

Summary
The CD73 ectonucleotidase catalyses the hydrolysis of AMP to adenosine, an immunosuppressive molecule. Recent evidence has demonstrated that this ectonucleotidase is up‐regulated in T helper type 17 cells when generated in the presence of transforming growth factor‐β (TGF‐β), and hence CD73 expression is related to the acquisition of immunosuppressive potential by these cells. TGF‐β is also able to induce CD73 expression in CD8+ T cells but the function of this ectonucleotidase in CD8+ T cells is still unknown. Here, we show that Tc17 cells present high levels of the CD73 ectonucleotidase and produce adenosine; however, they do not suppress the proliferation of CD4+ T cells. Interestingly, we report that adenosine signalling through A2A receptor favours interleukin‐17 production and the expression of stem cell‐associated transcription factors such as tcf‐7 and lef‐1 but restrains the acquisition of Tc1‐related effector molecules such as interferon‐γ and Granzyme B by Tc17 cells. Within the tumour microenvironment, CD73 is highly expressed in CD62L+ CD127+ CD8+ T cells (memory T cells) and is down‐regulated in GZMB + KLRG1+ CD8+ T cells (terminally differentiated T cells), demonstrating that CD73 is expressed in memory/naive cells and is down‐regulated during differentiation. These data reveal a novel function of CD73 ectonucleotidase in arresting CD8+ T‐cell differentiation and support the idea that CD73‐driven adenosine production by Tc17 cells may promote stem cell‐like properties in Tc17 cells.
Keywords: adenosine, CD73, cell differentiation, stem cell, Tc17, tumour immunology
Abbreviations
- A2AR
A2A adenosine receptor
- APC
antigen‐presenting cell
- FCS
fetal calf serum
- GzmB
granzyme B
- IFN‐ γ
interferon‐γ
- IL
interleukin
- MFI
mean fluorescence intensity
- OVA
ovalbumin
- PI
propidium iodide
- SCH
SCH 58261
- TGF‐β
transforming growth factor‐β
- Th17
T helper type 17
Introduction
Clinical studies have demonstrated the ability of tumour‐specific T cells to mediate the regression of established tumours.1, 2 However, cellular therapies have been limited by the loss of anti‐tumour function in vivo as T cells mature towards terminal differentiation.3, 4 Memory T cells share with stem cells the ability to self‐renew and give rise to progeny of multiple lineages, a feature known as multipotency.5 Hence, relying on these properties mentioned above, memory CD8+ T cells may be regarded as presenting a superior anti‐tumour capacity compared with other terminally differentiated T‐cell subsets. Indeed, pre‐clinical studies using adoptive transfer of purified CD8+ T‐cell subpopulations have revealed that less differentiated or memory T cells can mediate enhanced anti‐tumour responses compared with terminally differentiated T cells, mainly through increased proliferative potential and survival.6, 7
Recent evidence has pointed to a role of Wnt signalling in the formation of long‐term maintenance of memory CD8+ T cells. The group of Restifo has demonstrated that Wnt signalling arrests effector T‐cell differentiation and generates CD8+ memory stem cells with enhanced anti‐tumour function.7 Therefore, enforcing the acquisition of stem cell‐like properties on anti‐tumour CD8+ T cells by activating Wnt signalling may be pivotal to the development of more effective T‐cell‐based immunotherapies.
CD8+ T cells can be classified into type 1 (Tc1) and type 2 (Tc2) cytotoxic CD8+ T cells that are characterized by the production of interferon‐γ (IFN‐γ) and interleukin‐4 (IL‐4), respectively.8 In addition to Tc1 and Tc2 cells, a new subset of CD8+ T cells termed Tc17 cells was described. Tc17 cells produce IL‐17 and present a low cytotoxic capacity.9, 10 Interestingly, similar to their T helper type 17 (Th17) counterparts, Tc17 cells present active Wnt signalling, as suggested by the expression of tcf‐7 (a direct target of Wnt signalling) and β‐catenin accumulation.11 The capacity to self‐renew, persist in time and differentiate into IFN‐γ‐producing cells12 makes Tc17 cells interesting potential candidates for new T‐cell‐based therapies.
Purinergic mediators, such as ATP, can be released into the extracellular space constitutively and in response to stimulation where they can mediate an autocrine feedback signalling through several cell surface purinergic receptors. Following its release into the extracellular space, ATP is rapidly hydrolysed by the cooperative action of ectonucleotidases, such as CD39 and CD73.13 CD39 (E‐NTPDase1) converts ATP into AMP and then CD73 (Ecto5'NTase) dephosphorylates AMP into adenosine. Adenosine can signal through four G‐protein‐coupled adenosine receptors: A1, A2A, A2B and A3.14 The A2A receptor is a Gs‐protein‐coupled receptor, which is the predominant adenosine receptor expressed by CD4+ and CD8+ T cells and it is rapidly induced upon activation.15, 16 Adenosine signalling through the A2A receptor was previously related to the survival and persistence of T cells. This effect is mediated by preventing the down‐regulation of CD127 (IL‐7Rɑ) upon activation of CD4+ and CD8+ T cells through the down‐regulation of Akt activation.17, 18 Hence, in addition to its immunosuppressive properties, adenosine may be considered as having a role in restraining the progression of naive to effector CD4+ and CD8+ T cells in the absence of strong T‐cell receptor stimulation.
As it was demonstrated that transforming growth factor‐β (TGF‐β) can induce CD73 expression in CD8+ T cells19 and that Tc17 cells present an active Wnt signalling,11 we studied whether CD73‐mediated adenosine production has a role in the maintenance of stem cell‐like features by Tc17 cells. Our results demonstrate that Tc17 cells express high levels of lef‐1 and tcf‐7, two downstream transcription factors of the Wnt/β‐catenin‐signalling cascade. In agreement with this finding, Tc17 cells present stem‐cell‐like features such as persistence, reduced susceptibility to apoptosis‐induced cell death, and a high potential of differentiating into Tc1‐like cells within the tumour microenvironment. Interestingly, Tc17 cells express high levels of the CD73 ectonucleotidase and produce adenosine; however, they do not suppress CD4+ T‐cell proliferation. In contrast, we demonstrated that CD73‐mediated adenosine production may be involved in maintaining the stem‐cell programme on Tc17 cells and prevents the acquisition of a Tc1‐like phenotype. Finally, here we demonstrate that CD73 is expressed in memory/naive CD8+ T cells and is down‐regulated during effector differentiation within the tumour, suggesting that CD73 may have a role in restraining CD8+ T‐cell differentiation. Our results suggest that CD73‐driven adenosine production may be involved in arresting CD8+ T‐cell differentiation and promoting stem‐cell‐like properties on Tc17 cells.
Materials and methods
Mice, tumour cell lines and tumours
C57BL/6, B6.SJL‐PTPRC (CD45.1+) and OT‐I mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and kept in an animal facility under standard housing guidelines. Animal work was carried out under the institutional regulations of Fundacion Ciencia & Vida and Facultad de Ciencias, Universidad de Chile and was locally approved by an ethics review committee. The B16.F10 murine melanoma was obtained from the American Type Culture Collection (Manassas, VA). Dr Randolph Noelle (Dartmouth Medical School, Hanover, NH) kindly provided the B16.OVA cells. All cell lines were routinely tested for mycoplasma contamination. A total of 0·5 × 106 B16.OVA or 1 × 106 B16.F10 cells were injected into the intradermal layer of the right flank of mice. Tumours became visible at day 10, and the size was measured every 2–3 days. Two perpendicular measurements were made with a caliper, and the tumour area was estimated as the product of both measurements.
Generation of Tc17 and Tc1 cells
Antigen‐presenting cells (APC) were purified from the spleens of C57BL/6 mice. The spleen was mechanically disaggregated and resuspended in 5 ml RPMI + 10% fetal calf serum (FCS). Tissues were digested in the presence of Collagenase D (1 mg/ml) and DNase I (25 μg/ml) (Roche, Basel, Switzerland) for 45 min at 37° with constant agitation. Cell suspensions were filtered with a 70‐μm cell strainer. The APC were positively selected using anti‐CD11c MACS (clone n418; Miltenyi Biotec, Bergisch Gladbach, Germany) following the manufacturer's instructions.
CD8+ T cells were purified from the spleens of C57BL/6 or OT‐I mice. The spleens were perfused with RPMI + 10% FCS. CD8+ T cells were positively selected using anti‐CD8 MACS (Miltenyi Biotec) following the manufacturer's instructions. CD8+ T cells and APC were co‐cultured in a 5 : 1 ratio, respectively, (0·1 × 106 CD8+ T cells/well) in a 96‐well round‐bottom microplate. T cells were stimulated with 1 μg/ml α‐CD3 antibody (clone 145‐2C11; eBioscience, San Diego, CA) in the presence or absence of Tc17 polarizing conditions: 20 ng/ml IL‐6 (R&D Systems, Minneapolis, MN), 2 ng/ml TGF‐β (eBioscience) and 5 μg/ml α‐IFN‐γ (clone XMG1.2, BioLegend, San Diego, CA), or Tc1 polarizing conditions: 10 ng/ml IL‐2 (eBioscience) and 10 ng/ml IL‐12 (R&D Systems). Alternatively, Tc17 cells were generated in the absence of APC by stimulation with 1 μg/ml plate‐bound α‐CD3 (145‐2C11; eBioscience) and α‐CD28 antibodies (37.51; BioLegend) in the presence of polarizing cytokines.
Isolation of tumour‐infiltrating lymphocytes
Tumour‐infiltrating lymphocytes were isolated from intradermal melanoma tumours. Whole tumours were dissected and disaggregated mechanically. Minced tissues were resuspended in 5 ml of Hanks' balanced salt solution + 5% FCS and digested in the presence of 1 mg/ml Collagenase D (Roche) and 25 μg/ml DNase I (Roche) for 30 min at 37° with constant agitation. The cell suspension was filtered with a 70‐μm cell strainer (BD Falcon, Franklin Lakes, NJ). Leucocytes were resuspended in 40% Percoll (GE Healthcare, Chalfont St Giles, UK) and gently layered over 70% Percoll. The gradient was centrifuged at 750 g for 20 min at room temperature. Mononuclear cells were collected from the interphase and were washed and resuspended in RPMI‐1640 + 10% FCS.
Intracellular staining and flow cytometry
Tumour‐infiltrating lymphocytes, lymph node cells and polarized CD8 T‐cell subsets were re‐stimulated by incubation with 0·25 μm PMA (Sigma‐Aldrich, St Louis, MO) and 1 μg/ml ionomycin (Sigma‐Aldrich) or plate‐bound anti‐CD3 (145‐2C11, eBioscience) plus soluble anti‐CD28 (clone 37.51, BioLegend) in the presence of Golgi Plug (BD Biosciences, San Jose, CA) for 4 hr. Cells were first stained with antibodies against cell surface markers, CD8a (53‐6.7), CD45.2 (104), CD39 (24DMS1), CD73 (TY/11.8), and then were resuspended in a fixation/permeabilization solution (Cytofix/Cytoperm; BD Biosciences, San Jose, CA) and incubated with antibodies against IFN‐γ (XMG1.2), IL‐17 (eBio17B7) and GzmB (GB11) for 30 min at 4°. Cells were then washed with a permeabilization buffer and resuspended in PBS + 2% FCS for FACS analysis (FACSCanto II; BD Biosciences). In some cases, Fixable Viability Dye (eBioscience) was used to discard dead cells from the analysis. The analysis of FACS data was performed using the FLOWJO software (Tree Star Inc., Ashland, OR).
Adoptive transfer experiments
For adoptive transfer experiments, 1 × 106 ovalbumin (OVA) ‐specific Tc17 or Tc1 cells were transferred into CD45.1 congenic tumour‐bearing mice. Ten days after adoptive transfer, the mice were killed, and the tumour and tumour draining lymph nodes were dissected. Transferred cells (CD45.2+) were analysed for IL‐17, IFN‐γ and GzmB production by flow cytometry. For long‐term analysis of anti‐tumour activity of Tc1 and Tc17 cells, 1 × 106 OVA‐specific Tc17 or Tc1 cells were transferred into CD45.1 congenic mice. One day after the adoptive transfer, the mice were immunized intraperitoneally with OVA protein (1 mg). Two weeks later, mice were re‐immunized with OVA protein (0·5 mg) and 24 hr later, were challenged with 1 × 106 B16‐OVA cells. Tumour growth was monitored every 2 days.
Cytokine secretion measurements
Tc1 and Tc17 cells were activated for 4 hr at 1 × 106 cells/ml with 0·25 μm PMA (Sigma‐Aldrich) and 1 μg/ml ionomycin (Sigma‐Aldrich). Following activation, the supernatants were harvested and analysed using the mouse Th1/Th2/Th17 CBA Kit (BD Biosciences) following the manufacturer's instructions. Granulocyte–macrophage colony‐stimulating factor was analysed by ELISA using the BD OptEIA kit (BD Biosciences, 555167).
Activation‐induced cell death
Differentiated OVA‐specific Tc17 and Tc1 cells were re‐stimulated for 3 hr with plate‐bound α‐CD3 (145‐2C11) and α‐CD28 (37.51). Apoptosis was measured using Annexin V (BD Biosciences) and propidium iodide (PI; Sigma‐Aldrich) staining by flow cytometry.
HPLC analysis of AMP hydrolysis
CD73 enzymatic activity by Tc1 and Tc17 cells was evaluated as the percentage of AMP hydrolysis by HPLC. Briefly, Tc1 and Tc17 cells generated in vitro were diluted in Hanks' balanced salt solution and incubated in a 96‐well flat‐bottom plate at 0·5 × 105 cells/well in the presence of 10 μm AMP (Sigma‐Aldrich), with or without the CD73 inhibitor APCP (Adenosine 5'‐(α,β‐methylene)diphosphate) (50 μm) (Sigma‐Aldrich). After 1 hr, the cells were harvested, transferred to ice for 15 min, and then centrifuged at 1000 g for 10 min. Then supernatants were collected and stored at −20° until the analysis. The HPLC analysis was carried out in a Water Breeze system using an anion exchanger column (Mono Q; GE Healthcare, Chalfont St Giles, UK). The mobile phase used consisted of a linear gradient from buffer A (Tris–HCl 100 mm, pH 7·8) to buffer B (Tris–HCl 100 mm, NaCl 1·0 m, pH 7·8). The effluent was monitored at 257 nm using an online UV detector. The column was calibrated using AMP and adenosine as standards.
Real time PCR
After activation, CD8+ T cells were isolated, and RNA was extracted using the RNeasy Mini Kit (Qiagen, Hilden, Germany). First‐strand cDNA was synthesized from 1 μg of RNA using the First Strand Kit from Invitrogen (Carlsbad, CA). RT‐PCR was performed on a Stratagene Mx3000P machine following the manufacturer's protocol. The relative level of mRNA expression for each gene was normalized to the expression of HPRT (hypoxanthine guanine phosphoribosyl transferase). The following primers were used:
Gzmb forward 5′‐TGCTGCTAAAGCTGAAGAGTAAG‐3′;
reverse, 5′‐TGCTGCTAAAGCTGAAGAGTAAG‐3′
Prf‐1 forward, 5′‐GATGTGAACCCTAGGCCAGA‐3′;
reverse, 5′‐GGTTTTTGTACCAGGCGAAA‐3′
Tcf‐7 forward, 5′‐CAATCTGCTCATGCCCTACC‐3′;
reverse, 5′‐CTTGCTTCTGGCTGATGTCC‐3′
Lef‐1 forward, 5′‐TGAGTGCACGCTAAAGGAGA‐3′;
reverse, 5′‐CTGACCAGCCTGGATAAAGC‐3′
HPRT forward, 5′‐CTCCTCAGACCGCTTTTTG‐3′; reverse, 5′‐TAACCTGGTTCATCATCGC‐3′
Statistical analysis
Data are presented as mean ± SEM. Differences between groups were determined using a Mann–Whitney test or a two‐tailed t‐test. Differences between tumour growth rates were determined using two‐way analysis of variance. The statistical analysis and graphs were obtained using GRAPHPAD PRISM (GraphPad Software Inc., San Diego, CA).
Results
In vitro polarized Tc17 cells have stem cell properties
Interleukin‐17‐producing CD4+ T cells or Th17 cells have recently been considered as a less differentiated subset of T helper cells, presenting multipotency and self‐renewal properties, both features associated with stem cells.11, 20 To test whether stemness is a feature shared among all IL‐17‐producing T cells, we generated CD8+ IL‐17‐producing T cells (Tc17 cells) in vitro (see Supplementary material, Fig. S1a) and analysed markers related to their differentiation status. As previously reported,10 Tc17 cells present a dramatically reduced cytotoxic capacity compared with Tc1 cells (see Supplementary material, Fig. S1b,c). Importantly, Tc17 cells generated in vitro showed a higher expression of stemness‐associated genes, such as the Wnt targets tcf7 and lef1, and a lower expression of gzmB compared with Tc1 polarized T cells (Fig. 1a). Tc17 cells also produced higher levels of IL‐2, tumour necrosis factor‐α, and granulocyte–macrophage colony‐stimulating factor than did Tc1 cells when activated in vitro (Fig. 1b).
Figure 1.
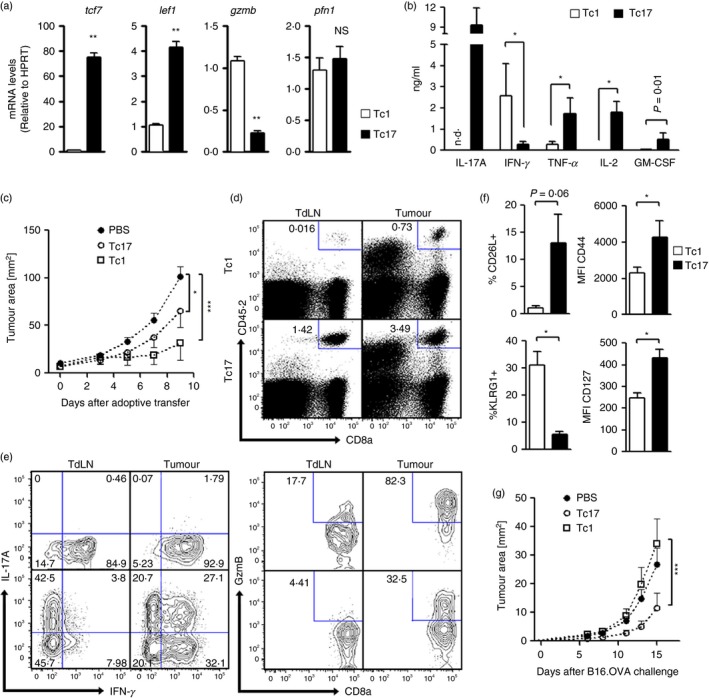
In vitro‐generated Tc17 cells present stem‐cell‐like properties and anti‐tumour activity. Ovalbumin (OVA) ‐specific Tc1 and Tc17 cells were generated by the co‐culture of CD8+ T cells (from OT‐I mice), with antigen‐presenting cells for 4 days in the presence of soluble α‐CD3 and polarizing cytokines [interleukin‐6 (IL‐6), transforming growth factor‐β 1 (TGF‐β 1) and α‐interferon‐γ (α‐IFN‐γ) for Tc17 cells and IL‐2 plus IL‐12 for Tc1 cells]. (a) Expression of tcf7, lef1, gzmb and pfn1 in Tc1 and Tc17 cells assayed by real time PCR (n = 3). (b) Cytokine production by Tc1 or Tc17 cells was measured by CBA or ELISA after stimulation with PMA plus ionomycin for 4 hr (n = 4). (c) Tc1 and Tc17 cells were adoptively transferred (1 × 106) into B16‐OVA‐bearing B6.SJL mice (CD45.1+). Tumour growth in Tc1, Tc17 or PBS‐treated mice was monitored every 2–3 days. (n = 6 mice per group). (d) Ten days after adoptive transfer, the mice were killed and the frequency of transferred (CD45.2+) cells in tumour‐draining lymph nodes (TdLN) and tumours was evaluated by flow cytometry. (e) The production of IFN‐γ, IL‐17 and GzmB was evaluated by flow cytometry on transferred cells (CD8+ CD45.2+) following in vitro activation with PMA plus ionomycin in the presence of Golgi Plug for 4 hr. (f) The expression of memory‐related markers (CD62L/CD127) and terminal differentiation‐related markers (CD44/KLRG1) was analysed on adoptively transferred Tc1 and Tc17 cells (n = 3). (g) 1 × 106 OVA‐specific Tc17 or Tc1 cells were transferred into B6.SJL CD45.1 congenic mice. One day after the adoptive transfer, the mice were immunized intraperitoneally with OVA protein (1 mg). Two weeks later, mice were re‐immunized with OVA protein (0·5 mg) and 24 hr later challenged with 1 × 106 B16‐OVA cells. Tumour growth was monitored every 2 days (n = 4 mice per group). Data in (d) and (e) are representative of two independent experiments (n = 3 per group). The percentage of cells within each gate or quadrant are presented in (d) and (e). n.d., not detected; *P < 0·05; **P < 0·01; ***P < 0·005 Mann–Whitney test.
As terminally differentiated or senescent T cells are prone to apoptosis‐induced cell death, which limits the therapeutic efficacy of T cells,21, 22 we next evaluated apoptosis‐induced cell death in Tc1 and Tc17 cells. In agreement with a less differentiated status, Tc17 cells suffered significantly lower apoptosis‐induced cell death than did Tc1 cells (4·9% versus 33·2% of apoptotic cells, respectively) (see Supplementary material, Fig. S1d).
Stem‐cell‐like features in T cells were related to better anti‐tumoral potential in adoptive transfer immunotherapy.6, 7 To evaluate the anti‐tumoral capacity of Tc17 cells, we generated Tc17 and Tc1 cells from OT‐I mice (CD45.2+) and transferred 1 × 106 Tc1 or Tc17 cells into B16‐OVA tumour‐bearing B6.SJL mice (CD45.1). As shown in Fig. 1(c), both Tc1 and Tc17 cells were able to reduce tumour growth, but Tc17 cells were less efficient compared with Tc1 cells. Both Tc1 and Tc17 cells were able to migrate to tumours and persist there until day 10. However, only Tc17 cells were able to migrate to tumour‐draining lymph nodes (Fig. 1d), a migration pattern that is related to less differentiated central memory T cells.23, 24 In addition, Tc17 cells were able to persist as IL‐17‐producing cells in the tumour‐draining lymph nodes and tumour, as long as 10 days after the adoptive transfer, providing evidence for the self‐renewal capacity of Tc17 cells (Fig. 1e). Moreover, within the tumour microenvironment, Tc17 cells were able to give rise to Tc1‐like IFN‐γ + GzmB+ effector T cells, showing their multi‐potency capacity (Fig. 1e). Interestingly, following adoptive transfer, Tc17 cells expressed higher levels of CD127 and CD62L and lower expression of KLRG1 when compared with Tc1 cells (Fig. 1f).
Next, we analysed long‐term protection by transferring Tc1 and Tc17 cells to normal mice and 2 weeks later challenging these mice with B16‐OVA cells. As shown in Fig. 1(g), Tc17 cells were more efficient than Tc1 cells in controlling tumour growth. All of these data support the idea that Tc17 cells are a less differentiated subset of CD8+ T cells that can persist in vivo, and differentiate into effector Tc1‐like T cells in the tumour microenvironment.
Tc17 cells express high levels of CD73 and produce adenosine
A previous report demonstrated that Th17 cells generated with IL‐6 and TGF‐β express CD39 and CD73 ectonucleotidases, leading to adenosine production and immunosuppression.25 Hence, we studied whether Tc17 cells, generated with the same combination of cytokines as Th17 cells, also express the CD39 and CD73 ectonucleotidases. Our results show that Tc17 cells generated in vitro are CD39– CD73+, whereas Tc1 cells are CD39+ CD73– (Fig. 2a). Endogenous Tc17 cells obtained from the lymph nodes of wild‐type mice also express higher levels of the CD73 ectonucleotidase compared to Tc1 cells (Fig. 2b).
Figure 2.
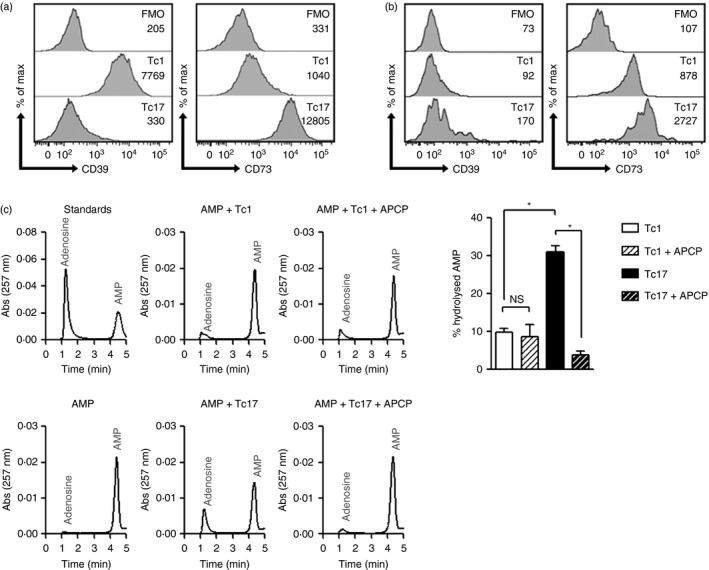
In vitro‐generated Tc17 cells express high levels of CD73 and hydrolyse AMP to adenosine. (a) CD39 and CD73 expression by Tc1 and Tc17 cells generated in vitro (obtained from C57BL/6 mice), as evaluated by flow cytometry (representative of seven independent experiments). (b) CD39 and CD73 expression by endogenous Tc1 and Tc17 cells obtained from peripheral lymph nodes of C57BL/6 mice (n = 3). The geometric mean of CD39 and CD73 is presented in each histogram. (c) 5 × 104 Tc1 or Tc17 cells were incubated with exogenous AMP (10 μm) for 1 hr and the supernatant was analysed by HPLC for the presence of AMP and adenosine using anion exchange chromatography (n = 4). Standards were prepared from the stock solution the same day of the analysis using HPLC. Results are expressed as % of hydrolysed AMP and calculated as (100 × Adenosine peak area)/(Adenosine peak area + AMP peak area). ns, non‐significant; *P < 0·05; Mann–Whitney test.
CD73 is an extracellular GPI‐anchored 5′‐nucleotidase that catalyses the hydrolysis of AMP to adenosine. In agreement with our previous observation, in‐vitro‐generated Tc17, but not Tc1, cells were able to hydrolyse AMP to adenosine, as measured by HPLC (Fig. 2c). This activity is CD73‐dependent because the addition of APCP, a specific inhibitor of CD73 activity, abolished adenosine production by CD4+Tc17 cells (Fig. 2c). We next tested the immunosuppressive capacity of Tc17 cells. Although Tc17 cells were able to delay CD4+ T‐cell proliferation in a contact‐dependent manner, this inhibition is abolished when Tc17 cells and effector cells were cultured in Transwell chambers even in the presence of exogenous AMP (see Supplementary material, Fig. S2). These data suggest that CD73‐mediated adenosine production by Tc17 cells is not involved in delaying T‐cell proliferation.
Adenosine induces stem‐cell‐like properties in Tc17 cells
As Akt inhibition enhances the expansion of lymphocytes with memory cell features26 and A2A receptor signalling blocks Akt activation,18 we studied whether adenosine might support the stem cell‐like phenotype of Tc17 cells. For this, we generated Tc17 cells in the presence of SCH 58261, a specific inhibitor of A2A adenosine receptor. As shown in Fig. 3(a), the addition of SCH 58261 during Tc17 cell differentiation induced the down‐regulation of tcf‐7 and lef‐1 and slightly up‐regulated gzmb expression in these cells. Other memory T‐cell‐associated markers such as CD127, CD44 and CD62L were not affected in Tc17 cells after the addition of SCH 58261 during their differentiation (Fig. 3b). Interestingly, the addition of SCH 58261 reduced IL‐17 production and enhanced IFN‐γ production during Tc17 cell differentiation (Fig. 3c). This effect is not only mediated by the blockade of A2A receptors on dendritic cells, as the addition of SCH 58261 during Tc17 cell differentiation using α‐CD3 and α‐CD28 antibodies also resulted in a reduced IL‐17 production and enhanced IFN‐γ production (see Supplementary material, Figure S3).
Figure 3.
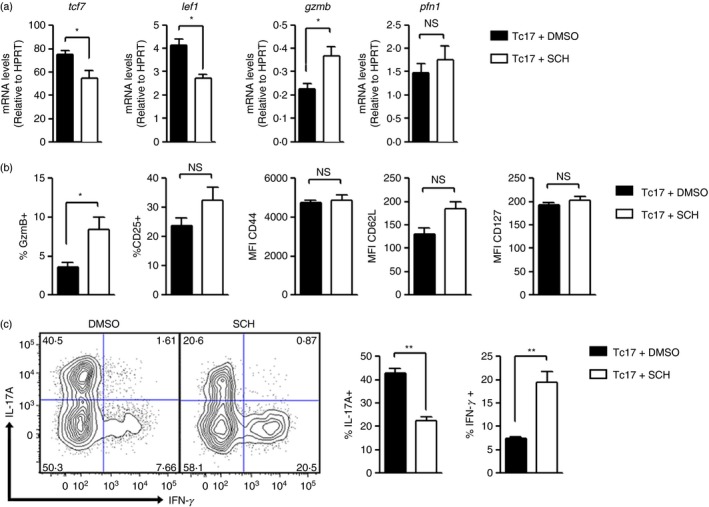
Adenosine induces stem cell markers in Tc17 cells. CD8+ T cells from C57BL/6 mice were co‐cultured for 4 days with antigen‐presenting cells and were activated with soluble α‐CD3 and polarizing cytokines [interleukin‐6 (IL‐6), transforming growth factor‐β 1 (TGF‐β 1) and α‐interferon‐γ (α‐IFN‐γ)] in the presence of the A2AR antagonist, SCH 58261(5 μm) or DMSO as the vehicle (n = 3). (a) The expression of tcf7, lef1, gzmb and pfn1 was measured using real time PCR. (b) The expression of surface markers was analysed by flow cytometry. (c) IFN‐γ and IL‐17 intracellular staining of Tc17 cells differentiated in the presence of SCH 58261 (5 μm) or DMSO and stimulated with PMA and ionomycin in the presence of Golgi Plug for 4 hr. SCH, SCH 58261; *P < 0·05; **P < 0·01 two‐tailed t‐test.
All of these results suggest that CD73‐mediated adenosine production sustains Tc17 stem cell‐like phenotype by restraining the acquisition of an effector Tc1‐like phenotype.
CD73 is down‐regulated following conversion to Tc1 cells
Next, we studied whether CD73 expression in Tc17 cells is down‐regulated following re‐activation and differentiation into effector Tc1 cells. For this, we generated Tc17 cells and then the cells were reactivated in the absence of Tc17 polarizing cytokines (TGF‐β and IL‐6) to induce conversion to Tc1 cells. As shown in Fig. 4(a), when reactivated in the absence of polarizing cytokines, Tc17 cells generated in vitro rapidly produce GzmB, acquiring a Tc1‐like phenotype and at the same time down‐regulate CD73 expression.
Figure 4.
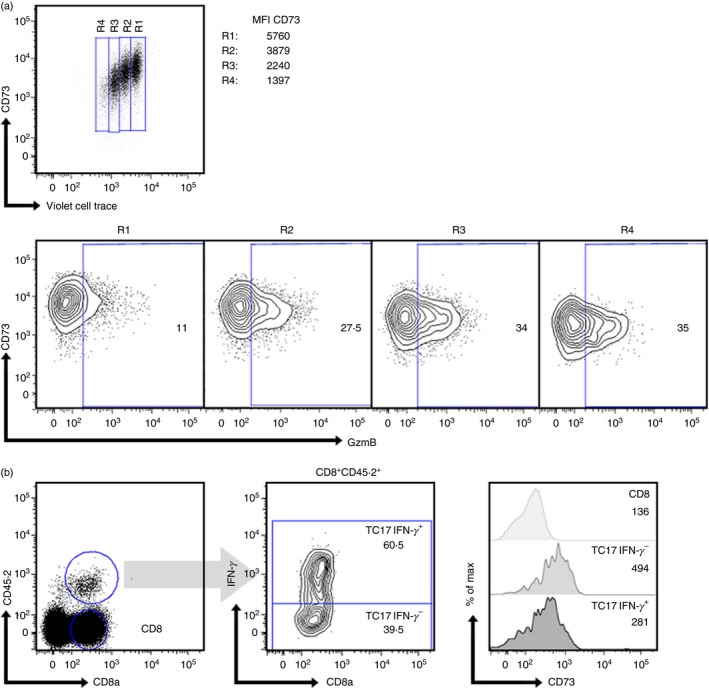
CD73 is down‐regulated in Tc17 cells during differentiation to Tc1‐like cells. (a) In vitro‐generated Tc17 cells (obtained from C57BL/6 mice) were stained with Violet cell‐trace dye and then re‐stimulated with plate‐bound α‐CD3 and α‐CD28 for 2 days in the absence of polarizing cytokines. CD73 expression and GzmB production was analysed by flow cytometry (n = 1). Numbers inside the contour plots represent the percentage of GzmB+ cells obtained for each region. (b) Tc17 cells generated in vitro from OT‐I mice were adoptively transferred to CD45.1+ B16‐OVA tumour‐bearing mice. Ten days after the adoptive transfer, CD45.2+ transferred cells were analysed for interferon‐γ (IFN‐γ) production and CD73 expression (n = 3). CD73 geometric mean was analysed in IFN‐γ + and IFN‐γ – transferred populations and compared with endogenous total CD8+ T cells (histograms).
As we have demonstrated that adoptively transferred Tc17 cells are able to convert to Tc1‐like cells in the tumour microenvironment (Fig. 1e), we studied whether these cells also down‐regulate CD73 expression during their differentiation to Tc1‐like cells in vivo. As shown in Fig. 4(b), adoptively transferred Tc17 cells that convert to IFN‐γ‐producing Tc1‐like cells in the tumour microenvironment down‐regulate CD73 expression. These data suggest that CD73 is down‐regulated in Tc17 cells during the conversion to Tc1‐like cells.
CD73 expression in CD8+ T cells is related to a memory T‐cell phenotype
To further understand the role of CD73 and adenosine production by CD8+ T cells, we analysed the expression of this ectonucleotidase in CD8+ T‐cell subsets obtained from the spleen of wild‐type mice. As shown in Fig. 5(a), naive (CD44lo CD62Lhi) CD8+ T cells express high levels of CD73 and lack CD39 expression. In contrast, effector/memory (CD44hi CD62Llo) CD8+ T cells can be divided into two subpopulations presenting high and low CD73 expression (CD73+ CD39– and CD73lo CD39+).
Figure 5.
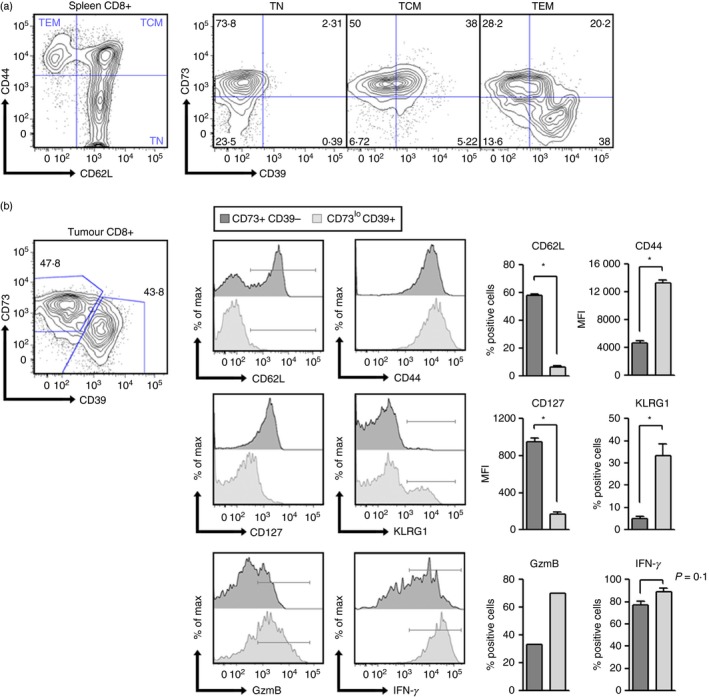
CD73 expression in CD8+ T cells is down‐regulated during effector differentiation. (a) CD39 and CD73 expression was analysed on naive (TN), central memory (TCM) and effector memory (TEM) splenic CD8+ T cells from C57BL/6 mice (n = 4). (b) Total CD8+ tumour‐infiltrating lymphocytes were analysed based on CD39 and CD73 expression. The expression of memory‐related markers (CD62L/CD127) and terminal differentiation‐related markers (CD44/KLRG1/IFN‐γ/GzmB) was analysed on CD39‐CD73+ (grey histograms) and CD39+ CD73– subsets (light grey histograms) (n = 4). TN, naive T cell; TCM, central memory T cell; TEM, effector memory T cell. The percentages of cells within each gate or quadrant are presented in (a) and (b). *P < 0·05 Mann–Whitney test.
Next, we studied CD73 expression in tumour‐infiltrating CD8+ T‐cell subsets obtained from B16 tumour‐bearing mice. Interestingly, we again observed two subpopulations of CD8+ T cells presenting high and low CD73 expression within the tumour (Fig. 5b). When analysing CD73+ and CD73lo CD8+ T‐cell subpopulations, we observed that CD73+ T cells had a naive/memory‐like phenotype characterized by high expression of CD62L and CD127 (IL‐7Rɑ) and absence of KLRG1 expression. In contrast, CD73lo T cells resemble a terminally differentiated phenotype because they lack CD62L expression and present low levels of CD127 (Fig. 5b). All of these data suggest that CD73 is expressed in naive/memory CD8+ T cells and is down‐regulated upon differentiation.
Discussion
Several reports have highlighted the role of adenosine release in the tumour microenvironment as being detrimental to the anti‐tumour immune response. High concentrations of adenosine, such as the levels achieved in the tumour microenvironment, can inhibit natural killer cell function,27 suppress Th1 and Th2 responses28 and modulate macrophage function.29 In agreement with these findings, the expression of adenosine‐generating ecto‐enzymes was also related to tumour evasion of host immunity.30 CD39 and CD73 are two major nucleotide‐metabolizing enzymes that act in tandem to catabolize ATP to adenosine.14 These enzymes are expressed in regulatory T cells and are now considered as part of the suppressive arsenal of these cells.31, 32, 33 Hence, CD39 and CD73 expression in tumours was proposed not only as prognostic markers in cancer patients but also as clinically relevant targets in anti‐tumour therapies.34 Accordingly, therapies using blocking antibodies against CD73 were shown to be effective in reducing tumour growth in mice.35, 36, 37, 38, 39
It has been reported that Th17 cells express the CD39 and CD73 ectonucleotidases, produce adenosine and present an immunosuppressive phenotype. In that study, Chalmin and collaborators describe that TGF‐β and IL‐6, both cytokines involved in inducing the Th17 programme, up‐regulate CD39 and CD73 expression in CD4+ T cells. Moreover, Th17 cells were able to reduce CD4+ and CD8+ T‐cell effector functions, such as IFN‐γ and granzyme B (GzmB) production.25 To study whether Tc17 cells also express these ectonucleotidases and produce adenosine, we generated Tc17 cells using TGF‐β and IL‐6 and observed that these cells express high levels of CD73 but low levels of CD39. In agreement with this observation, Tc17 cells produced adenosine in the presence of AMP, but were not able to reduce CD4+ T‐cell proliferation in suppression assays in vitro. However, we observed that Tc17 cells were highly proliferative and that at high Tc17/T effector ratios (1 : 1 and above), they could inhibit CD4+ T‐cell proliferation in a contact‐dependent fashion. This effect may occur through cold target inhibition, in which T‐cell contact with stimulatory APC is sterically limited by the third‐party cell population at high ratios, as previously described by others.40 In spite of this, adenosine produced by Tc17 cells may endow Tc17 cells with an immunosuppressive potential, as it reduces IFN‐γ production and GzmB expression, so restraining the acquisition of the cytotoxic phenotype by CD8+ T cells.
New evidence supports the idea that adenosine may regulate CD8+ and CD4+ T‐cell homeostasis and peripheral maintenance. In contrast to the general belief of adenosine only as an immunosuppressive factor, it was demonstrated that global deletion of A2A receptors increases tumour growth and impairs CD8+ T‐cell differentiation and accumulation in tumours.17 Moreover, the group of Linden demonstrated that adenosine favours naive CD8+ and CD4+ T‐cell survival by preventing IL‐7Rɑ (CD127) down‐regulation following T‐cell receptor stimulation.18 This effect is mediated by adenosine‐induced activation of protein kinase A, followed by downregulation of the phosphoinositide 3‐kinase‐Akt pathway. Interestingly, Crompton and collaborators recently reported that Akt inhibition enhances the expansion of CD8+ T cells with a memory phenotype.26 Conversely, sustained Akt activation enhances effector functions, drives differentiation into terminal effector cells and reduces CD8+ T‐cell potential to survive and differentiate into memory cells.41, 42 All this evidence point towards a role of adenosine in down‐regulating the Akt pathway, so mediating an arrest of the progressive differentiation cascade of CD8+ T cells from naive to effector cells.
In line with all of this evidence supporting an autocrine role of adenosine in restraining CD8+ T‐cell differentiation, here we show that CD73 expression is down‐regulated during CD8+ T‐cell progression from naive to effector T cells. Moreover, CD8+ T cells that lack CD73 expression, also express markers of terminal differentiation such as KLRG1 and down‐regulate markers associated with memory T cells such as CD127 and CD62L. Since CD73 can generate adenosine by the hydrolysis of AMP, these data suggest that CD73‐driven adenosine production mediates an autocrine loop that arrests T‐cell differentiation. Hence, CD73 may have a role in regulating the transition from naive to effector cells, an idea that has been already proposed by other groups.30 Further studies are needed to demonstrate that adenosine signalling is shut‐down in CD8+ T cells presenting a terminally differentiated phenotype.
A recent report has shown that Th17 cells differentiated in the presence of TGF‐β, at concentrations that induce high CD39 and CD73 expression, present a less‐differentiated phenotype. This phenotype is characterized by lower expression of effector molecules, lower glycolytic metabolism, high expression of stem‐cell‐associated genes and high persistence in vivo.43 In agreement with this finding, we observed that Tc17 cells that were generated in the presence of TGF‐β express higher levels of CD73 and present a stem‐cell‐like phenotype when compared with highly differentiated Tc1 cells. Also, when re‐stimulated in the absence of TGF‐β, Tc17 cells generated in vitro rapidly down‐regulated CD73 expression as they progressed to a more differentiated phenotype, as shown by GzmB production. Moreover, using a specific inhibitor of A2A adenosine receptor (SCH 58261), we report that adenosine prevents Wnt signalling shut‐down during Tc17 differentiation, allowing these cells to maintain a stem cell‐like phenotype. Although the role of CD73 in mediating this effect has to be further investigated, our results suggest that Tc17 stem cell‐like programme is maintained by CD73‐mediated adenosine production.
Interestingly, the addition of SCH 58261 during Tc17 cell differentiation reduced IL‐17 production and favoured the generation of IFN‐γ‐producing cells, even in the presence of blocking antibodies against IFN‐γ. This observation suggests that adenosine promotes Tc17 cell differentiation by directly activating A2A receptors on T cells (Fig. 6). In line with our observations, Wilson and collaborators have demonstrated that adenosine promotes Th17 cell differentiation by stimulating IL‐6 production by dendritic cells.44 Although we did not investigate the effect of adenosine on APC, our data showing that SCH 58261 interferes with Tc17 cell differentiation in absence of APC suggest that adenosine is also playing a direct role on T cells favouring the Tc17 programme. Further studies are needed to unravel the pathways underlying the effect of adenosine signalling on Tc17 cell programme.
Figure 6.
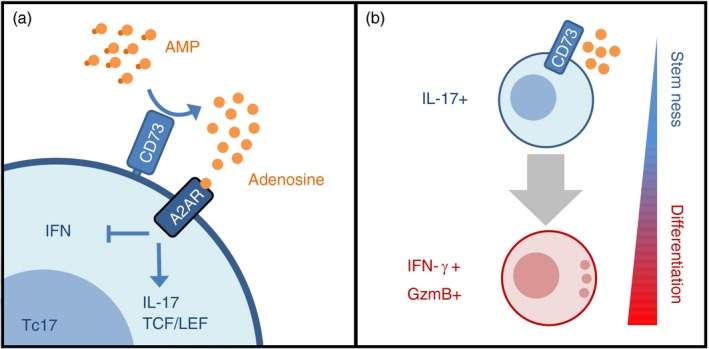
CD73‐driven adenosine production by Tc17 acts in an autocrine loop to sustain stem‐cell programme and restrain effector differentiation. (a) CD73 expression by Tc17 cells enables the production of extracellular adenosine that signals through adenosine receptor A2A (A2AR) in an autocrine fashion. Adenosine signalling sustains Tc17 phenotype [interleukin‐17 (IL‐17) production and TCF/LEF expression] and inhibit the acquisition of Tc1‐like effector molecules [interferon‐γ (IFN‐γ) and GzmB]. (b) CD73‐mediated adenosine production sustains the stem‐cell‐like programme on Tc17 cells, allowing these cells to survive and persist as long‐lived memory T cells. Upon antigen encounter, some Tc17 cells lose CD73 expression and adenosine production. In the absence of autocrine adenosine signalling, Tc17 cells can differentiate into short‐lived effector Tc1‐like cells (IFN‐γ + GzmB+).
In summary, our data provide evidence that suggests that CD73‐driven adenosine production mediates an autocrine loop that arrests T‐cell differentiation by preventing Wnt signalling shutdown. These results endow CD73 ectonucleotidase with a role, not only in maintaining stem cell‐like properties and IL‐17 production by Tc17 cells, but also in regulating the transition from naive to effector CD8+ T cells.
Authors' contributions
F.F. contributed to design of the work, development of methodology, acquisition, analysis and interpretation of data and writing the manuscript.
D.F., D.M., G.T. and L.V. contributed to development of methodology, acquisition and analysis of data. L.V.N and V.G. contributed to development of methodology, interpretation of data and revision of the manuscript. S.A. contributed to acquisition and analysis of data. M.R. and M.R.B contributed to design of the work, development of methodology and revision of the manuscript. D.S. contributed to the conception and design of the work, development of methodology, interpretation of data, writing and revision of the manuscript.
Disclosure
No potential conflict of interests were disclosed.
Supporting information
Figure S1. Phenotypic characterization of Tc17 cells generated in vitro. Total splenic CD8+ T cells from OT‐I mice were isolated and co‐cultured with antigen‐presenting cells and soluble α‐CD3 in the presence of interleukin‐6 (IL‐6), transforming growth factor‐β (TGF‐β) and α‐interferon‐γ (IFN‐γ) for Tc17 polarization, or IL‐2 and IL‐12 for Tc1 polarization. (a) IFN‐γ and IL‐17 intracellular staining of Tc1 or Tc17 cells generated in vitro stimulated with PMA and ionomycin in the presence of Golgi Plug for 4 hr (representative of seven independent experiments). (b) In vitro‐generated Tc1 and Tc17 cells were intracellularly stained for GzmB expression (n = 4). (c) Ovalbumin (OVA) ‐specific Tc1 or Tc17 cells were co‐cultured for 4 hr with OVA257–264‐loaded B16.F10 melanoma cells at different ratios. Apoptosis of the B16.F10 cells (gated as CD45– cells) was evaluated by flow cytometry [AnnexinV/propidium iodide (PI) co‐staining] (n = 1). (d) Apoptosis‐induced cell death of Tc1 and Tc17 cells was measured using AnnexinV/PI co‐staining after 3 hr of re‐activation with plate‐bound α‐CD3 and α‐CD28 (n = 1). **P < 0·01 Mann–Whitney test. Specific lysis was calculated as 100 × (% sample lysis − % basal lysis)/(100 − % basal lysis).
Figure S2. Tc17 cells do not present immunosuppressive properties. (a) Effector CD4+ T cells from C57BL/6 mice were stained with Violet Cell Trace dye, activated with antigen‐presenting cells in the presence of soluble α‐CD3 and were co‐cultured with Tc17 or Tc1 cells generated in vitro at different ratios in contact or in transwell chambers. Proliferation of CD4+ T cells was measured 4 days later as Violet Cell Trace dilution (n = 3). (b) The effect of Tc17 cells on CD4+ T‐cell proliferation and interferon‐γ (IFN‐γ) production was evaluated in the presence or absence of exogenous AMP (n = 2). (c) Effector CD4+ T cells from C57BL/6 mice were stained with Violet Cell Trace dye, activated with antigen‐presenting cells in the presence of soluble α‐CD3 and varying doses of 5′‐(N‐Ethylcarboxamido)adenosine (NECA). Four days later, T‐cell proliferation and IFN‐γ production by CD4+ T cells was evaluated (n = 1).
Figure S3. The A2AR antagonist SCH 58261 reduces interleukin‐17 (IL‐17) production by Tc17 cells generated in vitro. CD8+ T cells from C57BL/6 mice were co‐cultured for 4 days with plate bound α‐CD3 and α‐CD28 and polarizing cytokines [IL‐6, transforming growth factor‐β 1 (TGF‐β 1) and α‐interferon‐γ (α‐IFN‐γ)] in the presence of the A2AR antagonist, SCH 58261(5 μm) or DMSO as the vehicle. Cells were then stimulated with PMA plus ionomycin in the presence of Golgi Plug for 4 hr, stained for IFN‐γ and IL‐17 production and analysed by flow cytometry. SCH, SCH 58261; (n = 1).
Acknowledgements
This work was supported by FONDECYT 11121478, 1140431 and PFB‐16.
References
- 1. Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol 2012; 12:269–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rosenberg SA, Yang JC, Sherry RM, et al Durable complete responses in heavily pretreated patients with metastatic melanoma using T‐cell transfer immunotherapy. Clin Cancer Res 2011; 17:4550–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Crompton JG, Sukumar M, Restifo NP. Uncoupling T‐cell expansion from effector differentiation in cell‐based immunotherapy. Immunol Rev 2014; 257:264–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Klebanoff CA, Gattinoni L, Palmer DC, et al Determinants of successful CD8+ T‐cell adoptive immunotherapy for large established tumors in mice. Clin Cancer Res 2011; 17:5343–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gattinoni L, Klebanoff CA, Restifo NP. Paths to stemness: building the ultimate antitumour T cell. Nat Rev Cancer 2012; 12:671–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gattinoni L, Lugli E, Ji Y, et al A human memory T cell subset with stem cell‐like properties. Nat Med 2011; 17:1290–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gattinoni L, Zhong XS, Palmer DC, et al Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med 2009; 15:808–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Croft M, Carter L, Swain SL, Dutton RW. Generation of polarized antigen‐specific CD8 effector populations: reciprocal action of interleukin (IL)‐4 and IL‐12 in promoting type 2 versus type 1 cytokine profiles. J Exp Med 1994; 180:1715–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hamada H, Garcia‐Hernandez Mde L, Reome JB, et al Tc17, a unique subset of CD8 T cells that can protect against lethal influenza challenge. J Immunol 2009; 182:3469–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huber M, Heink S, Grothe H, et al A Th17‐like developmental process leads to CD8+ Tc17 cells with reduced cytotoxic activity. Eur J Immunol 2009; 39:1716–25. [DOI] [PubMed] [Google Scholar]
- 11. Muranski P, Borman ZA, Kerkar SP, et al Th17 cells are long lived and retain a stem cell‐like molecular signature. Immunity 2011; 35:972–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hinrichs CS, Kaiser A, Paulos CM, et al Type 17 CD8+ T cells display enhanced antitumor immunity. Blood 2009; 114:596–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Junger WG. Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol 2011; 11:201–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Antonioli L, Pacher P, Vizi ES, Hasko G. CD39 and CD73 in immunity and inflammation. Trends Mol Med 2013; 19:355–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lappas CM, Rieger JM, Linden J. A2A adenosine receptor induction inhibits IFN‐γ production in murine CD4+ T cells. J Immunol 2005; 174:1073–80. [DOI] [PubMed] [Google Scholar]
- 16. Su AI, Wiltshire T, Batalov S, et al A gene atlas of the mouse and human protein‐encoding transcriptomes. Proc Natl Acad Sci USA 2004; 101:6062–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cekic C, Linden J. Adenosine A2A receptors intrinsically regulate CD8+ T cells in the tumor microenvironment. Cancer Res 2014; 74:7239–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cekic C, Sag D, Day YJ, Linden J. Extracellular adenosine regulates naive T cell development and peripheral maintenance. J Exp Med 2013; 210:2693–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Regateiro FS, Howie D, Nolan KF, et al Generation of anti‐inflammatory adenosine by leukocytes is regulated by TGF‐β . Eur J Immunol 2011; 41:2955–65. [DOI] [PubMed] [Google Scholar]
- 20. Kryczek I, Zhao E, Liu Y, et al Human TH17 cells are long‐lived effector memory cells. Sci Transl Med 2011; 3:104ra0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chhabra A. Mitochondria‐centric activation induced cell death of cytolytic T lymphocytes and its implications for cancer immunotherapy. Vaccine 2010; 28:4566–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gattinoni L, Klebanoff CA, Palmer DC, et al Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest 2005; 115:1616–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Klebanoff CA, Gattinoni L, Torabi‐Parizi P, et al Central memory self/tumor‐reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci USA 2005; 102:9571–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mueller SN, Gebhardt T, Carbone FR, Heath WR. Memory T cell subsets, migration patterns, and tissue residence. Annu Rev Immunol 2013; 31:137–61. [DOI] [PubMed] [Google Scholar]
- 25. Chalmin F, Mignot G, Bruchard M, et al Stat3 and Gfi‐1 transcription factors control Th17 cell immunosuppressive activity via the regulation of ectonucleotidase expression. Immunity 2012; 36:362–73. [DOI] [PubMed] [Google Scholar]
- 26. Crompton JG, Sukumar M, Roychoudhuri R, et al Akt inhibition enhances expansion of potent tumor‐specific lymphocytes with memory cell characteristics. Cancer Res 2015; 75:296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Williams BA, Blay J, Hoskin DW. 2‐Chloroadenosine stimulates granule exocytosis from mouse natural killer cells: evidence for signal transduction through a novel extracellular receptor. Exp Cell Res 1997; 233:187–97. [DOI] [PubMed] [Google Scholar]
- 28. Csoka B, Himer L, Selmeczy Z, et al Adenosine A2A receptor activation inhibits T helper 1 and T helper 2 cell development and effector function. FASEB J 2008; 22:3491–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hasko G, Kuhel DG, Chen JF, et al Adenosine inhibits IL‐12 and TNF‐α production via adenosine A2a receptor‐dependent and independent mechanisms. FASEB J 2000; 14:2065–74. [DOI] [PubMed] [Google Scholar]
- 30. Regateiro FS, Cobbold SP, Waldmann H. CD73 and adenosine generation in the creation of regulatory microenvironments. Clin Exp Immunol 2013; 171:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Borsellino G, Kleinewietfeld M, Di Mitri D, et al Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood 2007; 110:1225–32. [DOI] [PubMed] [Google Scholar]
- 32. Deaglio S, Dwyer KM, Gao W, et al Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med 2007; 204:1257–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol 2008; 8:523–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Beavis PA, Stagg J, Darcy PK, Smyth MJ. CD73: a potent suppressor of antitumor immune responses. Trends Immunol 2012; 33:231–7. [DOI] [PubMed] [Google Scholar]
- 35. Stagg J, Beavis PA, Divisekera U, et al CD73‐deficient mice are resistant to carcinogenesis. Cancer Res 2012; 72:2190–6. [DOI] [PubMed] [Google Scholar]
- 36. Stagg J, Divisekera U, McLaughlin N, et al Anti‐CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc Natl Acad Sci USA 2010; 107:1547–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stagg J, Smyth MJ. Extracellular adenosine triphosphate and adenosine in cancer. Oncogene 2010; 29:5346–58. [DOI] [PubMed] [Google Scholar]
- 38. Wang L, Fan J, Thompson LF, et al CD73 has distinct roles in nonhematopoietic and hematopoietic cells to promote tumor growth in mice. J Clin Invest 2011; 121:2371–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yegutkin GG, Marttila‐Ichihara F, Karikoski M, et al Altered purinergic signaling in CD73‐deficient mice inhibits tumor progression. Eur J Immunol 2011; 41:1231–41. [DOI] [PubMed] [Google Scholar]
- 40. Szymczak‐Workman AL, Workman CJ, Vignali DA. Cutting edge: regulatory T cells do not require stimulation through their TCR to suppress. J Immunol 2009; 182:5188–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hand TW, Cui W, Jung YW, et al Differential effects of STAT5 and PI3K/AKT signaling on effector and memory CD8 T‐cell survival. Proc Natl Acad Sci USA 2010; 107:16601–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim EH, Sullivan JA, Plisch EH, et al Signal integration by Akt regulates CD8 T cell effector and memory differentiation. J Immunol 2012; 188:4305–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chatterjee S, Thyagarajan K, Kesarwani P, et al Reducing CD73 expression by IL1β‐programmed Th17 cells improves immunotherapeutic control of tumors. Cancer Res 2014; 74:6048–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wilson JM, Kurtz CC, Black SG, et al The A2B adenosine receptor promotes Th17 differentiation via stimulation of dendritic cell IL‐6. J Immunol 2011; 186:6746–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Phenotypic characterization of Tc17 cells generated in vitro. Total splenic CD8+ T cells from OT‐I mice were isolated and co‐cultured with antigen‐presenting cells and soluble α‐CD3 in the presence of interleukin‐6 (IL‐6), transforming growth factor‐β (TGF‐β) and α‐interferon‐γ (IFN‐γ) for Tc17 polarization, or IL‐2 and IL‐12 for Tc1 polarization. (a) IFN‐γ and IL‐17 intracellular staining of Tc1 or Tc17 cells generated in vitro stimulated with PMA and ionomycin in the presence of Golgi Plug for 4 hr (representative of seven independent experiments). (b) In vitro‐generated Tc1 and Tc17 cells were intracellularly stained for GzmB expression (n = 4). (c) Ovalbumin (OVA) ‐specific Tc1 or Tc17 cells were co‐cultured for 4 hr with OVA257–264‐loaded B16.F10 melanoma cells at different ratios. Apoptosis of the B16.F10 cells (gated as CD45– cells) was evaluated by flow cytometry [AnnexinV/propidium iodide (PI) co‐staining] (n = 1). (d) Apoptosis‐induced cell death of Tc1 and Tc17 cells was measured using AnnexinV/PI co‐staining after 3 hr of re‐activation with plate‐bound α‐CD3 and α‐CD28 (n = 1). **P < 0·01 Mann–Whitney test. Specific lysis was calculated as 100 × (% sample lysis − % basal lysis)/(100 − % basal lysis).
Figure S2. Tc17 cells do not present immunosuppressive properties. (a) Effector CD4+ T cells from C57BL/6 mice were stained with Violet Cell Trace dye, activated with antigen‐presenting cells in the presence of soluble α‐CD3 and were co‐cultured with Tc17 or Tc1 cells generated in vitro at different ratios in contact or in transwell chambers. Proliferation of CD4+ T cells was measured 4 days later as Violet Cell Trace dilution (n = 3). (b) The effect of Tc17 cells on CD4+ T‐cell proliferation and interferon‐γ (IFN‐γ) production was evaluated in the presence or absence of exogenous AMP (n = 2). (c) Effector CD4+ T cells from C57BL/6 mice were stained with Violet Cell Trace dye, activated with antigen‐presenting cells in the presence of soluble α‐CD3 and varying doses of 5′‐(N‐Ethylcarboxamido)adenosine (NECA). Four days later, T‐cell proliferation and IFN‐γ production by CD4+ T cells was evaluated (n = 1).
Figure S3. The A2AR antagonist SCH 58261 reduces interleukin‐17 (IL‐17) production by Tc17 cells generated in vitro. CD8+ T cells from C57BL/6 mice were co‐cultured for 4 days with plate bound α‐CD3 and α‐CD28 and polarizing cytokines [IL‐6, transforming growth factor‐β 1 (TGF‐β 1) and α‐interferon‐γ (α‐IFN‐γ)] in the presence of the A2AR antagonist, SCH 58261(5 μm) or DMSO as the vehicle. Cells were then stimulated with PMA plus ionomycin in the presence of Golgi Plug for 4 hr, stained for IFN‐γ and IL‐17 production and analysed by flow cytometry. SCH, SCH 58261; (n = 1).