

Abstract
Secreted toxin B (TcdB) substantially contributes to the pathology observed during Clostridium difficile infection. To be successfully incorporated into a vaccine, TcdB-based immunogens must stimulate the production of neutralizing antibody (Ab)-encoding memory B cells (Bmem cells). Despite numerous investigations, a clear analysis of Bmem cellular responses following vaccination against TcdB is lacking. B6 mice were therefore used to test the ability of a nontoxigenic C-terminal domain (CTD) fragment of TcdB to induce Bmem cells that encode TcdB-neutralizing antibody. CTD was produced from the historical VPI 10463 strain (CTD1) and from the hypervirulent strain NAP1/BI/027 (CTD2). It was then demonstrated that CTD1 induced strong recall IgG antibody titers, and this led to the development of functional Bmem cells that could be adoptively transferred to naive recipients. Bmem cell-driven neutralizing Ab responses conferred protection against lethal challenge with TcdB1. Further experiments revealed that an experimental adjuvant (Imject) and a clinical adjuvant (Alhydrogel) were compatible with Bmem cell induction. Reactivity of human Bmem cells to CTD1 was also evident in human peripheral blood mononuclear cells (PBMCs), suggesting that CTD1 could be a good vaccine immunogen. However, CTD2 induced strong Bmem cell-driven antibody titers, and the CTD2 antibody was neutralizing in vitro, but its protection against lethal challenge with TcdB2 was limited to delaying time to death. Therefore, CTD from different C. difficile strains may be a good immunogen for stimulating B cell memory that encodes in vitro neutralizing Ab but may be limited by variable protection against intoxication in vivo.
INTRODUCTION
The role of immune cell memory in Clostridium difficile infection (CDI) remains poorly understood. CDI is complicated by a high frequency of recurrence, often after disease has apparently resolved, and can be associated with progressively worsening pathology and ultimately death (1). However, patients that develop antibodies (Ab) capable of neutralizing two toxins secreted by C. difficile (TcdA and TcdB) are less likely to experience recurrence (reviewed in reference 2). This suggests that memory B (Bmem) cells may contribute to resistance to reinfection by encoding toxin-neutralizing Ab. Bmem cells have typically undergone affinity maturation and Ab class switch in the germinal centers of secondary lymphoid organs (3). Bmem cells are therefore poised to respond rapidly to booster vaccinations or infection, by rapidly differentiating into plasma cells that secrete class-switched, high-affinity Ab (4). Such plasma cells may display a range of short-to-extreme longevity, be associated with transient or sustained Ab titers, or secrete neutralizing or nonneutralizing Ab (5–7).
In earlier studies, a correlation between bacterial load and advanced age was observed during CDI, with older individuals lacking toxin-neutralizing Ab (8). In more recent work, the probability of HIV-positive patients experiencing CDI increased as their CD4+ T-cell counts declined (9), which could be partly attributable to altered CD4+ T-cell-dependent B cell function (10). Indeed, there is growing concern about CDI in a variety of immunocompromised individuals, including organ transplant recipients (reviewed in reference 11). These observations highlight the well-recognized importance of B cell responses and production of toxin-neutralizing antibodies in resisting CDI. However, the underlying characteristics of the Bmem cellular response and their contributions to production of toxin-neutralizing Ab have not been described.
CDI is best known as a disease of the gastrointestinal tract, causing diarrhea, and this infection may progress to a severe pathological condition in which pseudomembranous colitis and toxic megacolon are evident (1, 2). However, CDI-associated mortality may be attributable to systemic sequelae of the disease (12). Although large-scale epidemiological studies are lacking, reported systemic complications include hepatic abscesses (13), ascites (14), pleural effusion with acute respiratory distress (15, 16), and severe sepsis and multiorgan dysfunction (17).
TcdA and TcdB are large clostridial toxins that contribute substantially to enteric and systemic pathology associated with CDI (18, 19). Blood-borne TcdB and TcdA can be detected in some patients presenting with CDI, and diluted toxemic patient sera can kill target cells in vitro (20). C. difficile strains lacking TcdA and TcdB are less virulent than their toxin-expressing counterparts and are associated with attenuated disease or asymptomatic colonization (21). However, TcdA-negative strains can still be highly virulent, resulting in severe CDI (22, 23). Systemic effects of TcdA and TcdB have been observed in mice and piglets, with toxemia correlating with disease severity and mortality (24, 25). TcdA and TcdB cause cardiac arrest in rabbits (26), and more recently, TcdB was shown to be cardiotoxic in a zebrafish model (27).
While TcdA appears to be nearly identical among various strains of C. difficile, TcdB varies among these strains. TcdB1 (previously termed TcdBHIST and TcdB003), derived from clinical strains such as CD630, shares 92% identity with TcdB2 (previously termed TcdBHV and TcdB027) (27). TcdB2 is produced by recently emerged hypervirulent strains of C. difficile such as strains NAP1/027/BI (27). Toxemia is also a problem with TcdB2, which is more potent than TcdB1 in in vitro assays (27, 28). Furthermore, TcdB2 is associated with a more aggressive form of CDI than TcdB1 and represents a growing threat to public health (29–31).
TcdB1 and TcdB2 are single-chain 269-kDa proteins with at least four functional domains (32). Unless administered in the formaldehyde-inactivated form, TcdB1 and TcdB2 are not suitable vaccine candidates, due to their in vivo 100% lethal dose (LD100) of 5 μg/kg (or less) of body weight (27). Therefore, recombinant fragments of TcdB or specific-domain containing fusion proteins may be better candidates than whole toxin (33, 34). This includes the 70-kDa C-terminal domains of TcdB1 and TcdB2 (CTD1 and CTD2) (28).
Given the dearth of available information on memory responses to TcdB1 and TcdB2, we analyzed whether CTD1- and CTD2-specific Bmem cells encoded TcdB1- and TcdB2-neutralizing IgG. Using a tractable mouse model, we explored the fundamental characteristics of Bmem cell-driven responses to the two forms of TcdB. We report that recombinant CTD1 stimulated Bmem cell-driven TcdB1-neutralizing Ab responses that also protected in vivo. In contrast, CTD2 induced high Bmem cell-driven TcdB2-specific Ab titers that neutralized TcdB2 in vitro but demonstrated limited protection in vivo by delaying time to death. These findings provide new and important insights into the Bmem cellular response to a critical C. difficile virulence factor and suggest key differences that could impact neutralizing Ab responses due to vaccination or infection.
MATERIALS AND METHODS
Ethics.
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All animal procedures were approved by the OUHSC Institutional Animal Care and Use Committee (Protocol 12-120). The human subject studies were approved by the OUHSC Institutional Review Board (Protocol 2275). Written informed consent was given by study participants.
Expression and purification of TcdB and CTD.
C. difficile was cultured and TcdB purified as previously described (27, 35). The CTD-encoding region of tcdb gene (YP_001087135.1, nucleotides 4961 to 7111) from strains VPI-10463 and NAP1/BI/027 were codon optimized and cloned into pET15b (Genscript). The VPI-10463 and NAP1/BI/027 CTD genes were amplified using primers 5′-GATCATATGCTGTATGTGGGTAACCG-3′ and 5′-AACGGATCCTTATTCGCTAATAACCA-3′ containing BamHI and NdeI sites for cloning into pET15b. VPI-10463 CTD and NAP1/BI/027 CTD (representing VPI-10463 TcdB1651–2366 and NAP1/BI/027 TcdB1651–2366) were expressed in Escherichia coli BL21 star DE3 (Invitrogen) and purified by Ni2+ affinity chromatography (HisTrap; GE Life Sciences). VPI-10463 CTD and TcdB are referred to as “CTD1” and “TcdB1,” respectively, while the designations “CTD2” and “TcdB2” are used for NAP1/BI/027.
Immunizations, blood collection, and experimental schedule.
Female C57BL/6 mice were purchased from the National Cancer Institute (Bethesda, MD) and housed in a specific-pathogen-free facility. Mice were anesthetized with a vaporized 4% isoflurane–96% oxygen mixture for procedures. Mice were immunized subcutaneously (s.c.) at 6 to 10 weeks of age with the dosage divided evenly over both flanks. Vaccines consisted of 25, 50, or 100 μg of CTD in 200 μl sterile phosphate-buffered saline (PBS) adsorbed to Imject Alum (Thermo Scientific, Rockford, IL) or Alhydrogel Alum (Invivogen, San Diego, CA). Two vaccination schedules were employed. (i) The initial vaccine was administered on day 0, and the booster consisting of one-half of the original dose of CTD in PBS (no adjuvant) was administered on day 60. Mice were bled retro-orbitally using heparinized capillary tubes on days 0, 28, 60, 67, 74, and 180. (ii) The initial vaccine was administered on day 0, and the booster was administered on day 180. Blood samples were collected on days 180, 187, and 194.
Splenocyte isolation.
Spleens were harvested into RPMI medium and mechanically disrupted before ammonium chloride lysis of erythrocytes. Cells were enumerated using a Nexcelom Cellometer (Lawrence, MA). Viability was >98% by trypan blue exclusion.
B cell isolation and adoptive transfer.
B-lineage cells were enriched from preimmunized IgHb B6 donors by incubating splenocytes on ice with 10 μg of anti-Thy1.2, anti-CD4 monoclonal antibody (MAb) per spleen. The splenocytes were washed, and then T-lineage cells were removed by complement-mediated lysis. Cells were incubated with 66 μg/ml of LoTox rabbit complement (Cedarlane Laboratories, Burlington, NC) for 45 min at 37°C. The cells were then washed in sterile PBS and counted. This method was reported by us previously and gives B cell enrichment comparable to that of magnetic sorting (36, 37). B-cell-enriched splenocytes contained >95% B cells by flow cytometry of CD19+ cells (data not shown). Twenty-five million cells in 100 μl sterile PBS were injected paraorbitally into IgHa congenic recipient mice. The recipient mice received a boost of 50 μg/ml CTD 24 h after the adoptive transfer. Sera were collected at 14 days following administration of the booster vaccine. This is a standard method for confirming establishment of antigen (Ag)-specific B cell memory (38).
ELISA.
Immulon 4 enzyme-linked immunosorbent assay (ELISA) plates (Dynex, Chantilly, VA) were coated with CTD1 or CTD2, and specific endpoint IgG1, IgG2b, IgG2c, and IgG3 titers were determined as described previously (39). In adoptive transfers, IgHb was also detected by ELISA as described previously (37).
In vitro neutralization.
CHO cells (5 × 104 cells in 100 μl of medium consisting of F-12K, 4 μM l-glutamine, and 10% fetal calf serum [FCS]) were seeded into 96-well plates and cultured overnight (5% CO2, 37°C). Sera were diluted 1/500 in medium, mixed with TcdB1 or TcdB2 (final concentration, 2.5 nM unless indicated otherwise), and incubated for 1 h at room temperature. Concentrations of toxins were previously calibrated to cause 85 to 95% CHO cell death. Medium was removed from cell cultures and replaced with the serum-toxin-medium mixture. Plates were cultured for 24 h before addition of Cell Counting kit-8 (CCK-8) reagent (Dojindo Technologies, Rockville, MD) and an additional 2 h of incubation. A450 was then measured, and percent survival was calculated as follows: (A450 of sample − A450 of toxin-treated cells)/(A450 of untreated cells − A450 of toxin-treated cells × 100).
Toxin challenge.
Mice were tail vein injected with PBS containing 500 ng TcdB1, 75 ng TcdB2, or 200 ng TcdB2. Survival was monitored at 1- to 4-h intervals. Terminally moribund animals were euthanized, and time to death was recorded. Passive serum transfer was achieved by intraperitoneal (i.p.) injection of 400 μl of serum from naive or CTD1-immunized mice 16 h prior to TcdB1 challenge.
Peripheral blood mononuclear cell preparation.
Peripheral venous blood was drawn from healthy volunteers into heparinized vacuum tubes and layered onto 20 ml of lymphocyte separation medium (Lonza, Allendale, NJ) and centrifuged (700 relative centrifugal force [RCF], 35 min, room temperature). PBMCs were removed, washed with PBS, and resuspended in medium (RPMI, 10% FCS, 40 μg/ml gentamicin). Cell viability was routinely 80 to 90%.
Polyclonal stimulation of Bmem cells.
The standard method for stimulating human Bmem cells to differentiate into Ab-secreting plasma cells was employed by culturing PBMCs with polyclonal stimuli (40). In brief, CpG 2006 (Invivogen), Pokeweed Mitogen (a kind gift from Emory University Vaccine Center), and Staphylococcus aureus protein A (Sigma, St. Louis, MO) were added at 6 μg/ml, 1/100,000 dilution of stock, and 1/10,000 dilution of stock and cultured for 6 days.
ELISPOT assay.
Enzyme-linked immunospot (ELISPOT) assay plates (Millipore, Bedford, MA) were coated with CTD1, and Ab-secreting cells were detected as described previously (39), with the exception that horseradish peroxidase (HRP)-conjugated anti-human IgG was used for detection (Southern Biotech, Birmingham, AL).
Statistics.
A two-tailed Mann-Whitney U test and an analysis of variance (ANOVA) with Bonferroni's posttest were used to compare two and multiple experimental groups, respectively. For challenge experiments, survival data were subjected to Kaplan-Meier analysis with the log rank test.
RESULTS
The C-terminal domain of VPI 10463 TcdB (CTD1) induces TcdB-neutralizing protective Ab recall responses.
C57BL/6 mice were immunized with 25, 50, or 100 μg of CTD1 adsorbed to Imject Alum adjuvant (Fig. 1). Primary serum CTD1-specific IgG1 titers were dependent on the amount of protein administered. A booster administered 60 days after the initial vaccine led to an approximate 10-fold increase in IgG1 titer regardless of primary IgG1 titer. IgG1 titers were sustained for at least 180 days, at which time a nonsignificant decline in titer was observed. IgG2b, IgG2c, and IgG3 titers were determined for samples obtained before and after the booster (days 60 and 67, respectively) (Fig. 2). IgG2b and IgG2c titers were enhanced at least 10-fold by the booster. IgG3 was not induced. IgG2a was not measured because C57BL/6 mice have a γ2A deletion in the Ig locus (41). Therefore, CTD1 induced significant Ab recall responses that were stimulated 60 days after the initial immunization. This is consistent with induction of CTD1-specific Bmem cells.
FIG 1.
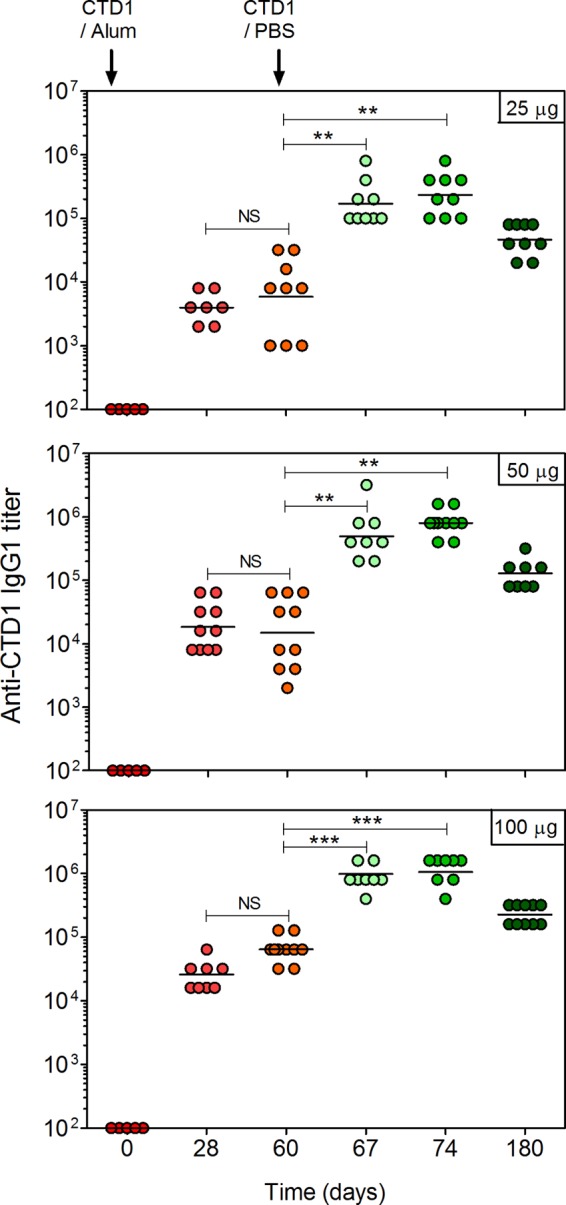
Imject Alum-adsorbed CTD1 stimulates primary and recall IgG1 responses. C57BL/6 mice were immunized s.c. with Imject-adsorbed CTD1. Mice received a booster vaccine consisting of CTD1 in PBS on day 60. Sera were collected at the times indicated, and endpoint CTD1-specific IgG1 titers were determined by ELISA for mice immunized with 25 μg (top), 50 μg (middle), or 100 μg (bottom) of CTD1. Each data point represents an individual mouse. Statistical significance was determined by ANOVA and is indicated by asterisks: **, P = 0.01 to 0.001; ***, P < 0.001. The day 60 titers were compared for each immunization by ANOVA, and titers were significantly higher for the 100-μg dose than the 50-μg or 25-μg dose (P < 0.001).
FIG 2.
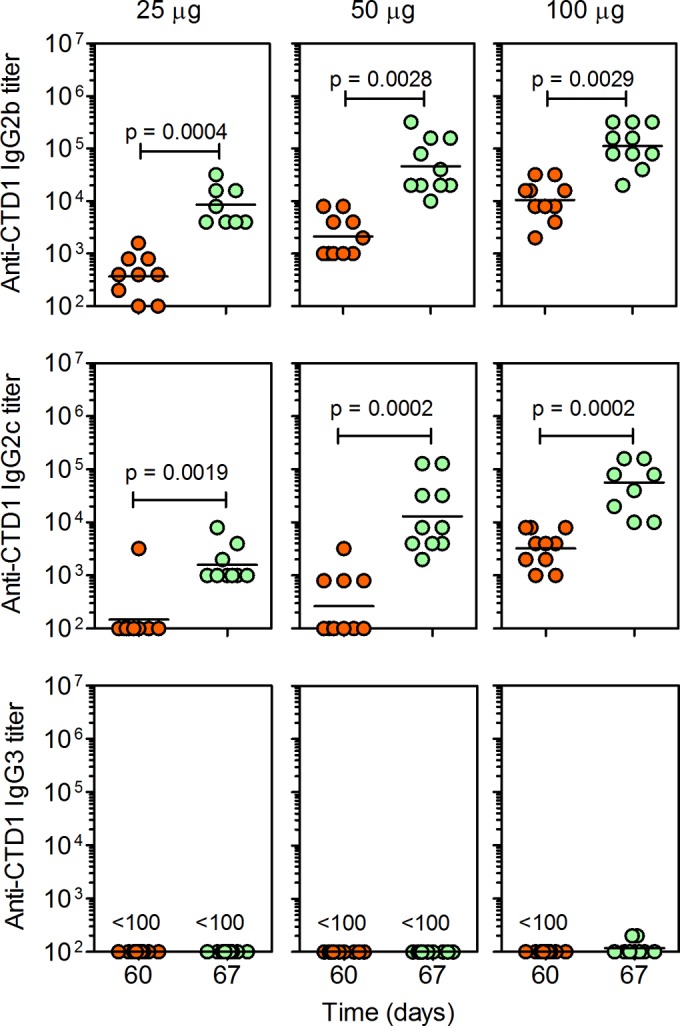
Imject Alum-adsorbed CTD1 stimulates primary and recall IgG2 responses. Endpoint CTD1-specific IgG2b (top), IgG2c (middle), and IgG3 (bottom) titers in the day 60 and 67 sera from the experiment whose results are shown in Fig. 1 were measured by ELISA (note that the booster was administered on day 60). Each data point represents an individual mouse. Statistical significance was determined by Mann-Whitney U-test, and P values are shown.
CHO cells were incubated with TcdB1 in the absence or presence of sera obtained before and after the booster (Fig. 3). Immunization with 25 μg of CTD1 did not induce measurable neutralization of TcdB1 by the day 60 sera, because CHO cells died under those conditions (Fig. 3A). The day 67 sera variably neutralized TcdB1 as evidenced by CHO cell survival. As the immunization dose increased to 50 μg and 100 μg, neutralization conferred by day 60 sera also increased. Neutralization was further enhanced by day 67 sera from mice immunized and boosted with higher doses of CTD1. The results were confirmed by repeating the assays with a range of serum dilutions (Fig. 3B; see also Fig. S1 in the supplemental material). Under these conditions, the stark differences between TcdB1 neutralization by the day 60 and 67 sera were still observed.
FIG 3.
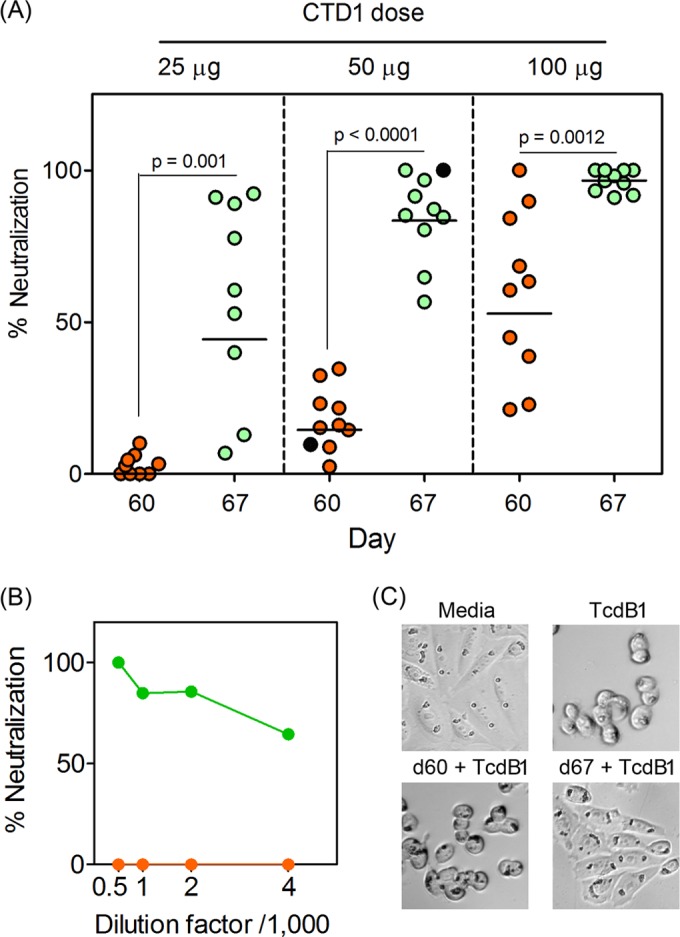
Bmem-encoded Ab neutralizes TcdB1 in vitro. Sera from the experiment whose results are shown in Fig. 1 were incubated with active TcdB1 before adding in triplicate to CHO cell cultures. The final concentration of sera was 1/500, and the final concentration of TcdB1 was 2.5 nM (sufficient to cause 85 to 95% cell death in the culture). After 24 h, the CCK-8 cell viability reagent was added and A405 was measured after a further 3 h. (A) Percent neutralization when comparing sera obtained before and after the booster vaccine (day 60 versus day 67). Black-filled symbols indicate serum number 487, used to generate the data in panels B and C. Statistical significance between neutralization in the day 60 and day 67 groups was determined by Mann-Whitney U test, and P values are shown. Administration of Alum alone did not induce Ab titers or neutralization responses (data not shown). (B) Effects of dilution of sera on CHO cell viability for selected sera (mouse ID number 487, immunized with 50 μg CTD1). Green symbols depict day 67 sera, and orange symbols depict day 60 sera. Serum titrations for every mouse that received the 50-μg dose of CTD1 are shown in Fig. S1 in the supplemental material. (C) Representative images from a cell-rounding assay whereby cells were treated as described for panel A and visualized by light microscopy 2 h after application of TcdB1 and/or sera (from mouse ID number 487).
To confirm the results, CHO TcdB1-induced cell rounding was analyzed (Fig. 3C). The day 60 sera did not substantially alter TcdB1-induced cell rounding, but the day 67 sera prevented cell rounding, such that the flattened adherent morphology observed in untreated cells was observed. Therefore, Ab recall responses led to enhanced neutralization of TcdB1 and were consistent with Bmem cells encoding TcdB1-neutralizing Ab.
CTD1-specific bone marrow plasma cells were detected by ELISPOT assay in immunized mice, and the response was significantly increased by the booster vaccine (data not shown). Partial neutralization of TcdB1 by primary sera was therefore attributable to serum IgG secreted by a small number of existing plasma cells. However, stimulation of Bmem cells was necessary to generate sufficient plasma cell-derived Ab to efficiently neutralize TcdB1.
Sustained TcdB1 neutralization was observed in immunized mice because sera obtained 180 days after immunization neutralized TcdB1 (Fig. 4A). Indeed, the neutralization observed after 180 days was comparable to that observed for sera obtained after 67 days. The CTD1-immunized mice were then challenged in vivo by the intravenous (i.v.) route with a 2× LD100 dose of TcdB1 (Fig. 4B). Naive mice all succumbed to TcdB1 and died within 20 h. Immunized mice were protected. Mice immunized with the lowest dose of CTD1 did not succumb to TcdB1 challenge despite variable in vitro neutralization. This suggests that the in vitro assays underestimate the amount of protection afforded in vivo. Mice immunized with higher doses were also protected, with 80% survival in the 50 μg group and 100% survival in the 100 μg group. Immunization with Alum alone followed by challenge was not performed due to the lack of Ab or neutralization in the absence of CTD1 (data not shown) and because it is well documented that Alum does not confer Ag-specific immune responses unless that Ag is adsorbed to it (reviewed in reference 42).
FIG 4.
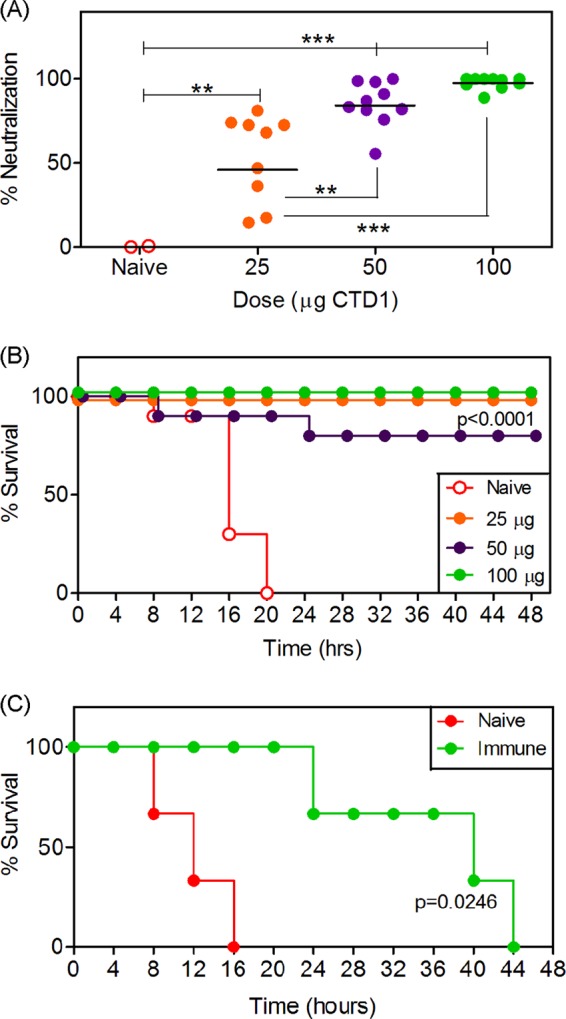
Bmem-encoded neutralizing Ab is durable and protects against in vivo challenge with TcdB. (A) Six months after the initial immunization (4 months after booster), mice from the experiment whose results are shown in Fig. 1 were bled again, and neutralization assays were repeated. Statistical significance was determined by ANOVA and is indicated by asterisks: **, P = 0.01 to 0.001; ***, P < 0.001. (B) Mice were then challenged i.v. with TcdB1 (20 ng/g of body weight). The graph shows survival of naive mice (n = 5) and those immunized with 25 μg (n = 9), 50 μg (n = 10), or 100 μg (n = 10) of Imject-adsorbed CTD1. (C) Naive B6 mice were transferred (i.p. route) with 400 μl of serum from naive or immunized donor mice. After 24 h, mice were challenged with TcdB1 (20 ng/g of body weight). The graph depicts percent survival over time. Three mice per group were used. Statistical significance in panels B and C was determined by Kaplan-Meier analysis with log rank test, and P values comparing immunized or serum-transferred groups to naive controls are shown.
To show that Ab-mediated mechanisms contributed to in vivo protection, naive mice received a passive transfer of serum from naive or immunized mice and then were challenged with TcdB1. Mice receiving serum from naive donors succumbed rapidly to TcdB1, displaying 0% survival after 16 h (Fig. 4C). Mice receiving serum from immunized mice succumbed over a period spanning 44 h, consistent with the temporary protection conferred by passive transfer (Fig. 4C).
The data presented in Fig. 1 through 4 therefore show that stimulation of CTD1-specific Ab recall responses led to durable and sustained in vivo protection against TcdB1. This is consistent with CTD1-specific Bmem cells having the capacity to differentiate into long-lived plasma cells that secrete TcdB1-neutralizing Ab.
CTD1 induces specific and functional Bmem cells.
Mice were immunized with Imject-adsorbed CTD1 and rested for 2 months. This was followed by isolating splenocytes from naive and immunized IgHb-congenic mice and subjecting them to depletion of all T-lineage cells before adoptive transfer into IgHa congenic recipients (Fig. 5, right panel). After immunization of recipient mice with a mock booster (CTD1 in PBS), serum IgHb Ab titers were observed only in those mice receiving cells from immunized donors. IgHa titers were not detected, showing that the Ab response was donor derived. This demonstrates that CTD immunization generated transferable Bmem cells that were stimulated with Ag to differentiate into productive Ab-secreting plasma cells. Neutralization and challenge experiments were not performed because this method does not generate sufficient Ab titers.
FIG 5.

CTD1 induces functional Bmem IgHb C57BL/6 mice were immunized with 100 μg Imject-adsorbed CTD1 and rested for 2 months. Splenocytes were then subjected to T-cell depletion before adoptive transfer of cells to naive IgHa C57BL/6 congenic recipient mice. Recipient mice were then immunized with 50 μg CTD1 in PBS. All recipient mice were immunized because transferred cells die if not stimulated with Ag. IgHb-specific Abs were then used in an ELISA to detect donor Bmem-derived anti-CTD1 Ab. The graph shows means ± standard deviations (SD) of A405 at a 1/200 dilution of sera (n = 2 for naive cell transfer, n = 5 for immune cell transfer). Statistical significance between the two groups was determined by Mann-Whitney U test, and the P value is shown.
Alhydrogel-adsorbed CTD1 induces long-term B cell memory.
To determine if long-term B cell memory could be stimulated by CTD1 adsorbed to a clinically relevant adjuvant, mice were immunized with CTD1 alone, CTD1 adsorbed to Imject Alum, or CTD1 adsorbed to Alhydrogel Alum. Mice were rested for 6 months before administration of a booster vaccine (Fig. 6). Imject or Alhydrogel was necessary to stimulate high CTD1-specific IgG1 titers (Fig. 6A) as well as IgG2b and IgG2c titers (Fig. 6B). After a 6-month resting period, administration of a booster led to Ab titers that were at least 10-fold higher than preboost titers. This was observed for Alhydrogel, which stimulated larger IgG1, IgG2b, and IgG2c titers than did Imject.
FIG 6.

Imject- and Alhydrogel Alum-adsorbed CTD1 stimulates long-term Bmem responses. Mice were not immunized (naive) or were immunized with 50 μg CTD1 in PBS, CTD1 adsorbed to Imject Alum, or CTD1 adsorbed to Alhydrogel. Mice were rested for 180 days before boosting with 25 μg CTD1 in PBS. Sera were collected before the booster and 7 and 14 days after the booster (days 187 and 194). (A) Endpoint CTD1-specific IgG1 titers. (B) IgG2b and IgG2c titers. (C) Neutralization of TcdB1 in vitro. (D) Results of neutralization assay in which sera were diluted 1/100 instead of 1/500. (E) Survival of mice following challenge with TcdB1 in vivo. (F) Mice were immunized with 50 μg CTD1 adsorbed to Alhydrogel and rested for 4 months. Mice were immunized i.p. with 100 ng of TcdB1. Sera were collected before and after TcdB1 administration as indicated. The graph shows CTD1-specific IgG1 titers, and each symbol represents an individual mouse. Statistical significance was determined in panels A, C, D, and F by ANOVA and is indicated by asterisks: *, P = 0.05 to 0.01; **, P = 0.01 to 0.001; ***, P < 0.001. Statistical significance in panel B was determined by Mann-Whitney U test, and P values are shown. Statistical significance in panel F was determined by Kaplan-Meier analysis with log rank test, and P values comparing CTD/adjuvant groups to naive controls and CTD1 only are shown.
In vitro assays revealed that CTD1 immunization alone led to minimal neutralization of TcdB (Fig. 6C). Neutralization in the CTD1/Imject group was variable and did not differ significantly between pre- and postbooster sera (Fig. 6C). In contrast, nearly complete and sustained TcdB1 neutralization was observed in postbooster sera in the CTD1/Alhydrogel group (Fig. 6C). Due to the apparent absence of neutralization in the CTD1 and CTD1/Imject groups, the assay was repeated using a higher concentration of serum, revealing the presence of neutralizing Ab in the postbooster sera from the CTD1/Imject group (Fig. 6D). The observation that neutralization was significantly higher in sera obtained after the boosters demonstrates that Bmem cells could be stimulated to differentiate into plasma cells and secrete TcdB1-neutralizing Ab 6 months after the initial immunization.
Following in vivo challenge with TcdB1, all naive mice died within 16 h. Of the CTD1 group, 50% survived. Of the CTD1/Imject and CTD1/Alhydrogel groups, 100% survived (Fig. 6E). In a further experiment, B6 mice were immunized with CTD/Alhydrogel, rested for 4 months, and then “immunized/challenged” with a sublethal dose of TcdB1 (Fig. 6F). CTD1-specific IgG1 titers were boosted by the toxin, suggesting that during natural infection, exposure to the active toxin may result in stimulation of CTD1-specific Bmem and production of more neutralizing Ab. Therefore, CTD1 adsorbed to a clinically relevant adjuvant stimulated long-term B cell memory against C. difficile toxin B.
Detection of CTD1-specific Bmem cells in human subjects.
PBMCs were obtained from healthy volunteers over age 55 that were unaware of previous C. difficile infection or from a cohort who had a positive diagnosis of CDI within the 5 years before sample collection. PBMCs were cultured with polyclonal stimuli that drive differentiation of Bmem cells into Ab-secreting plasma cells (Fig. 7). ELISPOT analysis showed that 2/19 individuals with no known history of C. difficile infection had peripheral blood Bmem that encoded CTD1-specific IgG. CTD1-specific Bmem cells were also observed in 4/8 individuals with a previous C. difficile infection. The number of CTD1-specific spots detected ranged from 0.01 to 0.1% of total IgG-secreting cells (of all specificities) and is consistent with recent observations by another laboratory (43). Therefore, Bmem cells in some human subjects encoded CTD1-specific IgG.
FIG 7.
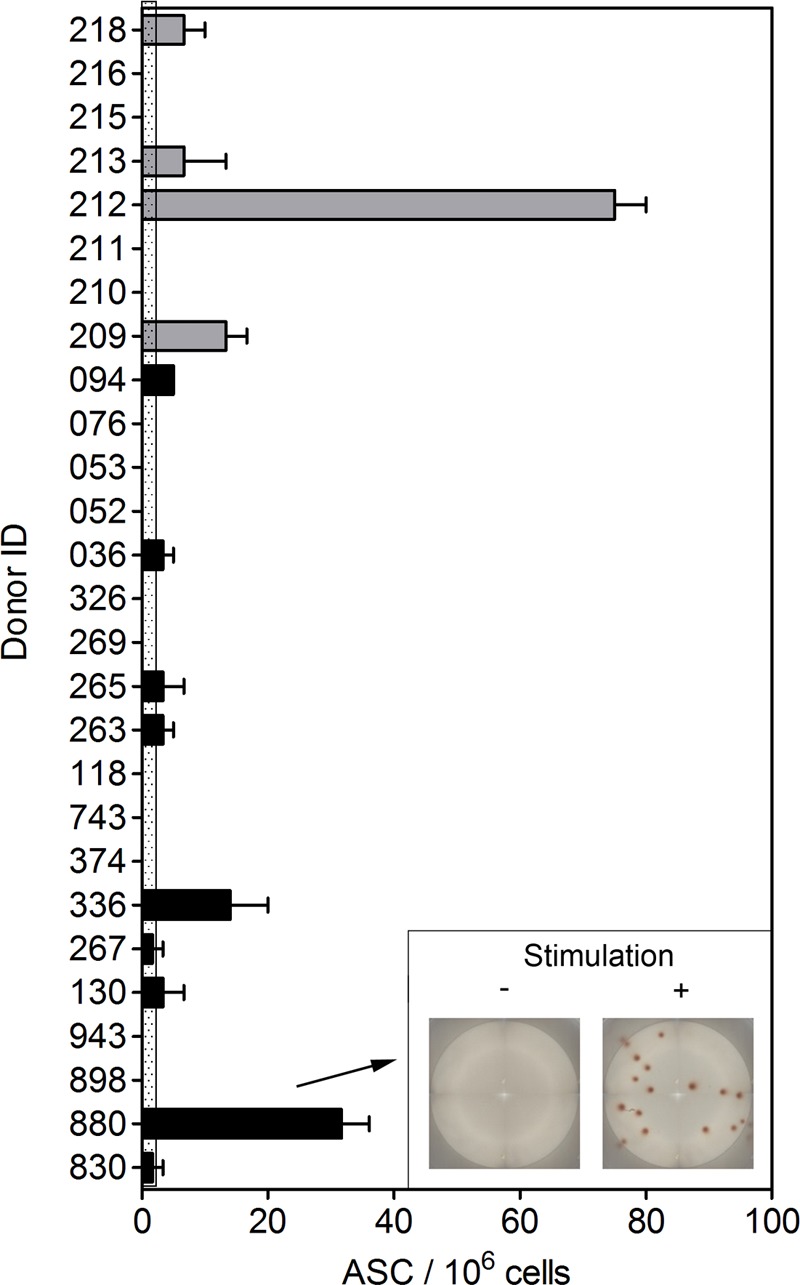
Detection of CTD1-specific Bmem in human subjects. PBMCs were obtained from healthy individuals age 55 years and older that had previously been diagnosed with C. difficile infection (gray bars) or were not aware of previous infection (black bars). PBMCs were cultured with polyclonal stimuli as described in Materials and Methods. Cells were then cultured on ELISPOT assay plates coated with CTD1. IgG reactive to CTD1 was then detected using HRP-conjugated anti-human IgG. The graph shows the number of Ab-secreting cells (ASC) per million total cells for each individual in the cohort. Data are represented as means ± standard errors of the means (SEM) for triplicate samples. The dotted area depicts background (from unstimulated samples) plus 2 standard deviations. The inset image shows raw ELISPOT assay data from donor 880.
Immunization with CTD2 leads to Bmem cell-driven production of Ab that neutralizes CTD2 in vitro but provides limited protection in vivo.
Since CTD1 appeared to be a good vaccine candidate, we then examined CTD2 from the “hyper-virulent” strain NAP1/BI/027. B6 mice were immunized with Alhydrogel-adsorbed CTD2 and boosted with CTD2 alone after 60 days (Fig. 8). Sera were collected before (day 60) and after (days 67 and 74) the booster vaccine and examined by ELISA. It was evident that CTD2 caused a significant IgG1, IgG2b, and IgG2c recall response consistent with Bmem cell restimulation by the booster vaccine (Fig. 8A). Sera obtained after the booster vaccine were able to neutralize TcdB2 in vitro, while sera obtained before the booster were not (Fig. 8B). However, a modest, but statistically significant drop in neutralization was observed between the day 67 and day 74 sera, showing that neutralizing titers were not sustained. Titration of sera was also performed and showed that neutralization could not be detected when the sera were further diluted (Fig. 8C). Titrations of sera for all mice in the study are shown in Fig. S2 in the supplemental material. A cell-rounding assay was also performed, in which cell morphology was analyzed 2 h after treatment of CHO cells with TcdB2 (Fig. 8D). The TcdB2 treatment resulted in previously flattened cells adopting a shrunken rounded morphology. Sera from mice obtained before the CTD2 booster did not change the shrunken appearance, but sera obtained after the booster limited the changes induced by TcdB2. The cells treated with TcdB2 plus postbooster sera had a rounded appearance but were not shrunken, indicating partial protection from toxicity. Mice were also challenged with TcdB2, and despite the good in vitro neutralization observed, 100% of mice ultimately succumbed to TcdB2, although time to death was significantly delayed (Fig. 8E). These data show the CTD2-specific Bmem cell-encoded Ab that could partially neutralize TcdB2 in vitro but not afford sustained protection in vivo.
FIG 8.

Immunization with CTD2 leads to Bmem-driven production of Ab that neutralizes CTD2 in vitro but does not protect in vivo. Mice were immunized with 50 μg CTD2 adsorbed to Alhydrogel and rested for 60 days before boosting with 25 μg CTD2 in PBS. Sera were collected before the booster and 7 and 14 days after the booster (days 67 and 74). (A) Endpoint CTD2-specific IgG1 titers, IgG2b, and IgG2c titers. Geometric mean titers are indicated for the 15 mice in each group, and statistical significance was determined by one-way ANOVA followed by Bonferroni's posttest (*, P = 0.05 to 0.01; ***, P < 0.001). (B) Neutralization of TcdB2 in vitro using sera obtained before and after booster vaccine administration. Statistical significance was determined by ANOVA and Bonferroni's posttest (**, P = 0.01 to 0.001; ***, P < 0.001). (C) Representative titration of serum in the neutralization assay (from mouse number 273). Results from all other sera are shown in Fig. S2 in the supplemental material. (D) Images from cell-rounding assay, in which TcdB2 was applied to CHO cells in the absence or presence of sera. Images depict sera from mouse number 264, where in vitro neutralization was most representative of the average for the group. (E) Survival of mice following challenge with 200 ng (left) or 75 ng (right) of TcdB2/mouse. Kaplan-Meier analysis with log rank test was used to determine significant differences between naive and immunized mice (**, P = 0.01 to 0.001). Data are representative of two similar experiments.
DISCUSSION
Here, we demonstrate that the C-terminal domain of TcdB from C. difficile strain VPI 10463 (CTD1) stimulated long-term Ag-specific B cell memory that encoded toxin-neutralizing Ab. Ag-specific IgG1, IgG2b, and IgG2c titers were boosted at least 10- to 40-fold by administration of CTD1 alone to B6 mice that had previously been immunized with CTD1 adsorbed to Imject Alum. The ability of CTD1 to stimulate an enhanced Ab response 60 days after a primary immunization was consistent with induction of Bmem cells (44). Importantly, Bmem cell-encoded IgG1, IgG2b, and IgG2c have the potential to invoke a diverse range of effector functions in vivo. Future experiments to determine whether IgG1, IgG2b, and IgG2c are singly or collectively protective in vivo are warranted. The Bmem cell-driven IgG titers were sustained and conferred protection against an in vivo toxin challenge some 4 months later. It is unlikely that the IgG detected by ELISA was derived from newly differentiated plasma cells at this later time point (6 months after immunization); it is more likely that it was derived from bone marrow-resident long-lived Ab-secreting plasma cells that were established at an earlier time (5–7). Interestingly, immunization of B6 mice with higher doses of CTD1 led to variable but measurable in vitro neutralization in serum obtained prior to administering a booster vaccine. This suggests that in the vaccine setting, a single immunization may afford limited protection but at least one booster would be necessary to achieve good protection.
TcdB-specific serum Ab are well documented to protect against CDI in patients and animal models by reducing the severity of enteric disease, recurrence of disease, and mortality (reviewed in reference 2). Furthermore, circulating TcdB-specific IgG is a correlate of protection against recurrent CDI (45). The mechanism by which serum IgG can protect against enteric CDI is not entirely clear, but mucosal TcdB-specific IgG can be detected in human intestines (43). The neonatal IgG Fc receptor (FcRn) mediates bidirectional transport of IgG across intestinal epithelia (46). Indeed, such transport of Citrobacter rodentium-specific IgG protects the gut of infected mice (47). The epithelial transport pathway therefore represents a strong possibility for a mechanism by which TcdB-specific IgG could limit the pathology associated with CDI.
The role of mucosal IgA in protection against CDI is less clear, but fecal IgA concentrations were reported to be lower in individuals with recurrent CDI than in healthy controls or in patients with a single occurrence (48, 49). Studies in mice have also shown that mucosal IgA was induced during infection and was protective in the absence of serum IgG responses, but mucosal IgA was also dispensable since mice lacking the poly-Ig receptor had outcomes similar to those of wild-type controls (50).
Passive transfers in this study confirmed that IgG-mediated mechanisms contributed to protection in vivo but did not confer full protection. Passive transfer is unlikely to provide the recipient mouse with as much IgG as that produced by the endogenous humoral immune response and may be one reason why it did not afford complete protection. Another reason may be that other IgG-independent mechanisms could contribute to protection, and this could also explain why the in vitro neutralization assay was better able to discriminate between experimental groups than the in vivo challenge assay. Indeed, α-defensins were recently shown to contribute to neutralization of TcdB1 in a manner dependent on binding to the N terminus of the protein, thus suggesting that neutralization could be complementary to that provided by C terminus-targeting IgG (51, 52).
In our in vivo challenge experiments, mice received 500 ng of CTD1 from the VPI 10463 strain. Directly relating this dosage to human disease is not straightforward, especially as toxemia presents only in a subset of patients, methods to measure toxin concentrations in serum vary in sensitivity, and serum Ab in infected individuals mask toxin detection (20). However, a 1/10 dilution of serum from toxemic patients was reported to be sufficient to kill, or cause 100% rounding up in, cultured Vero cells, similar to what was observed by adding TcdB1 at a 25-pg/ml concentration (20). Arguably, sera from toxemic patients could contain TcdB1 at a concentration exceeding the estimated 250-pg/ml concentration because neutralizing anti-TcdB1 IgG causes a 125-fold loss of sensitivity of detection and perhaps intoxication capacity (20). The true concentration of TcdB1 in toxemic sera could therefore be higher than 31 ng/ml (125 × 250 pg/ml). Our challenge dose of 500 ng/mouse equates to approximately 20 ng/ml if distributed evenly throughout the mouse, or 250 ng/ml if confined to a closed 2-ml circulatory system. Our challenge assays therefore reasonably recapitulate the likely concentrations observed during toxemia in patients.
As Imject is not used clinically, we also measured Bmem cell responses with Alhydrogel, which is used in the clinic. Longer-term experiments were also performed to determine if the Bmem cell response was durable. Alhydrogel led to higher Bmem cell-driven neutralizing Ab titers than Imject. CTD1 alone led to poor Ab titers and poor neutralization but was associated with partial (50%) protection in the in vivo challenge assays. Protection in vivo was the same for the Imject and Alhydrogel groups (100%). This further suggests that in addition to Ab-mediated protection, there is an Ab-independent component, which should be pursued in follow-up studies.
Polyclonal stimulation of human PBMCs showed that CTD1-specific Bmem cells were present in the blood of healthy individuals with no awareness of previous C. difficile infection or with a confirmed previous infection. This was consistent with a recent study in which reactivity to TcdB was detected in human PBMCs (43). Therefore, human Bmem cells can encode CTD1-specific IgG and indicate that the protein used in our mouse studies could be a good candidate for stimulating TcdB1-neutralizing Bmem cells in patients. There are several reasons why some individuals with no knowledge of prior infection had TcdB1-specific Bmem cells. Approximately 60% of individuals are colonized with C. difficile as infants, and it is maintained as a gut commensal (2). Some individuals may have had a mild infection and not be aware of it, or natural infection may not adequately immunize some individuals. Alternatively, the Bmem cells generated by infection may not be long-lived. The cohort in this study was diagnosed up to 5 years before obtaining the blood sample. Larger-scale studies will therefore be necessary to better understand Bmem cell responses to infection and immunization. However, we propose that CTD1 has the potential to be exploited as a vaccine Ag that stimulates long-lived Bmem cell responses that encode TcdB-specific Ab.
Due to the emergence of C. difficile strain NAP1/B/027, whose hypervirulence is attributable in part to secretion of a TcdB variant (referred to as TcdB2 here), we tested the ability of the CTD from TcdB2 (CTD2) to stimulate B cell memory. We observed that CTD2 induced a somewhat different response from that of CTD1. Both CTD1 and CTD2 induced Bmem restimulation, but the latter appeared to stimulate a slower recall response, reaching maximal IgG1 and IgG2b titers after 2 weeks rather than 1 week. CTD1 resulted in production of neutralizing Ab with responses that were observed up to 4 months after administration of the booster vaccine. In contrast, CTD2 resulted in Bmem cell-driven neutralizing Ab that was not sustained and diminished within 2 weeks of the booster vaccine. In vitro neutralization of CTD1 was able to withstand a 4,000-fold dilution of the sera. When anti-CTD2 sera were similarly titrated, in vitro neutralization was lost with one further dilution of sera. In vivo challenge experiments revealed that more or less complete protection against TcdB1 could be achieved, but with TcdB2, all mice died, albeit with time to death significantly delayed.
Ab titers showed that CTD2 was at least as immunogenic as CTD1, so differences in in vitro neutralization and in vivo protection were attributable to differences in Bmem cells encoding neutralizing Ab. We also immunized mice with a mutant CTD2 in which an amino acid sequence, SINKVIST, corresponding to residues 1791 to 1798 was substituted for an AAAAAIAA sequence. Following immunization with mutant CTD2, mice were challenged with wild-type TcdB2. We observed that survival was similar whether mice were immunized with CTD2 or mutant CTD2 (data not shown). The mutant CTD2 has a conformation that results in exposure of neutralizing epitopes (53), suggesting that immunogenicity of CTD2 is not a limitation in our experiments.
It should be noted that Ab raised to TcdB from one strain of C. difficile may not be effective at cross-neutralization of TcdB from other strains. CTD1- and CTD2-specific Ab raised in rabbits and in mice did not cross-neutralize TcdB1 and TcdB2 (27, 53). Ab raised against CTD1 neutralized TcdB1 but not TcdB2. Surprisingly, Ab raised in rabbit against CTD2 did not neutralize TcdB1 or TcdB2. Differences in the overall make-up of neutralizing epitopes in the CTD1 and CTD2 may in part explain the absence of cross-neutralization. This does not, however, explain why CTD2 did not stimulate a neutralizing response to TcdB2 itself. In the present study, we observed some CTD2-induced neutralization of TcdB2, suggesting that there are some species-related differences in response but that CTD1-specific Bmem cells encode neutralizing Ab to a greater degree than do CTD2-specific Bmem cells. Therefore, the two forms of the toxin may be fundamentally different in how they stimulate an effective Bmem cell-driven neutralizing Ab response. Collectively, our data demonstrate that CTD1 may be a useful immunogen, but engineering of CTD2 to improve establishment of Bmem cells that better encode neutralizing IgG is necessary. Further research to understand why such closely related immunogens result in such disparate immunological outcomes is also warranted.
Supplementary Material
ACKNOWLEDGMENTS
We thank Virginia Roberts, OMRF Clinical Coordinator, for recruiting and enrolling volunteers and procuring volunteer blood samples.
We have no conflicts of interest to declare.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00011-15.
REFERENCES
- 1.Bartlett JG. 2010. Clostridium difficile: progress and challenges. Ann N Y Acad Sci 1213:62–69. doi: 10.1111/j.1749-6632.2010.05863.x. [DOI] [PubMed] [Google Scholar]
- 2.Kaslow DC, Shiver JW. 2011. Clostridium difficile and methicillin-resistant Staphylococcus aureus: emerging concepts in vaccine development. Annu Rev Med 62:201–215. doi: 10.1146/annurev-med-051109-101544. [DOI] [PubMed] [Google Scholar]
- 3.McHeyzer-Williams LJ, McHeyzer-Williams MG. 2005. Antigen-specific memory B cell development. Annu Rev Immunol 23:487–513. doi: 10.1146/annurev.immunol.23.021704.115732. [DOI] [PubMed] [Google Scholar]
- 4.Amanna IJ, Slifka MK. 2010. Mechanisms that determine plasma cell lifespan and the duration of humoral immunity. Immunol Rev 236:125–138. doi: 10.1111/j.1600-065X.2010.00912.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Slifka MK, Ahmed R. 1998. Long-lived plasma cells: a mechanism for maintaining persistent antibody production. Curr Opin Immunol 10:252–258. doi: 10.1016/S0952-7915(98)80162-3. [DOI] [PubMed] [Google Scholar]
- 6.Slifka MK, Antia R, Whitmire JK, Ahmed R. 1998. Humoral immunity due to long-lived plasma cells. Immunity 8:363–372. doi: 10.1016/S1074-7613(00)80541-5. [DOI] [PubMed] [Google Scholar]
- 7.McHeyzer-Williams MG, Ahmed R. 1999. B cell memory and the long-lived plasma cell. Curr Opin Immunol 11:172–179. doi: 10.1016/S0952-7915(99)80029-6. [DOI] [PubMed] [Google Scholar]
- 8.Nakamura S, Mikawa M, Nakashio S, Takabatake M, Okado I, Yamakawa K, Serikawa T, Okumura S, Nishida S. 1981. Isolation of Clostridium difficile from the feces and the antibody in sera of young and elderly adults. Microbiol Immunol 25:345–351. doi: 10.1111/j.1348-0421.1981.tb00036.x. [DOI] [PubMed] [Google Scholar]
- 9.Haines CF, Moore RD, Bartlett JG, Sears CL, Cosgrove SE, Carroll K, Gebo KA. 2013. Clostridium difficile in a HIV-infected cohort: incidence, risk factors, and clinical outcomes. AIDS 27:2799–2807. doi: 10.1097/01.aids.0000432450.37863.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moir S, Fauci AS. 2013. Insights into B cells and HIV-specific B-cell responses in HIV-infected individuals. Immunol Rev 254:207–224. doi: 10.1111/imr.12067. [DOI] [PubMed] [Google Scholar]
- 11.Collini PJ, Bauer M, Kuijper E, Dockrell DH. 2012. Clostridium difficile infection in HIV-seropositive individuals and transplant recipients. J Infect 64:131–147. doi: 10.1016/j.jinf.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 12.Karas JA, Enoch DA, Aliyu SH. 2010. A review of mortality due to Clostridium difficile infection. J Infect 61:1–8. doi: 10.1016/j.jinf.2010.03.025. [DOI] [PubMed] [Google Scholar]
- 13.Sakurai T, Hajiro K, Takakuwa H, Nishi A, Aihara M, Chiba T. 2001. Liver abscess caused by Clostridium difficile. Scand J Infect Dis 33:69–70. doi: 10.1080/003655401750064112. [DOI] [PubMed] [Google Scholar]
- 14.Tsourous GI, Raftopoulos LG, Kafe EE, Manoleris EK, Makaritsis KP, Pinis SG. 2007. A case of pseudomembranous colitis presenting with massive ascites. Eur J Intern Med 18:328–330. doi: 10.1016/j.ejim.2006.09.034. [DOI] [PubMed] [Google Scholar]
- 15.Boaz A, Dan M, Charuzi I, Landau O, Aloni Y, Kyzer S. 2000. Pseudomembranous colitis: report of a severe case with unusual clinical signs in a young nurse. Dis Colon Rectum 43:264–266. doi: 10.1007/BF02236993. [DOI] [PubMed] [Google Scholar]
- 16.Jacob SS, Sebastian JC, Hiorns D, Jacob S, Mukerjee PK. 2004. Clostridium difficile and acute respiratory distress syndrome. Heart Lung 33:265–268. doi: 10.1016/j.hrtlng.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 17.Dobson G, Hickey C, Trinder J. 2003. Clostridium difficile colitis causing toxic megacolon, severe sepsis and multiple organ dysfunction syndrome. Intensive Care Med 29:1030. [DOI] [PubMed] [Google Scholar]
- 18.Ballard JD. 2010. Medical microbiology: a toxin contest. Nature 467:665–666. doi: 10.1038/467665a. [DOI] [PubMed] [Google Scholar]
- 19.Kuehne SA, Cartman ST, Heap JT, Kelly ML, Cockayne A, Minton NP. 2010. The role of toxin A and toxin B in Clostridium difficile infection. Nature 467:711–713. doi: 10.1038/nature09397. [DOI] [PubMed] [Google Scholar]
- 20.Yu H, Chen K, Wu J, Yang Z, Shi L, Barlow LL, Aronoff DM, Garey KW, Savidge TC, von Rosenvinge EC, Kelly CP, Feng H. 2015. Identification of toxemia in patients with Clostridium difficile infection. PLoS One 10:e0124235. doi: 10.1371/journal.pone.0124235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shim JK, Johnson S, Samore MH, Bliss DZ, Gerding DN. 1998. Primary symptomless colonisation by Clostridium difficile and decreased risk of subsequent diarrhoea. Lancet 351:633–636. doi: 10.1016/S0140-6736(97)08062-8. [DOI] [PubMed] [Google Scholar]
- 22.Drudy D, Fanning S, Kyne L. 2007. Toxin A-negative, toxin B-positive Clostridium difficile. Int J Infect Dis 11:5–10. doi: 10.1016/j.ijid.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 23.Lyras D, O'Connor JR, Howarth PM, Sambol SP, Carter GP, Phumoonna T, Poon R, Adams V, Vedantam G, Johnson S, Gerding DN, Rood JI. 2009. Toxin B is essential for virulence of Clostridium difficile. Nature 458:1176–1179. doi: 10.1038/nature07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Steele J, Chen K, Sun X, Zhang Y, Wang H, Tzipori S, Feng H. 2012. Systemic dissemination of Clostridium difficile toxins A and B is associated with severe, fatal disease in animal models. J Infect Dis 205:384–391. doi: 10.1093/infdis/jir748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steele J, Feng H, Parry N, Tzipori S. 2010. Piglet models of acute or chronic Clostridium difficile illness. J Infect Dis 201:428–434. doi: 10.1086/649799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siarakas S, Damas E, Murrell WG. 1995. Is cardiorespiratory failure induced by bacterial toxins the cause of sudden infant death syndrome? Studies with an animal model (the rabbit). Toxicon 33:635–649. [DOI] [PubMed] [Google Scholar]
- 27.Lanis JM, Barua S, Ballard JD. 2010. Variations in TcdB activity and the hypervirulence of emerging strains of Clostridium difficile. PLoS Pathog 6:e1001061. doi: 10.1371/journal.ppat.1001061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lanis JM, Heinlen LD, James JA, Ballard JD. 2013. Clostridium difficile 027/BI/NAP1 encodes a hypertoxic and antigenically variable form of TcdB. PLoS Pathog 9:e1003523. doi: 10.1371/journal.ppat.1003523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muto CA, Pokrywka M, Shutt K, Mendelsohn AB, Nouri K, Posey K, Roberts T, Croyle K, Krystofiak S, Patel-Brown S, Pasculle AW, Paterson DL, Saul M, Harrison LH. 2005. A large outbreak of Clostridium difficile-associated disease with an unexpected proportion of deaths and colectomies at a teaching hospital following increased fluoroquinolone use. Infect Control Hosp Epidemiol 26:273–280. doi: 10.1086/502539. [DOI] [PubMed] [Google Scholar]
- 30.Loo VG, Poirier L, Miller MA, Oughton M, Libman MD, Michaud S, Bourgault AM, Nguyen T, Frenette C, Kelly M, Vibien A, Brassard P, Fenn S, Dewar K, Hudson TJ, Horn R, Rene P, Monczak Y, Dascal A. 2005. A predominantly clonal multi-institutional outbreak of Clostridium difficile-associated diarrhea with high morbidity and mortality. N Engl J Med 353:2442–2449. doi: 10.1056/NEJMoa051639. [DOI] [PubMed] [Google Scholar]
- 31.McDonald LC, Killgore GE, Thompson A, Owens RC Jr, Kazakova SV, Sambol SP, Johnson S, Gerding DN. 2005. An epidemic, toxin gene-variant strain of Clostridium difficile. N Engl J Med 353:2433–2441. doi: 10.1056/NEJMoa051590. [DOI] [PubMed] [Google Scholar]
- 32.Pruitt RN, Chambers MG, Ng KK, Ohi MD, Lacy DB. 2010. Structural organization of the functional domains of Clostridium difficile toxins A and B. Proc Natl Acad Sci U S A 107:13467–13472. doi: 10.1073/pnas.1002199107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tian JH, Fuhrmann SR, Kluepfel-Stahl S, Carman RJ, Ellingsworth L, Flyer DC. 2012. A novel fusion protein containing the receptor binding domains of C. difficile toxin A and toxin B elicits protective immunity against lethal toxin and spore challenge in preclinical efficacy models. Vaccine 30:4249–4258. doi: 10.1016/j.vaccine.2012.04.045. [DOI] [PubMed] [Google Scholar]
- 34.Permpoonpattana P, Hong HA, Phetcharaburanin J, Huang JM, Cook J, Fairweather NF, Cutting SM. 2011. Immunization with Bacillus spores expressing toxin A peptide repeats protects against infection with Clostridium difficile strains producing toxins A and B. Infect Immun 79:2295–2302. doi: 10.1128/IAI.00130-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krivan HC, Wilkins TD. 1987. Purification of Clostridium difficile toxin A by affinity chromatography on immobilized thyroglobulin. Infect Immun 55:1873–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lang GA, Devera TS, Lang ML. 2008. Requirement for CD1d expression by B cells to stimulate NKT cell-enhanced antibody production. Blood 111:2158–2162. doi: 10.1182/blood-2007-10-117309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lang GA, Johnson AM, Devera TS, Joshi SK, Lang ML. 2011. Reduction of CD1d expression in vivo minimally affects NKT-enhanced antibody production but boosts B-cell memory. Int Immunol 23:251–260. doi: 10.1093/intimm/dxq477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Foy TM, Laman JD, Ledbetter JA, Aruffo A, Claassen E, Noelle RJ. 1994. gp39-CD40 interactions are essential for germinal center formation and the development of B cell memory. J Exp Med 180:157–163. doi: 10.1084/jem.180.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Devera TS, Shah HB, Lang GA, Lang ML. 2008. Glycolipid-activated NKT cells support the induction of persistent plasma cell responses and antibody titers. Eur J Immunol 38:1001–1011. doi: 10.1002/eji.200738000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crotty S, Aubert RD, Glidewell J, Ahmed R. 2004. Tracking human antigen-specific memory B cells: a sensitive and generalized ELISPOT system. J Immunol Methods 286:111–122. doi: 10.1016/j.jim.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 41.Martin RM, Brady JL, Lew AM. 1998. The need for IgG2c specific antiserum when isotyping antibodies from C57BL/6 and NOD mice. J Immunol Methods 212:187–192. doi: 10.1016/S0022-1759(98)00015-5. [DOI] [PubMed] [Google Scholar]
- 42.Lindblad EB. 2004. Aluminium compounds for use in vaccines. Immunol Cell Biol 82:497–505. doi: 10.1111/j.0818-9641.2004.01286.x. [DOI] [PubMed] [Google Scholar]
- 43.Monaghan TM, Robins A, Knox A, Sewell HF, Mahida YR. 2013. Circulating antibody and memory B-cell responses to C. difficile toxins A and B in patients with C. difficile-associated diarrhoea, inflammatory bowel disease and cystic fibrosis. PLoS One 8:e74452. doi: 10.1371/journal.pone.0074452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Benson MJ, Elgueta R, Schpero W, Molloy M, Zhang W, Usherwood E, Noelle RJ. 2009. Distinction of the memory B cell response to cognate antigen versus bystander inflammatory signals. J Exp Med 206:2013–2025. doi: 10.1084/jem.20090667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leav BA, Blair B, Leney M, Knauber M, Reilly C, Lowy I, Gerding DN, Kelly CP, Katchar K, Baxter R, Ambrosino D, Molrine D. 2010. Serum anti-toxin B antibody correlates with protection from recurrent Clostridium difficile infection (CDI). Vaccine 28:965–969. doi: 10.1016/j.vaccine.2009.10.144. [DOI] [PubMed] [Google Scholar]
- 46.Yoshida M, Claypool SM, Wagner JS, Mizoguchi E, Mizoguchi A, Roopenian DC, Lencer WI, Blumberg RS. 2004. Human neonatal Fc receptor mediates transport of IgG into luminal secretions for delivery of antigens to mucosal dendritic cells. Immunity 20:769–783. doi: 10.1016/j.immuni.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 47.Yoshida M, Kobayashi K, Kuo TT, Bry L, Glickman JN, Claypool SM, Kaser A, Nagaishi T, Higgins DE, Mizoguchi E, Wakatsuki Y, Roopenian DC, Mizoguchi A, Lencer WI, Blumberg RS. 2006. Neonatal Fc receptor for IgG regulates mucosal immune responses to luminal bacteria. J Clinical Invest 116:2142–2151. doi: 10.1172/JCI27821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Warny M, Vaerman JP, Avesani V, Delmee M. 1994. Human antibody response to Clostridium difficile toxin A in relation to clinical course of infection. Infect Immun 62:384–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johal SS, Lambert CP, Hammond J, James PD, Borriello SP, Mahida YR. 2004. Colonic IgA producing cells and macrophages are reduced in recurrent and non-recurrent Clostridium difficile associated diarrhoea. J Clin Pathol 57:973–979. doi: 10.1136/jcp.2003.015875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Johnston PF, Gerding DN, Knight KL. 2014. Protection from Clostridium difficile infection in CD4 T cell- and polymeric immunoglobulin receptor-deficient mice. Infect Immun 82:522–531. doi: 10.1128/IAI.01273-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Furci L, Baldan R, Bianchini V, Trovato A, Ossi C, Cichero P, Cirillo DM. 2015. New role for human alpha-defensin 5 in the fight against hypervirulent Clostridium difficile strains. Infect Immun 83:986–995. doi: 10.1128/IAI.02955-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kudryashova E, Quintyn R, Seveau S, Lu W, Wysocki VH, Kudryashov DS. 2014. Human defensins facilitate local unfolding of thermodynamically unstable regions of bacterial protein toxins. Immunity 41:709–721. doi: 10.1016/j.immuni.2014.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Larabee JL, Krumholz A, Hunt JJ, Lanis JM, Ballard JD. 2015. Exposure of neutralizing epitopes in the carboxyl-terminal domain of TcdB is altered by a proximal hypervariable region. J Biol Chem 290:6975–6985. doi: 10.1074/jbc.M114.612184. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.