

Summary
An important role of transforming growth factor‐β (TGF‐β) in the development of regulatory T cells is well established. Although integrin‐mediated activation of latent TGF‐β 1 is considered essential for the induction of regulatory T (Treg) cells by antigen‐presenting cells (APCs), such an activation mechanism is not applicable to the TGF‐β 2 isoform, which lacks an integrin‐binding RGD sequence in its latency‐associated peptide. Mucosal and ocular tissues harbour TGF‐β 2‐expressing APCs involved in Treg induction. The mechanisms that regulate TGF‐β activation in such APCs remain unclear. In this study, we demonstrate that murine APCs exposed to TGF‐β 2 in the environment predominantly increase expression of TGF‐β 2. Such predominantly TGF‐β 2‐expressing APCs use thrombospondin‐1 (TSP‐1) as an integrin‐independent mechanism to activate their newly synthesized latent TGF‐β 2 to induce Foxp3+ Treg cells both in vitro and in vivo. Expression of Treg induction by TGF‐β 2‐expressing APCs is supported by a TSP‐1 receptor, CD36, which facilitates activation of latent TGF‐β during antigen presentation. Our results suggest that APC‐derived TSP‐1 is essential for the development of an adaptive regulatory immune response induced by TGF‐β 2‐expressing APCs similar to those located at mucosal and ocular sites. These findings introduce the integrin‐independent mechanism of TGF‐β activation as an integral part of peripheral immune tolerance associated with TGF‐β 2‐expressing tissues.
Keywords: antigen presentation, regulatory T cells, transforming growth factor‐β2, thrombospondin‐1
Introduction
Recent studies have revealed a significant role of transforming growth factor‐β (TGF‐β) in the induction, expansion and maintenance of regulatory immune responses,1 implicating a favoured generation of regulatory T (Treg) cells by resident antigen‐presenting cells (APCs) that are exposed to TGF‐β in tissue environments. As such, APCs in mucosal tissue such as gut‐associated lymphoid tissue are reported to induce peripheral Foxp3‐expressing CD4+ Treg cells under homeostatic conditions.2 Similarly, APCs that phagocytose apoptotic or necrotic cells during tissue homeostasis release TGF‐β, which not only prevents the activation of inflammatory effectors but also allows the expansion of natural Foxp3+ Treg cells.3, 4 Hence, the presence of TGF‐β during antigen presentation is known to aid the generation of Treg cells.
Several studies have reported increased expression of TGF‐β 2 in epithelia at mucosal surfaces under inflammatory conditions. For example, gastric epithelial cells produce TGF‐β 2 in response to Helicobacter pylori infection,5 bronchial epithelial cells in asthmatic patients,6 and conjunctival epithelial cells from patients with ocular surface inflammation express significantly increased TGF‐β 2.7 Moreover, predominant expression of TGF‐β 2 is detected in adult human intestinal epithelium and lamina propria as well as in mouse ocular mucosal epithelium.8, 9 Although activation of TGF‐β associated with Treg induction has been largely attributed to integrins expressed by epithelial cells (αv β 6) or dendritic cells (DC; αv β 8),10, 11 these studies are predominantly focused on the TGF‐β 1 isoform, which contains an integrin‐binding RGD sequence in its latency‐associated peptide (LAP). The absence of such a sequence in the TGF‐β 2 LAP isoform prevents activation of latent TGF‐β 2 by integrins.12 Therefore, at sites with predominant TGF‐β 2 expression, integrin‐independent mechanisms are likely to assume greater importance in influencing Treg induction.
We have previously reported significantly increased expression of thrombospondin‐1 (TSP‐1) by APCs exposed to TGF‐β 2.13 Thrombospondin‐1 is a large (450 000 MW) multi‐domain glycoprotein with a binding site for LAP and the ability to activate latent TGF‐β to its biologically active form in vitro as well as in vivo.14, 15 The TSP‐1 binding amino acid sequence in LAP is distinct from RGD and is well conserved in all three isoforms (LAP1, LAP2 and LAP3). As such, TSP‐1 has been reported to activate TGF‐β 2 efficiently14 in an integrin‐independent manner. Therefore, TSP‐1 expression by APCs potentially influences their ability to use TGF‐β 2 available in the environment for Treg induction.
In addition to TSP‐1, APCs exposed to TGF‐β 2 concomitantly increase their endogenous expression of TGF‐β 2.13 Thrombospondin‐1 derived from these APCs further facilitates conversion of their newly synthesized latent TGF‐β to its biologically active form.13, 16 It has been speculated that this TSP‐1‐mediated autocrine feedback allows continued expression of TGF‐β 2 by APCs as they migrate to local lymphoid organs distant from their tissues of origin, making TGF‐β 2 available at the time of Foxp3+ Treg induction. Consistent with such a possibility, a decline in Foxp3 expression was detected in lymph nodes draining the ocular mucosa of TSP‐1‐deficient mice, which was accompanied by spontaneous development of conjunctivitis associated with autoimmune Sjögren syndrome.17 These results suggest an important role of TSP‐1 in homeostatic regulation at the ocular mucosa that expresses TGF‐β 2. Similarly, studies reporting exacerbated inflammation in the lung and gastric mucosa of TSP‐1‐deficient mice as a result of bleomycin‐induced injury or dextran sodium sulphate‐induced colitis, respectively, broaden the scope of potential TSP‐1‐mediated homeostatic regulation to other mucosal surfaces.18, 19, 20 Together, these studies strongly implicate an important contribution of APC‐derived TSP‐1 in maintaining homeostasis at mucosal surfaces. In this study, we test the hypothesis that TSP‐1 expression in TGF‐β 2‐exposed APCs is essential for their ability to induce Foxp3+ Treg cells.
Our results indicate that, indeed, TGF‐β 2‐exposed APCs predominantly express the TGF‐β 2 isoform and depend on TSP‐1 to activate their newly synthesized latent TGF‐β. The expression of TSP‐1 by TGF‐β 2‐exposed APCs is critical for generation of Foxp3+ Treg cells from naive CD4+ CD25− T cells in vitro and in vivo. Further, we report that TSP‐1 binds its receptor CD36 on APCs to bring about an autocrine activation of latent TGF‐β, and as such, this receptor contributes significantly to the induction of TGF‐β‐secreting and functional Foxp3+ Treg cells. These results identify a critical contribution of TSP‐1 in immune regulation at mucosal surfaces and induction of peripheral tolerance.
Methods
Mice
C57BL/6 (H‐2b) mice, 6–8 weeks old, were purchased from Charles River Laboratories (Wilmington, MA). C57BL/6‐Tg(TcraTcrb)425Cbn/J (transgenic for T‐cell receptor specific for chicken ovalbumin 323–339 in the context of I‐Ab) and C57BL/6‐Foxp3 tm1Flv/J [lymphocytes expressing the Foxp3 gene with monomeric red fluorescent protein (mRFP)], 6–8 weeks old, were purchased from Jackson Laboratories (Bar Harbor, ME). TSP−/− mice (C57BL/6 background) were originally received from the laboratory of Dr J. Lawler (Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA). CD36−/− mice (C57BL/6 background) were obtained from the laboratory of Dr M. Freeman (Massachusetts General Hospital, Harvard Medical School, Boston, MA). These mice were subsequently bred and maintained at the pathogen‐free animal facility at Schepens Eye Research Institute (Boston, MA). The corresponding author later moved to Boston University School of Medicine, where these animals were transferred and bred. All animals were handled in accordance with the institutional guidelines and approved animal protocols from Schepens Eye Research Institute and Boston University School of Medicine.
Serum‐free medium
Serum‐free medium was used for in vitro assays. The medium contained RPMI‐1640, 10 mm HEPES, 0·1 mm Non‐essential amino acids (NEAA), 1 mm sodium pyruvate, 100 U/ml penicillin, 100 mg/ml streptomycin, 20 mm l‐glutamine (Lonza, Basel, Switzerland), 0·1% BSA, and ITS+ culture supplement [1 μg/ml iron‐free transferrin, 10 ng/ml linoleic acid, 0·3 ng/ml Na2Se, and 0·2 μg/ml Fe(NO3)3] (Sigma Chemical Co., St Louis, MO).
Antibodies and flow cytometry
The following antibodies were used for flow cytometric analysis of cells: anti‐CD16/CD32 (Biolegend, San Diego, CA), FITC‐labelled anti‐CD4 (BD Biosciences, San Jose, CA), phycoerythrin‐labelled anti‐CD25, phycoerythrin‐Cy5‐labelled anti‐Foxp3 (eBioscience, San Diego, CA). Intracellular staining for Foxp3 was performed on cells fixed and permeabilized using a buffer (eBioscience), followed by staining with α‐Foxp3 antibodies. For surface staining of TSP‐1, cells were first fixed with 4% paraformaldehyde for 20 min at room temperature and subsequently stained with biotinylated α‐TSP‐1 (Abcam, Boston, MA). All flow cytometric analysis was performed with the appropriate isotype controls using a BD LSR II flow cytometer and flowjo analysis software (Treestar Inc., Ashland, OR).
TGF‐β 2‐exposed APCs
Dendritic cells were generated by culturing bone marrow cells (10 × 106) with granulocyte–macrophage colony‐stimulating factor (20 ng/ml; Biolegend) in a Petri dish for 7 days. The medium in these cultures was replenished every 2 days, and maturation of DCs was induced by adding lipopolysaccharide (100 ng/ml) for 24 hr before cell harvesting. Macrophages were harvested from peritoneal fluid of mice that received an intraperitoneal injection of 2 ml of a 3% thioglycollate solution (Sigma) 3 days earlier. These cells contained > 95% F4/80+ cells. Antigen‐presenting cells (1 × 106 per well) were cultured overnight in a 24‐well culture plate in serum‐free medium in the presence or absence of TGF‐β 2 (5 ng/ml; R&D Systems, Minneapolis, MN) – the predominant isoform in the ocular environment. After overnight cultures, cells were washed three times with culture medium to remove exogenous TGF‐β 2 and non‐adherent cells. Adherent cells were used as APCs.
Assay for active TGF‐β detection
To determine the active TGF‐β content of culture supernatants, TGF‐β reporter murine fibroblast cells stably transfected with the Smad‐Binding Element‐SEcreted Alkaline Phosphatase plasmid (MFB‐F11) were used.21 These cells were cultured in serum‐free Dulbecco's modified Eagle's medium containing penicillin/streptomycin for 2 hr before addition of culture supernatants from APCs, either directly, to measure active TGF‐β, or after acid activation of latent TGF‐β (2·5 μl of 6 m HCl to 50 μl sample for 10 min at room temperature followed by neutralization with 6 m NaOH) to measure total TGF‐β. After 24 hr, culture supernatants were tested for SEAP activity using Great EscAPe SEAP Reporter System 3 (Clontech, Mountain View, CA).
Preparation of T cells
Lymphocytes were isolated from the spleen and lymph nodes of C57BL/6, and C57BL/6‐Tg (TcraTcrb) 425Cbn/J OT‐II mice. CD4+ CD25− T cells were purified using magnetic beads (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer's protocol. Routine purity of CD4+ cell preparations was > 99% CD25− as assessed by flow cytometric analysis.
In vitro T‐cell activation assay
Antigen‐presenting cells pulsed overnight with ovalbumin (100 μg/ml; Sigma) were co‐cultured with CD4+ CD25− OT‐II T cells (3 × 105 per well). After 48 hr of culture at 37°, T cells were isolated, washed and analysed further. To analyse TGF‐β 1 secretion, these T cells (0·7 × 106) were stimulated with plate‐bound anti‐CD3 (2C11, 1 μg/ml; BD Biosciences) in serum‐free RPMI media. Culture supernatants collected after 48 hr were tested for levels of total TGF‐β 1 by ELISA (eBioscience).
Suppression of proliferation assay
CD4+ CD25− T cells (responder cells) were labelled with carboxyfluorescein succinimidyl ester (CFSE 1 μm; Invitrogen) and stimulated with 1 μg/ml of anti‐CD3 (2C11) and 1 μg/ml of anti‐CD28 antibody in a 24‐well plate (5 × 105 cells/well) in the presence of equal numbers of Treg cell‐containing activated T cells harvested from APC–T‐cell co‐culture assay (1 : 1 ratio of Treg : T responder cells). After 48 hr, the proliferation of responders was assessed by analysis of CFSE dilution using flow cytometry.
Statistical analyses
Student's t‐test was used to calculate statistical significance for a difference in parameter evaluated between groups. A P‐value ≤ 0·05 was considered statistically significant.
Results
TGF‐β 2‐exposed APCs predominantly express TGF‐β 2 and depend on TSP‐1 to activate their endogenous TGF‐β
To determine if APCs that encounter TGF‐β 2 in the tissue environment themselves become a significant source of TGF‐β 2, we first assessed their expression of isoforms TGF‐β 1 and TGF‐β 2. Macrophages that infiltrate the tissue during an inflammatory response, and resident DCs can both serve as APCs. Therefore, we exposed thioglycollate‐elicited peritoneal macrophages and bone marrow‐derived dendritic cells (BMDCs) derived from C57BL/6 (wild‐type; WT) mice to TGF‐β 2 in culture and examined message levels for TGF‐β 1 and TGF‐β 2 by real‐time PCR. As indicated in Fig. 1(a), predominant expression of TGF‐β 2 was detected in both macrophages and BMDCs exposed to TGF‐β 2. These results indicate that APCs may serve as a source of TGF‐β 2 during antigen presentation.
Figure 1.
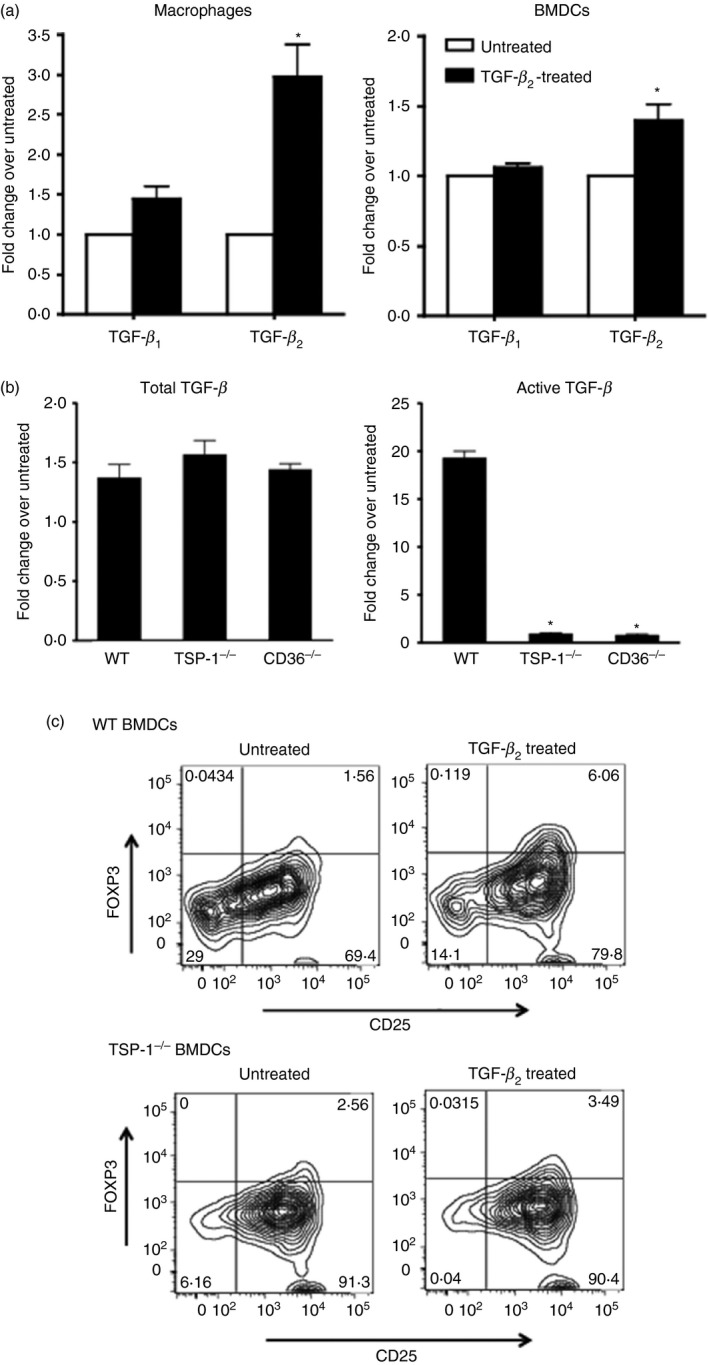
Transforming growth factor‐β 2 (TGF‐β 2) ‐exposed antigen‐presenting cells (APCs) predominantly express TGF‐β 2 and depend on thrombospondin‐1 (TSP‐1) to activate latent TGF‐β and induce Foxp3+ regulatory T (Treg) cells. (a) Expression of TGF‐β 1 and TGF‐β 2 in macrophages and bone marrow‐derived dendritic cells (BMDCs) cultured overnight in the presence of TGF‐β 2 as assessed by real‐time PCR. Changes in message levels relative to those in untreated APCs are presented. (b) Activation of TGF‐β by TGF‐β 2‐exposed wild‐type (WT), TSP‐1 null or CD36 knockout (KO) BMDCs detected using MFB‐F11 reporter fibroblast cells. Changes in the levels relative to the untreated APCs are presented. (c) Activation of CD4+ CD25− OT‐II T cells by ovalbumin (OVA) ‐pulsed WT or TSP‐1null untreated or TGF‐β 2‐treated BMDCs. Representative flow cytometry plots show percentage of activated Foxp3+ T cells (*P < 0·05 compared with TGF‐β 1 or total TGF‐β activated by WT cells). Data in panels a, b and c are as representative of three to four independent experiments.
Thrombospondin‐1 has been reported to activate TGF‐β 2 efficiently in an integrin‐independent manner.14, 22 We next tested the ability of BMDCs deficient in either TSP‐1 or its receptor CD36 in activating latent TGF‐β synthesized after their TGF‐β 2 exposure. Cultures of BMDCs treated with TGF‐β 2 were washed thoroughly to remove any residual exogenously added TGF‐β 2. These cells were further cultured in a serum‐free medium for 24 hr. Total and active TGF‐β in their culture supernatants was determined using TGF‐β‐SEAP reporter cells (MFB‐F11).21 As shown in Fig. 1(b), all three tested APCs produced comparable levels of total TGF‐β upon their TGF‐β 2 exposure. However, unlike WT BMDCs that responded to their TGF‐β 2 exposure with a nearly 20‐fold increase in active TGF‐β secretion, similarly treated APCs derived from both TSP‐1 and CD36‐deficient mice failed to increase their active TGF‐β secretion. These results indicate that APCs exposed to TGF‐β 2‐rich environments predominantly express TGF‐β 2, an isoform not activated by integrins and dependent on TSP‐1 for its activation. These results may also explain why αv integrin‐deficient BMDCs continue to induce Foxp3+ Treg cells similar to WT controls, as reported by others.23
TGF‐β 2‐exposed APCs promote de novo generation of Foxp3+ Treg cells in a TSP‐1‐dependent manner
A functionally distinct population of CD103+ DCs identified in the gut‐associated lymphoid tissue in mice was reported to predominantly express TGF‐β 2 and induce Foxp3+ Treg cells in a TGF‐β‐dependent manner.2 We sought to determine if TGF‐β 2‐exposed BMDCs in our experiments induce Foxp3+ Treg cells in vitro and, if so, whether it is dependent on their TSP‐1 expression. To determine this, we co‐cultured ovalbumin‐pulsed untreated or TGF‐β 2‐treated BMDCs (WT or TSP‐1‐deficient) with CD4+ CD25− OT‐II T cells expressing ovalbumin‐specific T‐cell receptors. After 48 hr, Foxp3 expression was analysed by flow cytometry. In the presence of untreated WT BMDCs, < 2% of activated T cells expressed Foxp3 (72 cells/4612 CD4+ cells), whereas TGF‐β 2‐exposed WT BMDCs induced a three‐fold increase in the proportion of Foxp3+ activated T cells (Fig. 1c, 222 cells/6351 CD4+ cells). These results establish the de novo Foxp3‐inducing ability of TGF‐β 2‐exposed APCs. However, TSP‐1‐deficient BMDCs exposed to TGF‐β 2 failed to induce such an increase in Foxp3+ Treg cells (untreated 197 cells/7697 CD4+ cells versus TGF‐β 2‐treated 406 cells/6702 CD4+ cells), indicating that their TSP‐1 expression is essential for the induction of Foxp3+ Treg cells. Furthermore, the comparison of absolute numbers rules out any possibility of selective increase in a Foxp3‐negative population as a basis for the noted difference. We noted similar diminished Foxp3 induction using a TSP‐1‐deficient adherent population harvested from BMDC cultures or a mixed population of adherent and non‐adherent BMDCs with a maximal difference detected using non‐adherent DCs as shown in Fig. 1(c). Interestingly, although T cells in our experiments were capable of expressing TSP‐1, it did not influence their Foxp3 expression. Hence, our results clearly demonstrate that TGF‐β 2‐exposed APC‐derived TSP‐1 is important in supporting the generation of Treg cells.
Deficiency of TSP‐1 receptor CD36 in TGF‐β 2‐exposed APCs affects expression of Foxp3 in activated T cells
Similar to BMDCs, predominant TGF‐β 2 expression was also detected in TGF‐β 2‐exposed macrophages (data not shown). Along with other investigators, we have previously reported TSP‐1‐dependent activation of macrophage‐derived TGF‐β.16, 24 As both tissue‐infiltrating macrophages and resident DCs are involved in presenting antigens, we evaluated the Foxp3+ Treg‐inducing ability of TGF‐β 2‐exposed macrophages (WT and TSP‐1‐deficient) in subsequent experiments in this study using an assay similar to that used to evaluate BMDCs. As described earlier, ovalbumin‐pulsed macrophages untreated or TGF‐β 2‐treated were co‐cultured with CD4+ CD25− OT‐II T cells followed by Foxp3 detection in activated T cells at 48 hr by flow cytometry. As shown in Fig. 2, similar to BMDCs, TGF‐β 2‐exposed WT macrophages induced a nearly three‐fold increase in Foxp3+ Treg cells compared with their untreated controls. In the absence of TSP‐1 expression, however, this Foxp3 induction was abolished. To rule out failure of antigen presentation, we used CFSE‐labelled CD4+ CD25− OT‐II T cells in our in vitro assay and assessed CFSE dilution by flow cytometry. Significantly increased proliferation in cells stimulated by TGF‐β 2‐exposed TSP‐1‐deficient APCs was detected compared with those stimulated by similarly treated WT APCs (% divided: 21·9 versus 12·7, Proliferation Index: 2·76 versus 2·22, respectively). These results are consistent with a previously reported inability of TSP‐1‐deficient macrophages to activate their endogenous TGF‐β.16 Together, our results indicate that, as in DCs, TSP‐1 is also essential for TGF‐β 2‐exposed macrophages to activate their endogenous TGF‐β and induce Foxp3+ Treg cells.
Figure 2.
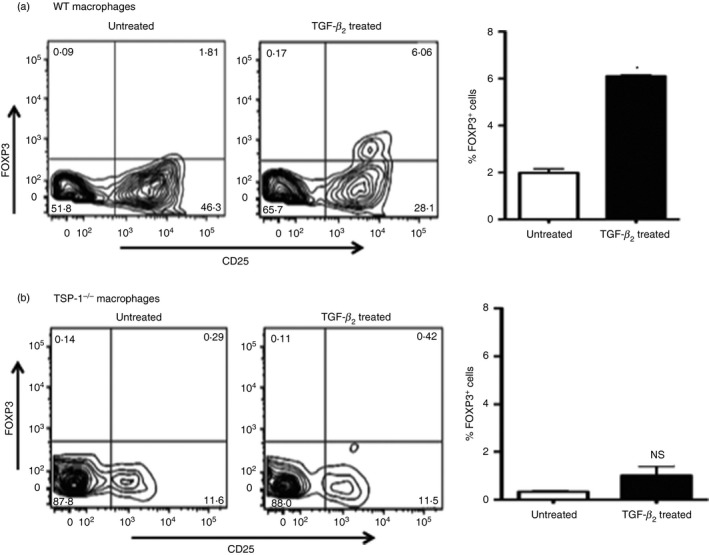
Wild‐type (WT) macrophages exposed to transforming growth factor‐β 2 (TGF‐β 2) induce Foxp3 expression in activated T cells in a thrombospondin‐1 (TSP‐1) ‐dependent manner. Activation of CD4+ CD25− OT‐II T cells by ovalbumin (OVA) ‐pulsed untreated or TGF‐β 2‐treated thioglycollate‐induced macrophages derived from WT (a) and TSP‐1−/− (b) mice. Representative flow cytometry and mean data plots of percentage of activated Foxp3+ T cells. These results are representative of two to three independent experiments (*P < 0·05).
Ligation of CD36 on macrophages by TSP‐1 has been reported to be essential for the activation of latent TGF‐β, and more recently it was reported to induce IL‐10 secretion in alveolar macrophages.19, 24 To determine if CD36 plays a key role in the induction of Foxp3+ Treg cells, we first confirmed that, similar to rat alveolar macrophages, peritoneal exudate mouse macrophages localize TSP‐1 and LAP on their surface in a CD36‐dependent manner. Flow cytometric analysis of WT and CD36‐deficient macrophages stained for surface TSP‐1 or LAP (that binds TSP‐1) indicated significantly reduced expression of both in CD36‐deficient macrophages compared with WT macrophages (Fig. 3a, CD36−/− versus WT, TSP‐1 MFI: 1045 ± 81·4 versus 1668 ± 14·7, P < 0·05 and LAP MFI: 1002 ± 111·4 versus 1436 ± 16·1, P < 0·05). We then assessed the effect of CD36 deficiency in macrophages on the induction of Foxp3+ Treg cells in our in vitro T‐cell activation assay. The CD36 deficiency of APCs and the resulting reduced surface TSP‐1 significantly diminished, but did not abolish, the induction of Foxp3+ Treg cells as noted, with TSP‐1 deficiency of APCs (Fig. 3b) compared with the WT controls. These results highlight a significant contribution of CD36‐bound TSP‐1 to the ability of TGF‐β 2‐exposed APCs to induce Foxp3+ Treg cells. Considering that both TSP‐1 and CD36 deficiency in APCs result in a loss of TGF‐β activation, the partial abrogation of Foxp3 induction by TGF‐β 2‐exposed CD36‐deficient APCs may suggest the existence of an as yet unappreciated TGF‐β‐independent mechanism of Treg cell induction, which requires further investigation.
Figure 3.
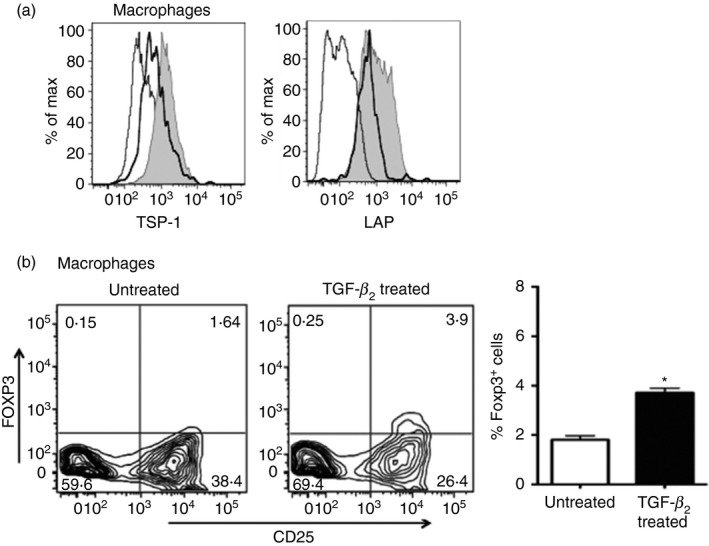
The absence of thrombospondin‐1 (TSP‐1) receptor CD36 on transforming growth factor‐β 2 (TGF‐β 2‐exposed antigen‐presenting cells (APCs) reduces their ability to induce Foxp3. (a) Flow cytometric analysis of the surface staining of TSP‐1 and latency‐associated peptide (LAP) on macrophages derived from wild‐type (WT; filled grey) and CD36−/− (thick black lines) mice presented as histograms. Isotype control staining is indicated by thin lines. (b) Representative flow cytometric and mean data plots of Foxp3 induction by untreated or TGF‐β 2‐treated CD36−/− macrophages in CD4+ CD25− OT‐II T cells (*P < 0·05). Data are representative of three independent experiments.
TSP‐1 and CD36‐deficient APCs fail to generate functional Foxp3+ Treg cells
It has been reported by others that Foxp3 expression in T cells may not be adequate for their differentiation into functional Treg cells.25 Secretion of TGF‐β by Foxp3+ Treg cells is well documented along with their ability to suppress T‐cell proliferation.26 To determine if TGF‐β 2‐exposed APCs generate functional Foxp3+ Treg cells in a TSP‐1‐dependent manner, we tested their ability to secrete TGF‐β 1 and suppress T‐cell proliferation. We first investigated TGF‐β 1 secretion by Foxp3+ Treg cells induced by TGF‐β 2‐exposed APCs and whether it is influenced by the absence of TSP‐1 or its receptors on APCs. From the in vitro assay described earlier, T cells activated by TGF‐β 2‐exposed WT, TSP‐1 null, and CD36−/− APCs were washed and stimulated with anti‐CD3/CD28 antibodies, and TGF‐β 1 released in 48‐hr culture supernatants was measured by ELISA. Although TGF‐β 2‐exposed WT APCs induced TGF‐β 1‐secreting activated T cells consistent with an increased proportion of Foxp3+ Treg cells, the APCs deficient in TSP‐1 and CD36 failed to do so (Fig. 4a). Next, we tested their ability to suppress the proliferation of naive CD4+ CD25− T cells used as responders in a CFSE dilution assay. In these experiments, CFSE‐labelled responders were stimulated with plate‐bound anti‐CD3 in the presence of putative Treg cells harvested from in vitro T‐cell activation assays set up with different APCs as described earlier. Cells from multiple wells (four per APC type) were pooled, and their CFSE dilution was assessed by flow cytometry to determine proliferation index. As shown in Fig. 4(b), TGF‐β 2‐treated WT APCs clearly induced functional Treg cells that suppressed proliferation of CD3‐stimulated naive T cells, but APCs lacking either TSP‐1 or CD36 expression failed to induce Treg cells with this suppressive property. Such a functional deficit was consistent with their inability to secrete TGF‐β. These results clearly demonstrate that during Treg induction, APCs serve as a critical source of TGF‐β that is important for preserving the functionality of Treg cells.
Figure 4.
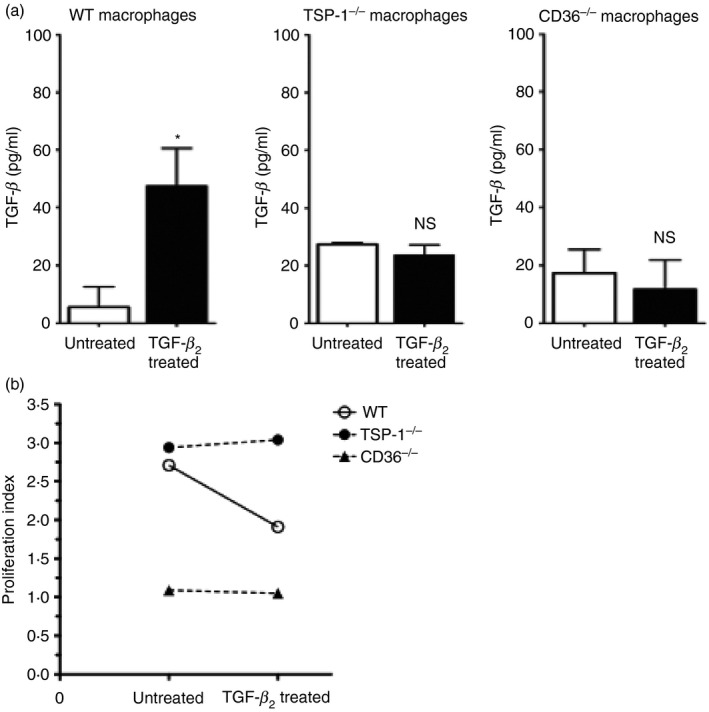
Functional loss of regulatory T (Treg) cells with the absence of either thrombospondin‐1 (TSP‐1) or CD36 on transforming growth factor‐β 2 (TGF‐β 2) ‐exposed antigen‐presenting cells (APCs). Activated T cells by the indicated APCs were assessed for their ability to secrete TGF‐β and suppress T‐cell proliferation. (a) Culture supernatants of activated T cells re‐stimulated with anti‐CD3/CD28 antibodies as described in Materials and methods were tested by ELISA for levels of TGF‐β. (b) Suppressor activity was assessed by analysing proliferation of anti‐CD3‐stimulated CFSE‐labelled CD4+ CD25− wild‐type (WT) T cells in the presence of Treg cells added at a 1 : 1 ratio. Flow cytometric analysis of CFSE dilution of one million cells in each experiment was used to determine the percentage of proliferating cells. The data represent two to three individual experiments (*P < 0·05).
Endogenous TSP‐1 and CD36 expression by APCs is necessary for in vivo Treg induction by TGF‐β 2‐exposed APCs
Although in our in vitro experiments T‐cell‐derived TSP‐1 did not contribute to Treg induction, TSP‐1 is expressed by many other cell types in the periphery, including vascular endothelial cells and activated platelets.15 To determine if peripheral expression of TSP‐1 or any other TGF‐β‐activating mechanism available in vivo can substitute for the APC‐derived TSP‐1 and support the induction of Treg cells, we infused antigen‐pulsed APCs (untreated or TGF‐β 2‐exposed) intravenously into Foxp3.mRFP transgenic mice and assessed numbers of Foxp3+ Treg cells. In these transgenic mice, mRFP fluorescence intensity is reported to faithfully mark Foxp3 expression.27 Previously, it was reported that TGF‐β 2‐exposed APCs preferentially localize to the spleen to induce Treg cells.28 Therefore, we tested induction of Foxp3+ Treg cells in the recipient spleens by determining CD4+ T cells that are also positive for mRFP fluorescence. A baseline Foxp3+ population, as reported originally,27 was detectable in Foxp3.mRFP transgenic control mice that did not receive any APCs (Fig. 5a). In this experiment, Foxp3+ Treg cells detected in the spleens of mice infused with APCs include expanded pre‐existing natural Treg cells as well as Treg cells induced from naive CD4+ T cells. Hence, Treg cells detectable in recipients of untreated APCs are likely to be the expanded population of natural Treg cells, while those detected in recipients of TGF‐β 2‐exposed APCs may contain additional Treg cells induced from naive T cells. Consistent with this possibility, a significantly increased proportion of Foxp3+ Treg cells in the recipients of TGF‐β 2‐treated WT APCs was detectable compared with control recipients of untreated WT APCs (Fig. 5b). However, such an increase was not detected with either TSP‐1 or CD36‐deficient APCs (Fig. 5c,d), just as noted in our in vitro experiments. This result clearly indicates that APC‐derived TSP‐1 is critical for Foxp3+ Treg induction, and that peripheral TSP‐1 in recipients could not substitute for this source of TSP‐1 during antigen presentation by TGF‐β 2‐exposed APCs in the spleen.
Figure 5.
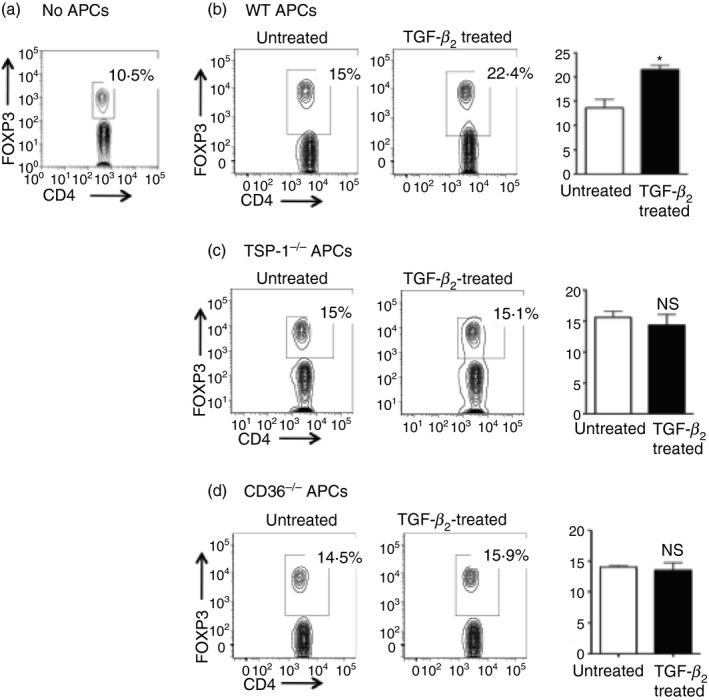
Transforming growth factor‐β (TGF‐β) ‐exposed antigen‐presenting cells (APCs) deficient in thrombospondin‐1 (TSP‐1) or its receptor CD36 fail to induce Treg cells in vivo. Foxp3.mRFP mice (n = 3/group) were injected intravenously with (a) no APCs or ovalbumin (OVA) ‐pulsed, untreated, or TGF‐β‐treated (b) wild‐type (WT), (c) TSP‐1−/− or (d) CD36−/− macrophages (104 cells/mouse) 12 days before determining the proportion of Foxp3‐expressing CD4 T cells in their spleens. The plots show analysis of pooled splenocytes harvested from mice in each group. Gates in each experiment were set based on respective untreated APC controls and in some include CD4 T cells expressing lower levels of Foxp3. The data represent three individual experiments.
Discussion
In this study, we demonstrate that APCs exposed to TGF‐β 2 predominantly express the same isoform and depend on their TSP‐1–CD36 interaction to activate it. We provide evidence that both TSP‐1 and CD36 expression by such APCs is essential for the subsequent induction of TGF‐β 1‐secreting Foxp3+ Treg cells. In particular, these results highlight the significance of TSP‐1‐mediated TGF‐β activation for the immunological tolerance induced by APCs located in the proximity of TGF‐β 2‐expressing parenchymal cells at mucosal surfaces.
In our experiments, DCs exposed to TGF‐β 2 induced increased expression of endogenous TGF‐β 2, as previously detected in similarly treated macrophages,9 but not TGF‐β 1. The lack of an integrin‐binding RGD site in the LAP of TGF‐β 2 prevents its activation by integrins,12 while RGD‐independent TSP‐1–LAP binding allows efficient activation of latent TGF‐β 2.14 More recent studies have reported a predominance of the TGF‐β 2 isoform in epithelial cells at mucosal surfaces like conjunctiva, lungs and gut.5, 6, 7, 8, 9 Hence it is possible that mucosal epithelial cell‐derived TGF‐β 2 induces endogenous expression of TGF‐β 2 in APCs located in the vicinity and contributes to their Foxp3+ Treg induction in a TSP‐1‐dependent manner. Interestingly, the absence of TSP‐1 or CD36 in APCs in our experiments resulted in a virtual loss of active TGF‐β, and residual active TGF‐β 1, if any, did not support the induction of Foxp3+ Treg cells by TSP‐1‐deficient APCs, highlighting the significance of APC‐derived TGF‐β 2 in Treg induction. In fact, consistent with our results, TSP‐1 deficiency in mice leads to spontaneous inflammation of the ocular mucosa (conjunctivitis), which correlates with a significant decline in Foxp3+ Treg cells in their draining lymph nodes and development of age‐related autoimmunity that targets the conjunctiva.17
Both macrophages and DCs express the TSP‐1 receptor CD36.29 Our results show that deficiency of CD36 in APCs results in a marked decline in cell‐surface‐bound LAP, which is also consistent with their lost ability to activate latent TGF‐β, clearly indicating a role of CD36 in facilitating activation of endogenous TGF‐β. We demonstrate that the absence of either TSP‐1 or CD36 on TGF‐β 2‐exposed APCs abrogates their ability to induce Foxp3+ Treg cells in vivo. These results not only confirm an important role of TSP‐1 and CD36 in Treg induction, but also underscore the importance of endogenous TSP‐1 expressed in TGF‐β 2‐exposed APCs as opposed to that available in the environment. Furthermore, our observations suggest that APCs, once exposed to TGF‐β 2, may retain their ability to induce Treg cells even when away from the site of TGF‐β 2 exposure.
In murine mesenteric lymph nodes, a subpopulation of APCs (CD103+ DCs) is believed to have arrived from the intestinal mucosa and is reported to express high levels of TGF‐β 2.2 In light of our results, their TGF‐β 2 expression may be attributed to their exposure to the same isoform expressed by intestinal epithelia.8 Such TGF‐β 2‐expressing CD103+ DCs are capable of inducing Foxp3+ Treg cells and are implicated in the maintenance of intestinal homeostasis. Although integrins have been suggested to play a role in TGF‐β activation in the intestinal environment,11, 23 this mechanism is applicable only to the TGF‐β 1‐expressing subpopulation of APCs. These APCs may represent a local population, as opposed to TGF‐β 2‐expressing CD103+ DCs, which probably represent a distinct migratory subset that reaches draining mesenteric lymph nodes. Based on our results, TGF‐β 2‐expressing APCs are equipped with TSP‐1 and CD36 that contribute to their Treg‐inducing ability. The presence of such distinct populations may also explain observations reported in mice with a conditional deletion of integrins in DCs,23 where selective reduction is detected in colonic but not splenic Treg proportions and the loss of integrins failed to impair Treg generation by BMDCs.11 Considering a common bone marrow‐derived origin of CD103+ DCs and splenic DCs,30 it is quite likely that both of these DC subsets represent TGF‐β 2‐expressing APCs similar to the BMDCs used in our experiments and depend on TSP‐1 to activate their endogenous TGF‐β, making integrins dispensable. Together, these studies suggest that the expression of TGF‐β 2 and TSP‐1 may indeed define the phenotype of a migratory APC population at mucosal surfaces.
Recently, we identified a significant correlation between a THBS1 polymorphism and reduced TSP‐1 expression in human ocular mucosal cells.31 The lowered TSP‐1 expression in these cells was also accompanied by significantly increased expression of pro‐inflammatory IL‐1β. Such changes correlated with the development of chronic ocular surface inflammation. During such inflammation, the expression of TGF‐β 2 in human conjunctival tissue is significantly increased.7 Our findings in the present study clearly demonstrate the significance of TSP‐1 in activation of newly synthesized TGF‐β 2 by APCs and its contribution to the development of regulatory immunity. It is conceivable that any imbalance in such regulation may therefore contribute to chronic inflammatory responses. Hence our results offer a potential mechanism underlying the development of chronic inflammation of the ocular mucosa in individuals with reduced TSP‐1 expression. Chronic ocular inflammation is also associated with autoimmune Sjögren syndrome and is noted in TSP‐1‐deficient mice.32 Hence, TSP‐1 clearly plays an important role in homeostatic regulation at the ocular mucosal surface.
In this study, we identify TSP‐1‐dependent Treg induction by TGF‐β 2‐exposed APCs as a mechanism potentially important in homeostatic regulation at mucosal surfaces. Overall, emerging evidence supports TSP‐1 as an important player in tolerance induction and immune regulation at sites where the TGF‐β 2 isoform predominates.
Disclosure
The authors have no competing interest to disclose.
Acknowledgements
This research was supported by National Eye Institute Grant EY015472.
During the course of this study authors moved from Schepens Eye Research Institute, Department of Ophthalmology, Harvard Medical School to the Boston University School of Medicine.
References
- 1. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol 2012; 30:531–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Coombes JL, Siddiqui KR, Arancibia‐Carcamo CV, Hall J, Sun CM, Belkaid Y et al A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGFβ and retinoic acid‐dependent mechanism. J Exp Med 2007; 204:1757–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGFβ, PGE2, and PAF. J Clin Invest 1998; 101:890–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kleinclauss F, Perruche S, Masson E, de Carvalho Bittencourt M, Biichle S, Remy‐Martin JP et al Intravenous apoptotic spleen cell infusion induces a TGF‐β‐dependent regulatory T‐cell expansion. Cell Death Differ 2006; 13:41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beswick EJ, Pinchuk IV, Earley RB, Schmitt DA, Reyes VE. Role of gastric epithelial cell‐derived transforming growth factor β in reduced CD4+ T cell proliferation and development of regulatory T cells during Helicobacter pylori infection. Infect Immun 2011; 79:2737–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chu HW, Balzar S, Seedorf GJ, Westcott JY, Trudeau JB, Silkoff P et al Transforming growth factor‐β2 induces bronchial epithelial mucin expression in asthma. Am J Pathol 2004; 165:1097–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Benito MJ, Calder V, Corrales RM, Garcia‐Vazquez C, Narayanan S, Herreras JM et al Effect of TGF‐β on ocular surface epithelial cells. Exp Eye Res 2013; 107:88–100. [DOI] [PubMed] [Google Scholar]
- 8. Maheshwari A, Kelly DR, Nicola T, Ambalavanan N, Jain SK, Murphy‐Ullrich J et al TGF‐β2 suppresses macrophage cytokine production and mucosal inflammatory responses in the developing intestine. Gastroenterology 2011; 140:242–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Takeuchi M, Kosiewicz MM, Alard P, Streilein JW. On the mechanisms by which transforming growth factor‐β2 alters antigen‐presenting abilities of macrophages on T cell activation. Eur J Immunol 1997; 27:1648–56. [DOI] [PubMed] [Google Scholar]
- 10. Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J et al The integrin αvβ6 binds and activates latent TGF β1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999; 96:319–28. [DOI] [PubMed] [Google Scholar]
- 11. Travis MA, Reizis B, Melton AC, Masteller E, Tang Q, Proctor JM et al Loss of integrin αvβ8 on dendritic cells causes autoimmunity and colitis in mice. Nature 2007; 449:361–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFβ activation. J Cell Sci 2003; 116:217–24. [DOI] [PubMed] [Google Scholar]
- 13. Masli S, Turpie B, Hecker KH, Streilein JW. Expression of thrombospondin in TGFβ‐treated APCs and its relevance to their immune deviation‐promoting properties. J Immunol 2002; 168:2264–73. [DOI] [PubMed] [Google Scholar]
- 14. Ribeiro SM, Poczatek M, Schultz‐Cherry S, Villain M, Murphy‐Ullrich JE. The activation sequence of thrombospondin‐1 interacts with the latency‐associated peptide to regulate activation of latent transforming growth factor‐β . J Biol Chem 1999; 274:13586–93. [DOI] [PubMed] [Google Scholar]
- 15. Crawford SE, Stellmach V, Murphy‐Ullrich JE, Ribeiro SM, Lawler J, Hynes RO et al Thrombospondin‐1 is a major activator of TGFβ1 in vivo . Cell 1998; 93:1159–70. [DOI] [PubMed] [Google Scholar]
- 16. Masli S, Turpie B, Streilein JW. Thrombospondin orchestrates the tolerance‐promoting properties of TGFβ‐treated antigen‐presenting cells. Int Immunol 2006; 18:689–99. [DOI] [PubMed] [Google Scholar]
- 17. Contreras‐Ruiz L, Regenfuss B, Mir FA, Kearns J, Masli S. Conjunctival inflammation in thrombospondin‐1 deficient mouse model of Sjogren's syndrome. PLoS One 2013; 8:e75937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ezzie ME, Piper MG, Montague C, Newland CA, Opalek JM, Baran C et al Thrombospondin‐1‐deficient mice are not protected from bleomycin‐induced pulmonary fibrosis. Am J Respir Cell Mol Biol 2011; 44:556–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhao Y, Xiong Z, Lechner EJ, Klenotic PA, Hamburg BJ, Hulver M et al Thrombospondin‐1 triggers macrophage IL‐10 production and promotes resolution of experimental lung injury. Mucosal Immunol 2014; 7:440–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Punekar S, Zak S, Kalter VG, Dobransky L, Punekar I, Lawler JW et al Thrombospondin 1 and its mimetic peptide ABT‐510 decrease angiogenesis and inflammation in a murine model of inflammatory bowel disease. Pathobiology 2008; 75:9–21. [DOI] [PubMed] [Google Scholar]
- 21. Tesseur I, Zou K, Berber E, Zhang H, Wyss‐Coray T. Highly sensitive and specific bioassay for measuring bioactive TGF‐β . BMC Cell Biol 2006; 7:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Annes JP, Rifkin DB, Munger JS. The integrin αVβ6 binds and activates latent TGFβ3. FEBS Lett 2002; 511:65–8. [DOI] [PubMed] [Google Scholar]
- 23. Lacy‐Hulbert A, Smith AM, Tissire H, Barry M, Crowley D, Bronson RT et al Ulcerative colitis and autoimmunity induced by loss of myeloid αv integrins. Proc Natl Acad Sci USA 2007; 104:15823–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yehualaeshet T, O'Connor R, Green‐Johnson J, Mai S, Silverstein R, Murphy‐Ullrich JE et al Activation of rat alveolar macrophage‐derived latent transforming growth factor β‐1 by plasmin requires interaction with thrombospondin‐1 and its cell surface receptor, CD36. Am J Pathol 1999; 155:841–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gavin MA, Torgerson TR, Houston E, DeRoos P, Ho WY, Stray‐Pedersen A et al Single‐cell analysis of normal and FOXP3‐mutant human T cells: FOXP3 expression without regulatory T cell development. Proc Natl Acad Sci USA 2006; 103:6659–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nakamura K, Kitani A, Fuss I, Pedersen A, Harada N, Nawata H et al TGF‐β1 plays an important role in the mechanism of CD4+CD25+ regulatory T cell activity in both humans and mice. J Immunol 2004; 172:834–42. [DOI] [PubMed] [Google Scholar]
- 27. Wan YY, Flavell RA. Identifying Foxp3‐expressing suppressor T cells with a bicistronic reporter. Proc Natl Acad Sci USA 2005; 102:5126–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wilbanks GA, Streilein JW. Macrophages capable of inducing anterior chamber associated immune deviation demonstrate spleen‐seeking migratory properties. Reg Immunol 1992; 4:130–7. [PubMed] [Google Scholar]
- 29. Febbraio M, Hajjar DP, Silverstein RL. CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J Clin Investig 2001; 108:785–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bogunovic M, Ginhoux F, Helft J, Shang L, Hashimoto D, Greter M et al Origin of the lamina propria dendritic cell network. Immunity 2009; 31:513–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Contreras‐Ruiz L, Ryan DS, Sia RK, Bower KS, Dartt DA, Masli S. Polymorphism in THBS1 gene is associated with post‐refractive surgery chronic ocular surface inflammation. Ophthalmology 2014; 121:1389–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Turpie B, Yoshimura T, Gulati A, Rios JD, Dartt DA, Masli S. Sjogren's syndrome‐like ocular surface disease in thrombospondin‐1 deficient mice. Am J Pathol 2009; 175:1136–47. [DOI] [PMC free article] [PubMed] [Google Scholar]