTris gels and transferred to a nitrocellulose membrane (Invitrogen). P85 PI3K was detected by using rabbit polyclonal anti‐p85 PI3K followed by anti‐rabbit peroxidase‐conjugated antibody (Cell Signaling, Danvers, MA) and ECL‐plus detection reagents (GE Healthcare, Pittsburgh, PA). Rabbit anti‐actin (Santa Cruz Biotechnology, Santa Cruz, CA) was used as a loading control.
Statistical analysis
Data are expressed as mean ± standard deviation (SD), unless otherwise stated. In experiments comparing multiple experimental groups, statistical differences between groups were analyzed using the one‐way ANOVA parametric and a Tukey's multiple comparison post‐test. In experiments comparing two experimental groups, statistical differences between groups were determined using a Student's t‐test. P < 0.05 was considered significant. Spearman's rank coefficients were used to quantify the relations between hemoconcentration and hypothermia. All analyses were performed with Prism 5.0 software (GraphPad Software Inc., San Diego, CA).
Results
Susceptibility of IL‐4RαY500F mice to food‐induced anaphylaxis
Previous studies in the IL‐4RαY500F mice have revealed that the Y500F mutation in the IL‐4Rα receptor and loss of IL‐4Rα‐mediated PI3K activation increased allergic inflammation and the asthmatic phenotype 29. To determine the effect of this mutation on susceptibility to food allergy, we assessed intestinal and systemic symptoms of anaphylaxis (diarrhea and hypothermia) in BALB/c WT and IL‐4RαY500F mice that were sensitized to OVA and then challenged with OVA via oral gavage 14 days later and then every other day for a total of seven challenges. We observed no significant difference in the occurrence of anaphylaxis between WT and IL‐4RαY500F mice (Fig. 1). After the fourth challenge, 34.6% of WT and 42.1% of IL‐4RαY500F mice demonstrated symptoms of anaphylaxis, which increased to 77.1% of WT and 84.3% of IL‐4RαY500F mice following the seventh challenge (Fig. 1A and B). Assessment of systemic symptom and disease severity (hypothermia) revealed no significant difference in the maximal shock response between WT and IL‐4RαY500F mice after the seventh challenge (Fig. 1B). During these analyses, we observed that the IL‐4RαY500F mice appeared to develop signs of anaphylaxis earlier than WT mice. Moreover, the IL‐4RαY500F mice demonstrated evidence of anaphylaxis (scratching and rubbing around the nose and head, decreased activity with an increasing respiratory rate and pilar erecti) earlier than WT mice following the seventh OVA challenge (results not shown). To quantitate these observations, we examined shock response (body temperature) of the WT and IL‐4RαY500F mice at 0, 15, 30, and 45 min after the seventh OVA oral gavage challenge. Indeed, the IL‐4RαY500F mice demonstrated a more rapid decrease in body temperature than the WT mice (Fig. 1C). Importantly, by 45 min, there was no significant difference in body temperature between groups (Fig. 1C). These datasets indicate that IL‐4RαY500F mice do not have increased susceptibility to food‐induced anaphylaxis or develop a more severe disease phenotype but rather experience an accelerated disease progression.
Figure 1.
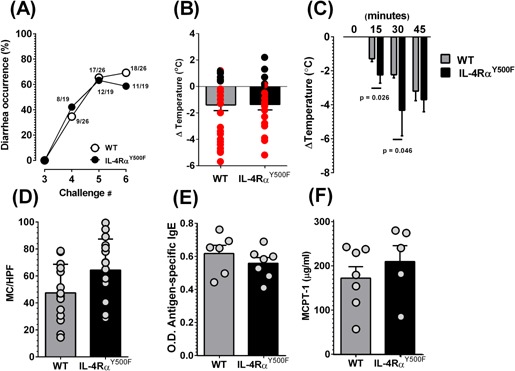
Loss of IL‐4Rα‐PI3K signaling accelerates progression of an anaphylactic reaction. A: Diarrhea occurrence in OVA‐sensitized, intragastric (i.g.) OVA‐challenged WT and IL‐4RαY500F mice. B: Temperature change from 0 to 60 min and C: 0, 15, 30, and 45 min following the seventh intragastric (i.g.) OVA challenge in OVA‐sensitized, OVA‐challenged WT and IL‐4RαY500F mice. D: Mast cell (MC) numbers per high power field (HPF) in the small intestine, OVA‐specific IgE (E) and mast cell protease 1 (MCPT‐1), (F) levels in the serum of OVA‐sensitized, OVA‐challenged WT and IL‐4RαY500F mice following the seventh challenge. A: Data are presented as a percentage of diarrhea occurrence after a number of OVA challenges. The fraction indicates the number of mice with diarrhea out of the total number of mice in that group. (B, D–F: Individual circles represent 1 mouse). B: Red circles represent identification of positive intestinal symptoms of anaphylaxis (diarrhea), and black circles represent no evidence of intestinal symptoms of anaphylaxis. B–F: Data represent mean ± SD; n = 4–18 mice per group; P values as indicated. O.D., optical density.
In previous studies, we have demonstrated that antigen‐specific IgE and intestinal mast cells are the critically important factors in the regulation of food‐induced experimental anaphylaxis 30. Assessing intestinal mast cell levels revealed no differences in number between WT and IL‐4RαY500F mice (Fig. 1D). Furthermore, we observed no significant difference in the level of mast cell activation (secreted MCPT 1) or of antigen‐specific and total IgE in WT and IL‐4RαY500F mice after the seventh oral gavage challenge (Fig. 1E and F and Fig. S1). We concluded from this that the observed accelerated disease progression cannot be explained by altered IgE and mast cell levels.
In mice, the shock organ is the capillary bed; IgE‐mediated, mast cell‐dependent anaphylaxis causes capillary bed dilatation and extravasation, leading to severe hypovolemia 32, 33. A consequence of the hypovolemia‐induced shock in mice is hypothermia 16, 34, 35. Consistent with this concept, we show a direct relationship between hypovolemia (fluid extravasation as measured by hemoconcentration) and severity of oral antigen‐induced anaphylaxis (hypothermia) in our mice (P < 0.0001), indicating a direct relationship between fluid extravasation and hypothermia associated with a food‐induced anaphylactic reaction (Fig. S2). To determine whether the increased progression of food allergy in the IL‐4RαY500F mice is associated with hypovolemic shock, we examined hypothermia and hemoconcentration in WT and IL‐4RαY500F mice 10 min following the seventh oral antigen challenge. We observed a significantly stronger hypothermic response in the IL‐4RαY500F mice than WT mice, and the increased temperature loss was associated with increased hemoconcentration, indicating that the IL‐4RαY500F mice experience a more accelerated hypovolemic shock response (Fig. 2A and B).
Figure 2.
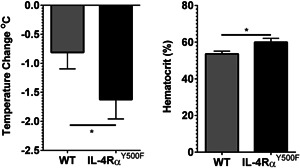
Loss of IL‐4Rα‐PI3K signaling accelerates progression of hypovolemic shock. A: Temperature change from 0 to 10 min and B: percentage hemacrit levels at 10 min following the seventh intragastric (i.g.) OVA challenge in OVA‐sensitized, OVA‐challenged WT and IL‐4RαY500F mice. Data represent mean ± SD; n = 5–8 mice per group; *P < 0.05.
Previous studies have demonstrated that the systemic manifestations of IgE/mast cell‐dependent anaphylaxis, particularly the hypothermic component of shock response, is mediated by histamine, as it can be blocked by histamine H1 and H2 receptor antagonism 36. Pretreatment of WT and IL‐4RαY500F mice with the histamine H1 and H2 receptor antagonist completely abrogated the oral antigen‐induced hypothermia in both WT and IL‐4RαY500F mice (Fig. S3), suggesting that the accelerated disease progression in the IL‐4RαY500F mice is a consequence of altered histamine‐induced hypothermic response. To determine whether histamine was sufficient to promote accelerated progression of the systemic manifestations of anaphylaxis, naive WT and IL‐4RαY500F mice received an i.v. injection of histamine, and hypothermia was assessed. Consistent with our OVA‐induced anaphylaxis experiments, IL‐4RαY500F mice experienced an accelerated progression of hypothermia in response to histamine compared to WT mice (Fig. 3). Importantly, we show that administration of equivalent amounts of histamine (25 μg) to WT and IL‐4RαY500F mice induced a more accelerated response in IL‐4RαY500F mice, suggesting 1) that histamine is sufficient to promote accelerated progression of the shock response in IL‐4RαY500F mice and 2) that the observed accelerated response is related in part to altered histamine responsiveness and not histamine levels.
Figure 3.
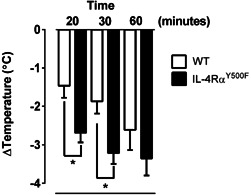
Loss of IL‐4Rα‐PI3K signaling accelerates progression of histamine‐induced hypothermia. Temperature change from 0 to 30 min after i.v. administration of histamine to WT and IL‐4RαY500F mice. Data represent n = 4 mice per group from three independent experiments and mean ± SD; *P < 0.01.
Increased rate of shock in IL‐4RαY500F mice in response to histamine
It is postulated that histamine‐induced hypothermia is a consequence of vascular endothelial leak and fluid shift into the periphery. Furthermore, previous studies have demonstrated that IL‐4 can modulate histamine‐induced hypothermia 15. The demonstration of 1) a direct link between fluid extravasation and hypothermic response in OVA‐challenged mice; 2) that fluid extravasation and hypothermic response in OVA‐challenged WT and IL‐4RαY500F mice were dependent on H1 and H2 receptor and 3) an accelerated hypothermic response in the IL‐4RαY500F mice compared to WT mice led us to speculate that the IL‐4Rα/PI3K signaling pathway negatively regulated histamine‐induced vascular endothelial leak. To begin to assess this possibility, we examined the effect of IL‐4RαY500F mutation on IL‐4/histamine–induced vascular leak and increased hemoconcentration. WT and IL‐4RαY500F mice were primed with IL‐4C and treated 24 h later with the vasoactive mediator histamine. Histamine treatment of WT mice induced a hypothermic response and increased hematocrit, with the former being amplified by pretreatment with IL‐4C (Fig. 4A and B; Average difference between WT Vehicle + Histamine and WT IL‐4C + Histamine: −1.05 ± 0.60 ΔTemperature (°C); mean ± SEM; indicated by gray pattern in column). Similarly, histamine treatment of IL‐4RαY500F mice induced hypothermia and increased hematocrit. The temperature change induced by histamine in the IL‐4RαY500F mice was significantly greater than that of WT mice (Fig. 4A). Importantly, combined IL‐4C and histamine treatment of IL‐4RαY500F mice caused a significantly greater hypothermic response and increase in hematocrit than that observed in histamine only‐treated IL‐4RαY500F mice or combined IL‐4C‐ and histamine‐treated WT mice (Fig. 4A and B; P < 0.05; Average difference between IL‐4RαY500F Vehicle + Histamine and IL‐4RαY500F IL4C + Histamine: −2.7 ± 0.66; mean ± SEM; indicated by gray pattern in column). The greater hypothermic and hematocrit response in the absence of PI3K signaling (IL‐4RαY500F) suggests that IL‐4Rα/PI3K signaling negatively regulates histamine‐induced vascular endothelial responses.
Figure 4.
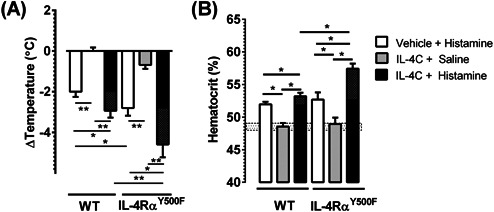
IL‐4 attenuation of histamine‐induced hypothermia is alleviated in IL‐4RαY500F mice. A: Temperature change from 0 to 30 min and B: hematocrit at 60 min after i.v. administration of IL‐4C and/or histamine to WT and IL‐4RαY500F mice. B: Hatched box indicates average hematocrit level of WT BALB/c mice. Grey checkered box within columns indicates difference between Vehicle + Histamine and IL‐4C + Histamine within the respective strains. Data represent mean ± SD. n = 6–18 mice per group. *P < 0.05; **P < 0.01.
PI3K signaling negatively regulates histamine‐induced hypothermic response
The observation of accelerated progression of OVA‐induced and IL‐4/histamine–induced vascular leak indicate that the absence of PI3K signaling (IL‐4RαY500F) accelerates and/or enhances histamine‐induced vascular endothelial responses. On the basis of these datasets, one would speculate that stimulating vascular endothelial PI3K signaling would attenuate histamine‐induced vascular leak. Dehydroepiandrosterone (DHEA), an adrenal steroid that acts as a precursor in the biosynthesis of testosterone and estrogen, has also been implicated in regulating vascular endothelial cell function 37, 38. Notably, DHEA‐mediated effects are predominantly induced via G‐protein coupled receptor (GPCR)‐stimulated, PI3K/AKT‐dependent activation of FOXO1 37. We therefore speculated that exposure of mice to DHEA would induce vascular endothelial cell PI3K activation and subsequently attenuate histamine‐induced vascular endothelial leak. Firstly, we confirmed that histamine and DHEA stimulation of endothelial cells induce PI3K activation. To do this, we examined phosphorylation of the p85 subunit of PI3K in the human vascular endothelial cell line EA.hy926, which is derived from A549 and HUVEC cells and used as a model of systemic endothelial cells 39, following histamine and DHEA stimulation. We demonstrate increased phosphorylation of the p85 subunit of PI3K between 5 and 15 min following histamine (100 μM) and DHEA (100 nM) stimulation (Fig. S4). Next, WT and IL‐4RαY500F mice were pretreated with vehicle or DHEA and IL‐4C and received i.v. histamine treatment 24 h later, after which hypothermia was evaluated. Histamine treatment induced a hypothermic response in WT and IL‐4RαY500F mice, with the response being significantly greater in the IL‐4RαY500F mice (Fig. 5; P < 0.05). Notably, pretreatment with DHEA did not significantly alter the level of hypothermia in histamine‐treated WT mice but did significantly attenuate the level of hypothermia in histamine‐treated IL‐4RαY500F mice (Fig. 5; P < 0.05). These data suggest that constitutive PI3K activation can attenuate the histamine‐induced increase in hypothermia. Furthermore, these data support the concept that IL‐4–induced PI3K activation attenuates histamine‐induced hypothermia.
Figure 5.
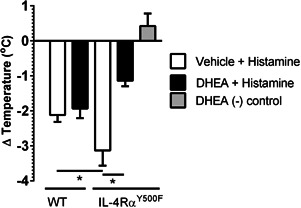
DHEA attenuates histamine‐induced hypothermia in IL‐4RαY500F mice. Temperature change from 0 to 30 min after i.v. administration of histamine to WT and IL‐4RαY500F mice after pretreatment with vehicle or DHEA (500 μg). Data represent mean ± SD; n = 3–8 mice per group from n = 2 experiments. *P < 0.01.
As these experiments were performed in IL‐4RαY500F global mice, all cells of the hematopoietic and non‐hematopoietic compartment were deficient in IL‐4Rα–mediated PI3K activation. Thus, we cannot determine whether IL‐4Rα–mediated PI3K signaling in endothelial cells directly or indirectly attenuates anaphylactic symptoms. To further elucidate the mechanism, we performed a similar experiment using the human vascular endothelial cell line EA.hy926 39. Histamine stimulation decreases TER of EA.hy926 cells (Fig. 6A). Notably, the decrease in endothelial TER was associated with increased flux of horseradish peroxidase (HRP) (40 kDa), indicating increased paracellular permeability and vascular endothelial leak (Fig. 6B). Stimulation of EA.hy926 cells with DHEA also induced a small decrease in TER and increase in paracellular permeability compared with unstimulated cells (Fig. 6A and B). Importantly, the histamine‐induced increase in endothelial cell permeability was attenuated by pretreatment with DHEA, supporting the concept that endothelial cell PI3K signaling reduces histamine‐induced endothelial permeability.
Figure 6.
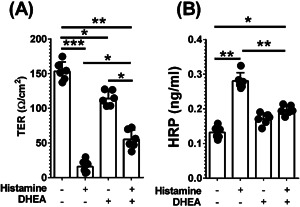
DHEA attenuates histamine‐induced paracellular leak in human vascular endothelial cell line EA.hy926. A: Transendothelial resistance (TER) and B: HRP flux in DHEA‐treated human vascular endothelial cell line (EA.hy926) after histamine stimulation. Confluent (>100 Ω/cm2) vascular endothelial cells treated with vehicle or DHEA (100 nM) were stimulated with histamine (100 μM) for 30 min, and TER and HRP flux were determined. Data are representative of 5 wells per treatment group from two independent experiments. Individual circles represent an individual well. Column represents mean ± SD from n = 2 experiments. *P < 0.05, **P < 0.01, ***P < 0.005.
Discussion
Previous clinical and murine studies have revealed a link between gain‐of‐function mutations in the IL‐4Rα chain and increased susceptibility to allergic inflammatory responses 26, 27. The majority of the mutations described are thought to drive atopy susceptibility via modulation of the effects of IL‐4/IL‐4Rα on hematopoietic cell function. In this study, we demonstrate that loss of IL‐4Rα/PI3K signaling, via a mutation in the IL‐4R motif necessary for the recruitment of IRS‐1 and IRS‐2, does not increase severity or susceptibility in allergic disease but rather accelerates IgE/mast cell‐mediated, food‐induced anaphylaxis progression in mice. We show that the increased rate of symptom development was not associated with dysregulation of IgE and mast cell function but rather was due to increased sensitivity of the vascular endothelium to mast cell‐derived histamine.
Clinical and murine‐based evidence indicate that the symptoms of food allergy are driven by allergen/IgE/FcϵRI‐mediated mast cell degranulation and release of mast cell mediators that act on target cells to promote the pathophysiologic features of disease, including urticaria, diarrhea, bronchoconstriction, respiratory and cardiovascular collapse, the latter of which reflects a decrease in intravascular volume resulting in decreased vital organ perfusion and shock 10, 11, 14, 15, 40, 41, 42, 43. Consistent with previous reports, we show that the fluid extravasation and decreased intravascular volume (increased hemoconcentration) is dependent on histamine, as pharmacologic antagonism of H1 and H2 receptors inhibited the hypothermic component of shock response 36.
The molecular basis of histamine‐mediated increase in vascular endothelial leak is not yet fully delineated. Histamine ligation to the H1 receptor leads to Gq‐protein–coupled and phospholipase C (PLC) activation, inositol phospholipid hydrolysis and increased intracellular Ca2+ 44, 45, which can lead to 1) reduced F‐actin focal attachment formation 46; 2) destabilization of the adherens junction VE‐cadherin and catenin interactions, leading to decreased intercellular tethering resulting in reduced endothelial cell adhesiveness and increased paracellular permeability 47. The IL‐4/IL‐4Rα pathway has previously been shown to magnify the histamine‐mediated effector phase of anaphylaxis 15. The mechanism by which IL‐4 modulates histamine responses is not fully elucidated; however, it is postulated that IL‐4 can magnify histamine responses via enhancement of histamine‐induced PAF synthesis and PGE2 release via IL‐4‐induced upregulation of the H1 receptor expression 48.
Unexpectedly, we show that loss of IL‐4Rα/PI3K signaling leads to an accelerated histamine‐induced hypothermic response and anaphylaxis progression. Murine‐based and in vitro studies indicate that the accelerated response could be attributed to increased responsiveness of the vascular endothelium to histamine. The molecular basis of IL‐4Rα/PI3K‐mediated negative regulation of histamine‐induced anaphylactic shock response is unclear; however, we speculate that the mechanism is related to IL‐4Rα/PI3K's negative regulation of Ca2+‐dependent responses. Recent investigations have reported that IL‐4 can attenuate carbachol‐ and caffeine‐induced Ca2+ mobilization from the sarcoplasmic reticulum (SR) in airway smooth muscle cells 49. Notably, the IL‐4–mediated inhibition of transient Ca2+ release was sensitive to PI3K antagonism, implicating IL‐4–induced PI3K activity in intracellular Ca2+ release. Since carbachol‐ and caffeine‐induced Ca2+ release in the SR is mediated by different Ca2+ release channels, the reduction in the transient Ca2+ release by IL‐4/PI3K is not by inhibition of Ca2+ release channels but rather by reduction in the amount of SR‐restricted Ca2+ levels. Importantly, in some cell types, including HUVECs, PI3K activation promotes PLCγ activation and inositol 1,4,5 triphosphate (IP3) metabolism 50, thus linking IL‐4R activation to PLCγ‐generated IP3 and Ca2+ release. In support of PI3K signaling's negative regulation of histamine‐induced shock, we show that constitutive activation of the endothelial PI3K pathway by DHEA attenuated histamine‐induced shock in IL‐4RαY500F mice and that DHEA reduced histamine‐induced endothelial paracellular leak in vitro. Demonstrating that DHEA can also suppress histamine responses eliminates concerns with respect to the possibilities of IL‐4RαY500F mice possessing an intrinsic defect in endothelial PI3K signaling or IL‐4Rα mediating suppression of Ca2+ channel expression or function. Interestingly, we show in vitro that DHEA alone decreased endothelial barrier function as compared with unstimulated endothelial cells. Notably, this baseline DHEA‐induced effect was not related to increased paracellular leak, as there were no differences in HRP flux between vehicle‐treated and DHEA only‐treated cells, suggesting that DHEA was stimulating altered ion secretion.
Previous studies in IL‐4RαY500F mice have demonstrated a role for the loss of IL‐4Rα/PI3K signaling in the exacerbation of allergic inflammatory responses 29. Moreover, in a pulmonary airway inflammation model, IL‐4RαY500F mice developed a more severe asthmatic phenotype as demonstrated by increased airway hyperresponsiveness, pulmonary eosinophilia and mucus hypersecretion 29. We did not observe increased severity of food‐induced anaphylaxis but rather the accelerated rate of symptom onset in IL‐4RαY500F mice. Importantly, the features of pulmonary allergic inflammation in this particular murine asthma model are not dependent on mast cell–mediated vascular endothelial permeability and fluid extravasation. However, Blaeser et al. reported that the IL‐4RαY500F mice had increased total IgE and allergen‐induced IgE production following OVA/Alum immunization 29. In contrast, we did not observe differences in total and antigen‐specific IgE following OVA/Alum immunization and challenge. One possible explanation for the observed differences between our studies and that of Blaeser et al. with respect to serum IgE is the intestinal microbial diversity 51. Recent mouse studies indicate that absence of microbial colonization or colonization with low‐diversity microbiota leads to increased serum IgE levels and enhancement of CD4+ T‐cell and IL‐4 responses 51.
We show that though MCPT‐1 levels were comparable between WT and IL‐4RαY500F mice 60 min following OVA challenge, IL‐4RαY500F mice experienced an accelerated progression of hypovolemia and hypothermia compared to WT mice, suggesting that the IL‐4/PI3K pathway alters histamine‐mediated responses. We cannot rule out the possibility of a more rapid activation of mast cells and increase in the level of mast cell mediators in the IL‐4RαY500F mice, which could accelerate progression of the oral antigen‐induced anaphylactic symptoms. Consistent with this argument, IL‐4 has been shown to amplify mast cell secretory function and release of preformed mediators such as serotonin and arachidonates 52, 53. However, we do show that administration of 25 μg of histamine to IL‐4RαY500F mice also lead to an accelerated progression of hypovolemia and hypothermia, suggesting that the altered response in IL‐4RαY500F mice can be attributed in part to altered sensitivity of the vascular endothelium to mast cell‐derived histamine.
A number of murine‐based studies have revealed that additional gain‐of‐function mutations in IL‐4Rα can enhance allergic inflammatory responses. IL‐4RαY709F mice, which have a tyrosine to phenylalanine mutation at position 709 within the ITIM of IL‐4Rα, have increased susceptibility to allergen‐induced airway inflammation and enhanced sensitivity to food allergens 26, 27. Similarly, mice that possess the glutamine to arginine substitution at position 576 (Q576R) of IL‐4Rα exhibited increased allergen‐induced inflammation and remodeling 28. To the best of our knowledge, this study is the first demonstration that an IL‐4Rα mutation can accelerate disease progression. Though no polymorphisms have been observed in the human equivalent tyrosine residue within the insulin: IL‐4 receptor motif (Y497), a human serine proline polymorphism six amino acids downstream of Y497 and within the IL‐4Rα ITIM motif (S503P) has been reported 54, 55. The impact of this polymorphism on the function of the PI3K motif of the human IL‐4Rα chain is currently unknown; however, the presence of polymorphic amino acid residues at this location (P503 and R576) are known to alter receptor polarity and secondary structure and affect IRS‐1 and IRS‐2 propagation of the IL‐4Rα signaling 54.
In the current manuscript, we show that loss of IL‐4Rα–mediated PI3K signaling accelerates the progression of oral antigen‐induced anaphylactic reactions. In vitro and in vivo studies suggest that IL‐4Rα PI3K signaling negatively regulates histamine‐mediated vascular endothelial leak and loss of this pathway leads to accelerated histamine‐mediated hypovolemic shock and hypothermia. These results define an unanticipated role for IL‐4Rα–mediated PI3K signaling in negative regulation of IgE‐mediated anaphylactic reactions.
Disclosures
All of the authors have declared that they have no conflict of interest.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Figure S1. No effect of loss of IL‐4Rα‐PI3K signaling on total IgE. Total IgE levels in the serum of OVA‐sensitized, intragastric (i.g.) OVA‐challenged WT and IL‐4RαY500F mice following the seventh challenge. Each filled circle represents an individual mouse. Data represent mean ± SD.
Figure S2. A positive relationship between vascular leak and shock response in murine oral antigen‐induced anaphylaxis. Correlation between hematocrit and systemic symptoms of oral antigen‐induced anaphylaxis. Spearman's rank correlation coefficient between hematocrit and temperature change from 0 to 60 min after the seventh intragastric (i.g.) OVA challenge in OVA‐sensitized WT mice. Individual symbols represent 1 mouse.
Figure S3. Systemic anaphylaxis in WT and IL‐4RαY500F mice is dependent on histamine. Temperature change from 0 to 30 min in OVA‐sensitized, intragastric (i.g.) OVA‐challenged (A) WT and (B) IL‐4RαY500F mice following the sixth and seventh intragastric (i.g.) OVA challenge. OVA‐sensitized WT and IL‐4RαY500F mice receive repeated i.g. OVA challenges, and temperature change from 0 to 30 min was determined following the sixth challenge. Prior to the seventh challenge, mice were administered the histamine Type 1 and type 2 receptor antagonists Triprolidine (200 μg) and Cimetidine (200 μg) intravenously (i.v.) (200 μL final volume) 30 min prior to OVA challenge. Each filled circle represents an individual mouse. Data represent the temperature change from 0 to 30 min following the sixth and seventh challenge; P values as indicated.
Figure S4. Histamine and DHEA‐induced PI3K activation in human vascular endothelial cell line EA.hy926. Representative Western blot analyses probing for PI3K p85 full‐length protein and actin in protein lysates from human vascular endothelial cell line EA.hy926 following 0, 1, 5, 15, 30 and 60 min stimulation with histamine (20 nM) or DHEA (100 nM).
Acknowledgments
We thank Dr. Patricia Fulkerson and Fred Finkelman and members of the Divisions of Allergy and Immunology and of Gastroenterology, Hepatology, and Nutrition, Cincinnati Children's Hospital Medical Center for critical review of the manuscript and insightful conversations. We would also like to thank Shawna Hottinger for editorial assistance and manuscript preparation.
Funding information This work was supported by NIH R01 AI073553, R01 DK090119, P30 DK078392 (S.P.H.); U19 A1070235, Crohns Colitis Foundation of America and a Food Allergy Research Education Award.
References
- 1. Sicherer, S. H. 2002. Clinical update on peanut allergy. Ann. Allergy Asthma Immunol. 88:350–361; quiz 61‐2, 94. [DOI] [PubMed] [Google Scholar]
- 2. Branum, A. M. , and Lukacs S. L.. 2009. Food allergy among children in the United States. Pediatrics 124:1549–1555. [DOI] [PubMed] [Google Scholar]
- 3. Sampson, H, A. 2004. Update on food allergy. J. Allergy Clin. Immunol. 113:805–819. [DOI] [PubMed] [Google Scholar]
- 4. Sicherer, S. H. 2011. Epidemiology of food allergy. J. Allergy Clin. Immunol. 127:594–602. [DOI] [PubMed] [Google Scholar]
- 5. Branum, A. M. , and Lukacs S. L.. 2008. Food allergy among U.S. children: trends in prevalence and hospitalizations. NCHS Data Brief 1–8. [PubMed] [Google Scholar]
- 6. Keet Ca, Fau , and Wood R. A.. 2007. Food allergy and anaphylaxis. Immunol. Allergy Clin. North Am. 27:193‐212, vi. [DOI] [PubMed] [Google Scholar]
- 7. Sampson, H. A. 1999. Food allergy. Part 2: diagnosis and management. J. Allergy Clin. Immunol. 103:981–989. [DOI] [PubMed] [Google Scholar]
- 8. Sampson, H. A. 1999. Food allergy. Part 1: immunopathogenesis and clinical disorders. J. Allergy Clin. Immunol. 103:717–728. [DOI] [PubMed] [Google Scholar]
- 9. Galli, S. J. , Kalesnikoff J., Grimbaldeston M. A., Piliponsky A. M., Williams C. M. M., and Tsai M.. 2005. Mast cells as “tunable” effector and immunoregulatory cells: recent advances. Annu. Rev. Immunol. 23:749–786. [DOI] [PubMed] [Google Scholar]
- 10. Strait, R. T. , Morris S. C., Yang M., Qu X. W., and Finkelman F. D.. 2002. Pathways of anaphylaxis in the mouse. J. Allergy Clin. Immunol. 109:658–668. [DOI] [PubMed] [Google Scholar]
- 11. Dombrowicz, D. , Flamand V., Brigman K. K., Koller B. H., and Kinet J. P.. 1993. Abolition of anaphylaxis by targeted disruption of the high affinity immunoglobulin E receptor alpha chain gene. Cell 75:969–976. [DOI] [PubMed] [Google Scholar]
- 12. Lorentz, A. , Schwengberg S., Mierke C., Manns M. P., and Bischoff S. C.. 1999. Human intestinal mast cells produce IL‐5 in vitro upon IgE receptor cross‐linking and in vivo in the course of intestinal inflammatory disease. Eur. J. Immunol. 29:1496–1503. [DOI] [PubMed] [Google Scholar]
- 13. Santos, J. , Benjamin M., Yang P. C., Prior T., and Perdue M. H.. 2000. Chronic stress impairs rat growth and jejunal epithelial barrier function: role of mast cells. Am. J. Physiol. Gastrointest. Liver Physiol. 278:G847–G854. [DOI] [PubMed] [Google Scholar]
- 14. Kelefiotis, D. , and Vakirtzi‐Lemonias C.. 1993. In vivo responses of mouse blood cells to platelet‐activating factor (PAF): role of the mediators of anaphylaxis. Agents Actions 40:150–156. [DOI] [PubMed] [Google Scholar]
- 15. Strait, R. T , Morris S. C., Smiley K., J. F. Urban, Jr. , and Finkelman F. D.. 2003. IL‐4 exacerbates anaphylaxis. J. Immunol. 170:3835–3842. [DOI] [PubMed] [Google Scholar]
- 16. Finkelman, F. D. 2007. Anaphylaxis: lessons from mouse models. J. Allergy Clin. Immunol. 120:506–515. [DOI] [PubMed] [Google Scholar]
- 17. Finkelman, F. D. , Rothenberg M. E., Brandt E. B., Morris S. C., and Strait R. T.. 2005. Molecular mechanisms of anaphylaxis: lessons from studies with murine models. J. Allergy Clin. Immunol. 115:449‐57; quiz 58. [DOI] [PubMed] [Google Scholar]
- 18. Nelms, K. , A. D. Keegan , Zamorano J., Ryan J. J., and Paul W. E.. 1999. The IL‐4 receptor: signaling mechanisms and biologic functions. Annu. Rev. Immunol. 17:701–738. [DOI] [PubMed] [Google Scholar]
- 19. Hershey, G. K. 2003. IL‐13 receptors and signaling pathways: an evolving Web. J. Allergy Clin. Immunol. 111:677–690. [DOI] [PubMed] [Google Scholar]
- 20. Chatila, T. A. 2004. Interleukin‐4 receptor signaling pathways in asthma pathogenesis. Trends Mol. Med. 10:493–499. [DOI] [PubMed] [Google Scholar]
- 21. Giallourakis, C. , Kashiwada M., Pan P. Y., Danial N., Jiang H., Cambier J., Coggeshall K. M., and Rothman P.. 2000. Positive regulation of interleukin‐4‐mediated proliferation by the SH2‐containing inositol‐5'‐phosphatase. J. Biol. Chem. 275:29275–29282. [DOI] [PubMed] [Google Scholar]
- 22. Kashiwada, M. , Giallourakis C. C., Pan P. Y., and Rothman P. B.. 2001. Immunoreceptor tyrosine‐based inhibitory motif of the IL‐4 receptor associates with SH2‐containing phosphatases and regulates IL‐4‐induced proliferation. J. Immunol. 167:6382–6387. [DOI] [PubMed] [Google Scholar]
- 23. Losman, J. , Chen X. P., Jiang H., Pan P. Y., Kashiwada M., Giallourakis C., Cowan S., Foltenyi K., and Rothman P.. 1999. IL‐4 signaling is regulated through the recruitment of phosphatases, kinases, and SOCS proteins to the receptor complex. Cold Spring Harb. Symp. Quant. Biol. 64:405–416. [DOI] [PubMed] [Google Scholar]
- 24. Hershey, G. K. , Friedrich M. F., Esswein L. A., Thomas M. L., and Chatila T. A.. 1997. The association of atopy with a gain‐of‐function mutation in the alpha subunit of the interleukin‐4 receptor. N. Engl. J. Med. 337:1720–1725. [DOI] [PubMed] [Google Scholar]
- 25. Brown, P. , Nair B., Mahajan S. D., Sykes D. E., Rich G., Reynolds J. L., Aalinkeel R., Wheeler J., and Schwartz S. A.. 2012. Single nucleotide polymorphisms (SNPs) in key cytokines may modulate food allergy phenotypes. Eur. Food Res. Technol. 235:971–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tachdjian, R. , Al Khatib S., Schwinglshackl A., Kim H. S., Chen A., Blasioli J., Mathias C., Kim H. Y., Umetsu D. T., Oettgen H. C., et al. 2010. In vivo regulation of the allergic response by the IL‐4 receptor alpha chain immunoreceptor tyrosine‐based inhibitory motif. J. Allergy Clin. Immunol. 125:1128–1136, e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mathias, C. B. , Hobson S. A., Garcia‐Lloret M., Lawson G., Poddighe D., Freyschmidt E. J., Xing W., Gurish M. F., Chatila T. A., and Oettgen H. C.. 2011. IgE‐mediated systemic anaphylaxis and impaired tolerance to food antigens in mice with enhanced IL‐4 receptor signaling. J. Allergy Clin. Immunol. 127:795–805, e1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tachdjian, R. , Mathias C., Al Khatib S., Bryce P. J., Kim H. S., Blaeser F., O'Connor B. D., Rzymkiewicz D., Chen A., Holtzman M. J., et al. 2009. Pathogenicity of a disease‐associated human IL‐4 receptor allele in experimental asthma. J. Exp. Med. 206:2191–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Blaeser, F. , Bryce P. J., Ho N., Raman V., Dedeoglu F., Donaldson D. D., Geha R. S., Oettgen H. C., and Chatila T. A.. 2003. Targeted inactivation of the IL‐4 receptor alpha chain I4R motif promotes allergic airway inflammation. J. Exp. Med. 198:1189–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ahrens, R. , Osterfeld H., Wu D., Chen C. Y., Arumugam M., Groschwitz K., Strait R., Wang Y. H., Finkelman F. D., and Hogan S. P.. 2012. Intestinal mast cell levels control severity of oral antigen‐induced anaphylaxis in mice. Am. J. Pathol. 180:1535–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Finkelman, F. D. , Madden K. B., Morris S. C., Holmes J. M., Boiani N., Katona I. M., and Maliszewski C. R.. 1993. Anti‐cytokine antibodies as carrier proteins. Prolongation of in vivo effects of exogenous cytokines by injection of cytokine‐anti‐cytokine antibody complexes. J. Immunol. 151:1235–1244. [PubMed] [Google Scholar]
- 32. Munoz, J. , and Bergman R. K.. 1965. Mechanism of anaphylactic death in the mouse. Nature 205:199–200. [DOI] [PubMed] [Google Scholar]
- 33. Bergmann, R. K. , and Munoz J.. 1965. Circulatory chnages in anaphylaxis and histamine toxicity in mice. J. Immunol. 95:1–8. [PubMed] [Google Scholar]
- 34. Fulton, J. D. , Harris W. E., and Craft C. E.. 1957. Hematocrit chnage as indication of anaphyactic shock in the mouse. Proc. Soc. Exp. Biol. Med. 95:625–627. [DOI] [PubMed] [Google Scholar]
- 35. Kind, L. S. 1955. Fall in rectal temperature as an indication of anaphylactic shock in the mouse. J. Immunol. 74:387–390. [PubMed] [Google Scholar]
- 36. Brandt, E. B. , Strait R. T., Hershko D., Wang Q., Muntel E. E., Scribner T. A., Zimmermann N., Finkelman F. D., and Rothenberg M. E.. 2003. Mast cells are required for experimental oral allergen‐induced diarrhea. J. Clin. Invest. 112:1666–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen, H. , Lin A. S., Li Y., Reiter C. E., Ver M. R., and Quon M. J.. 2008. Dehydroepiandrosterone stimulates phosphorylation of FoxO1 in vascular endothelial cells via phosphatidylinositol 3‐kinase‐ and protein kinase A‐dependent signaling pathways to regulate ET‐1 synthesis and secretion. J. Biol. Chem. 283:29228–29238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu, D. , Iruthayanathan M., Homan L. L., Wang Y., Yang L., and Dillon J. S.. 2008. Dehydroepiandrosterone stimulates endothelial proliferation and angiogenesis through extracellular signal‐regulated kinase 1/2‐mediated mechanisms. Endocrinology 149:889–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bouis, D. , Hospers G. A., Meijer C., Molema G., and Mulder N. H.. 2001. Endothelium in vitro: a review of human vascular endothelial cell lines for blood vessel‐related research. Angiogenesis 4:91–102. [DOI] [PubMed] [Google Scholar]
- 40. Stone, S. F. , and Brown S. G.. 2012. Mediators released during human anaphylaxis. Curr. Allergy Asthma Rep. 12:33–41. [DOI] [PubMed] [Google Scholar]
- 41. Brown, S. G. 2007. The pathophysiology of shock in anaphylaxis. Immunol. Allergy Clin. North Am. 27:165–175. [DOI] [PubMed] [Google Scholar]
- 42. Kemp, S. F. , and Lockey R. F.. 2002. Anaphylaxis: a review of causes and mechanisms. J. Allergy Clin. Immunol. 110:341–348. [DOI] [PubMed] [Google Scholar]
- 43. Peavy, R. D. , and Metcalfe D. D.. 2008. Understanding the mechanisms of anaphylaxis. Curr. Opin. Allergy Clin. Immunol. 8:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hill, S. J. , C. R. Ganellin , Timmerman H., Schwartz J. C., Shankley N. P., Young J. M., Schunack W., Levi R., and Haas H. L.. 1997. International Union of Pharmacology. XIII. Classification of histamine receptors. Pharmacol. Rev. 49:253–278. [PubMed] [Google Scholar]
- 45. Pantazaka, E. , Taylor E. J., Bernard W. G., and Taylor C. W.. 2013. Ca(2+) signals evoked by histamine H1 receptors are attenuated by activation of prostaglandin EP2 and EP4 receptors in human aortic smooth muscle cells. Br. J. Pharmacol. 169:1624–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. van Nieuw Amerongen, G. P. , Draijer R., Vermeer M. A., and van Hinsbergh V. W.. 1998. Transient and prolonged increase in endothelial permeability induced by histamine and thrombin: role of protein kinases, calcium, and RhoA. Circ. Res. 83:1115–1123. [DOI] [PubMed] [Google Scholar]
- 47. Andriopoulou, P. , Navarro P., Zanetti A., Lampugnani M. G., and Dejana E.. 1999. Histamine induces tyrosine phosphorylation of endothelial cell‐to‐cell adherens junctions. Arterioscler. Thromb. Vasc. Biol. 19:2286–2297. [DOI] [PubMed] [Google Scholar]
- 48. Wierzbicki, T , Iqbal S. M., Cuvelier S. L., Awong G., Tibbles L. A., and Patel K. D.. 2003. IL‐4 primes human endothelial cells for secondary responses to histamine. J. Leukoc. Biol. 74:420–427. [DOI] [PubMed] [Google Scholar]
- 49. Madison, J. M. , and Ethier M. F.. 2001. Interleukin‐4 rapidly inhibits calcium transients in response to carbachol in bovine airway smooth muscle cells. Am. J. Respir. Cell Mol. Biol. 25:239–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Maffucci, T. , Raimondi C., Abu‐Hayyeh S., Dominguez V., G. Sala , Zachary I., and Falasca M.. 2009. A phosphoinositide 3‐kinase/phospholipase Cgamma1 pathway regulates fibroblast growth factor‐induced capillary tube formation. PLoS ONE 4:e8285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cahenzli, J. , Y. Koller , Wyss M., Geuking M. B., and McCoy K. D.. 2013. Intestinal microbial diversity during early‐life colonization shapes long term IgE levels. Cell Host Microbe 14:559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Coleman, J. W. , Holliday M. R., Kimber I., Zsebo K. M., and Galli S. J.. 1993. Regulation of mouse peritoneal mast cell secretory function by stem cell factor, IL‐3 or IL‐4. J. Immunol. 150:556–562. [PubMed] [Google Scholar]
- 53. Holliday, M. R. , Banks E. M., Dearman R. J., Kimber I., and Coleman J. W.. 1994. Interactions of IFN‐gamma with IL‐3 and IL‐4 in the regulation of serotonin and arachidonate release from mouse peritoneal mast cells. Immunology 82:70–74. [PMC free article] [PubMed] [Google Scholar]
- 54. Kruse, S. , Japha T., Tedner M., Sparholt S. H., Forster J., Kuehr J., and Deichmann K. A.. 1999. The polymorphisms S503P and Q576R in the interleukin‐4 receptor alpha gene are associated with atopy and influence the signal transduction. Immunology 96:365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tam, E. K. , Jourdan‐LeSaux C., Stauder S., Bollt O., Reber B., Yamamoto F., and Haymer D.. 2003. Polymorphisms in the interleukin‐4 receptor alpha chain: association with traits of allergy and asthma in an admixed population in Hawaii. Cell. Mol. Biol. (Noisy‐le‐grand) 49:1345–1349. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Figure S1. No effect of loss of IL‐4Rα‐PI3K signaling on total IgE. Total IgE levels in the serum of OVA‐sensitized, intragastric (i.g.) OVA‐challenged WT and IL‐4RαY500F mice following the seventh challenge. Each filled circle represents an individual mouse. Data represent mean ± SD.
Figure S2. A positive relationship between vascular leak and shock response in murine oral antigen‐induced anaphylaxis. Correlation between hematocrit and systemic symptoms of oral antigen‐induced anaphylaxis. Spearman's rank correlation coefficient between hematocrit and temperature change from 0 to 60 min after the seventh intragastric (i.g.) OVA challenge in OVA‐sensitized WT mice. Individual symbols represent 1 mouse.
Figure S3. Systemic anaphylaxis in WT and IL‐4RαY500F mice is dependent on histamine. Temperature change from 0 to 30 min in OVA‐sensitized, intragastric (i.g.) OVA‐challenged (A) WT and (B) IL‐4RαY500F mice following the sixth and seventh intragastric (i.g.) OVA challenge. OVA‐sensitized WT and IL‐4RαY500F mice receive repeated i.g. OVA challenges, and temperature change from 0 to 30 min was determined following the sixth challenge. Prior to the seventh challenge, mice were administered the histamine Type 1 and type 2 receptor antagonists Triprolidine (200 μg) and Cimetidine (200 μg) intravenously (i.v.) (200 μL final volume) 30 min prior to OVA challenge. Each filled circle represents an individual mouse. Data represent the temperature change from 0 to 30 min following the sixth and seventh challenge; P values as indicated.
Figure S4. Histamine and DHEA‐induced PI3K activation in human vascular endothelial cell line EA.hy926. Representative Western blot analyses probing for PI3K p85 full‐length protein and actin in protein lysates from human vascular endothelial cell line EA.hy926 following 0, 1, 5, 15, 30 and 60 min stimulation with histamine (20 nM) or DHEA (100 nM).