

Abstract
Pneumonic plague represents the most severe form of disease caused by Yersinia pestis due to its ease of transmission, rapid progression, and high mortality rate. The Y. pestis outer membrane Pla protease is essential for the development of pneumonic plague; however, the complete repertoire of substrates cleaved by Pla in the lungs is not known. In this study, we describe a proteomic screen to identify host proteins contained within the bronchoalveolar lavage fluid of mice that are cleaved and/or processed by Y. pestis in a Pla-dependent manner. We identified peroxiredoxin 6 (Prdx6), a host factor that contributes to pulmonary surfactant metabolism and lung defense against oxidative stress, as a previously unknown substrate of Pla. Pla cleaves Prdx6 at three distinct sites, and these cleavages disrupt both the peroxidase and phospholipase A2 activities of Prdx6. In addition, we found that infection with wild-type Y. pestis reduces the abundance of extracellular Prdx6 in the lungs compared to that after infection with Δpla Y. pestis, suggesting that Pla cleaves Prdx6 in the pulmonary compartment. However, following infection with either wild-type or Δpla Y. pestis, Prdx6-deficient mice exhibit no differences in bacterial burden, host immune response, or lung damage from wild-type mice. Thus, while Pla is able to disrupt Prdx6 function in vitro and reduce Prdx6 levels in vivo, the cleavage of Prdx6 has little detectable impact on the progression or outcome of pneumonic plague.
INTRODUCTION
The respiratory tract of mammals serves as a major interface with the external environment, and as such, the host relies on a number of mechanisms to prevent and/or resolve infections by pathogenic microorganisms within the pulmonary compartment. These host defenses within the lungs include mucociliary clearance, phagocytic cells, pulmonary surfactant, and an array of proteins, including antimicrobial peptides and lysozyme (1–3). Furthermore, infiltrating inflammatory cells produce large amounts of reactive oxygen species (ROS) via the NADPH oxidase enzyme complex during infection. This not only results in the killing of invading pathogens but can also lead to the activation of various cytokines and growth factors that promote a proinflammatory environment while causing direct damage to the host itself (2, 4).
The alveoli are covered by a thin layer of pulmonary surfactant, which not only lowers surface tension at the air-liquid interface but also contributes to host defense and regulates immune cell function (5, 6). A major component of this surfactant consists of phospholipids; however, surfactant also contains the proteins SP-A and SP-D, which play crucial roles in innate immune function and enhance the clearance of pathogen by acting as collectins (7). Furthermore, several additional proteins found within the surfactant have antibacterial properties, such as lactoferrin, complement, and LL-37/CRAMP (2, 6, 8). Thus, changes in the composition of pulmonary surfactant can serve as biomarkers for infection and underlie the mechanisms for the establishment of multiple respiratory diseases (5).
Proteins within the peroxiredoxin family are emerging as important protectors in the response to ROS generated as part of the host defense during lung disease and infection (9). Particularly, peroxiredoxin 6 (Prdx6) has been shown to be a key antioxidant enzyme that plays an important role in lung function by regulating the cellular redox balance, particularly under stress conditions, acute lung injury (ALI)/acute respiratory disease (ARD), and lipopolysaccharide (LPS)-induced inflammation (10–13). Prdx6 is present and associated with surfactant proteins in the alveolar extracellular spaces of the lungs and is expressed at highest levels by alveolar epithelial type II cells, lamellar bodies, and alveolar macrophages (6, 14–18). This bifunctional protein is a glutathione peroxidase and is the only peroxiredoxin that also carries phospholipase A2 (PLA2) activity (12). Thus, the physiological roles of Prdx6 include protection of cell-membrane phospholipids against oxidative damage and the regulation of lung surfactant phospholipid turnover.
Increased Prdx6 levels are detected within the lungs during ALI/ARD, hyperoxia, and pneumonia (12, 19). In addition, recent findings have demonstrated that Prdx6 is upregulated in response to LPS and is necessary for the NADPH oxidase NOX2 activation pathway in pulmonary endothelial cells, alveolar macrophages, and polymorphonuclear leukocytes (PMN) (20–22). Investigations on the physiology of Prdx6 have shown increased lung injury in response to oxidant stress and altered lung surfactant turnover in Prdx6-deficient mice (10, 23), whereas transgenic mice overexpressing Prdx6 show increased resistance to oxidant stress (24). In addition, while several proteomic studies have revealed increased abundance of Prdx6 in the lungs in response to Klebsiella pneumoniae, Francisella tularensis, and H1N1 influenza virus pneumonia (25–27), it is not known if Prdx6 contributes to the protective response to pulmonary infections.
Yersinia pestis, the causative agent of plague, is reemerging as a significant human pathogen, and currently, there is no approved vaccine for plague in the United States. More recently, there have been reported outbreaks of plague in the United States and Madagascar (28, 29). The pneumonic form of plague represents the most severe form of the disease due to its rapid progression and ease of transmission by aerosols. The progression of pneumonic plague is characterized as biphasic; the beginning phase is relatively asymptomatic and noninflammatory, while the second phase is proinflammatory, as evidenced by the development of massive inflammatory lesions, upregulation of cytokine and chemokine levels, and bacterial dissemination (30). Through the mechanisms of its type III secretion system, Y. pestis limits its exposure to ROS and reactive nitrogen species (RNS) within the host early during the infection (31). Additionally, Y. pestis is known to manipulate additional innate immune responses of the lungs through the activities of multiple virulence determinants, thereby creating a protective environment in the lungs (32, 33).
One of the virulence factors of Y. pestis responsible for acute pathogenesis in mammals is the omptin family outer membrane protease Pla, which has a wide range of proteolytic, adhesive, and invasive properties (34–37). The protease activity of Pla is essential for the development of pneumonic plague, and its best-studied activity in vitro is the activation of host plasminogen (plg) into plasmin (38–40). Although Pla has been demonstrated to cleave a number of additional host substrates in vitro, including alpha2-antiplasmin (A2AP), plasminogen activator inhibitor-1 (PAI-1), urokinase plasminogen activator (uPA), complement component C3, and the apoptotic molecule Fas ligand (FasL) (39, 41–43), to date only the cleavage of FasL by Pla (43) has been shown to contribute to the progression of pneumonic plague. Thus, the precise mechanisms by which the Pla protease manipulates lung function and innate immunity are still largely unknown.
As Y. pestis infection is primarily extracellular in nature and localized to the small airways of the lung (44), in this study we sought to discover additional host factors degraded or cleaved by Pla specifically within the alveolar space that might contribute to the development of pneumonic plague. Here, we describe Prdx6 as a newly identified Pla substrate within the lungs of mice and show that the cleavage by Pla disrupts both the peroxidase and phospholipase activities of Prdx6. Furthermore, we demonstrate that following infection with Y. pestis, the levels of extracellular Prdx6 are reduced in a Pla-dependent manner; however, Prdx6-deficient mice infected with Y. pestis exhibit no significant difference from wild-type mice in bacterial burden, host immune response, or lung damage. These results suggest that while Pla alters Prdx6 levels in the lung and inactivates Prdx6 activities, these effects during pneumonic plague have little impact on the development of disease within the lungs.
MATERIALS AND METHODS
Reagents, bacterial strains, and culture conditions.
All reagents used in this work were obtained from Sigma-Aldrich or VWR unless otherwise stated. The bacterial strains and plasmids used in this work are listed in Table S1 in the supplemental material. Brain heart infusion (BHI) broth or agar (Difco) was used to maintain Y. pestis strains and derivatives. Luria-Bertani (LB) broth or agar was used to maintain all Escherichia coli strains. Experiments described in Fig. 1 to 3 and in Fig. S1 and Table S2 in the supplemental material used the pCD1− derivatives of CO92; all other experiments used the virulent Y. pestis CO92 and derivatives. Ampicillin (100 μg/ml) was added to the medium as needed. For animal infections, Y. pestis strains were cultured in BHI with the addition of 2.5 mM CaCl2 at 37°C to induce the type III secretion system, as previously described (35). All experiments using select agent strains of Y. pestis were conducted in a Centers for Disease Control and Prevention-approved biosafety level 3 (BSL-3)/animal biosafety level 3 (ABSL-3) facility at Northwestern University.
FIG 1.
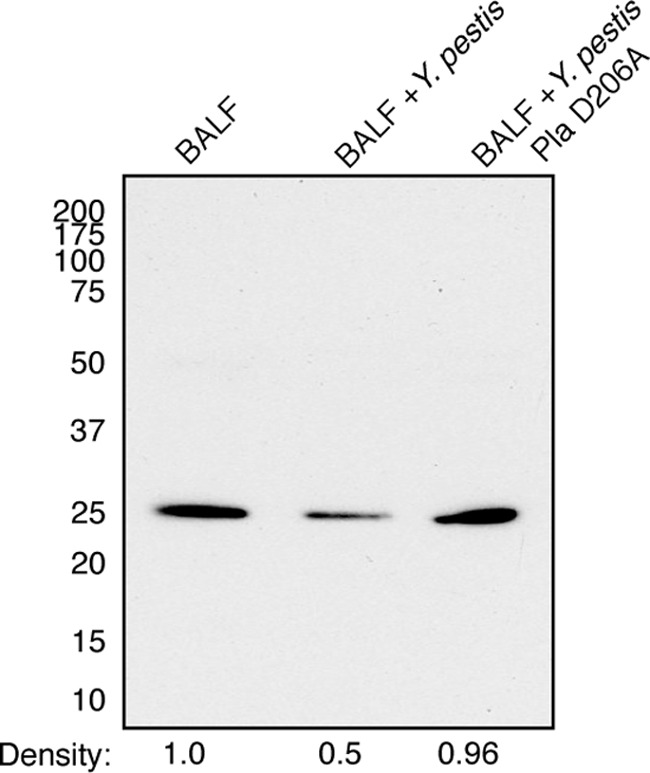
Validation of Pla-dependent Prdx6 degradation within BALF. Immunoblot analysis of Prdx6 from C57BL/6 mouse BALF only or BALF following incubation with wild-type Y. pestis or Y. pestis Pla D206A for 6 h at 37°C. The density of each band relative to BALF only is indicated beneath. Numbers to the left of the blot indicate molecular masses in kilodaltons. The blot is representative of 3 independent experiments.
FIG 3.
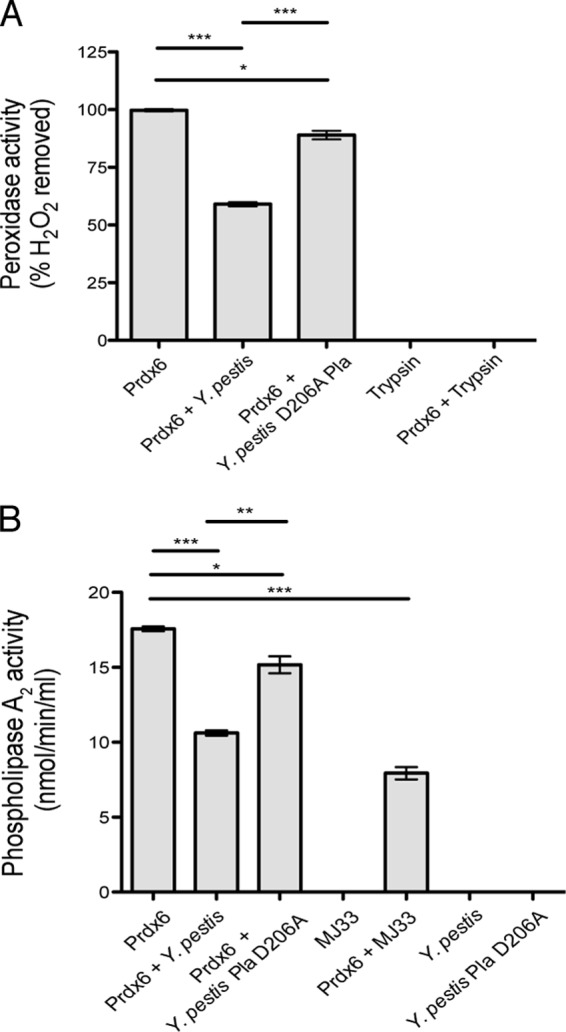
Cleavage of Prdx6 by Pla disrupts both phospholipase A2 and peroxidase activities. (A) Peroxidase activity of Prdx6 following incubation with Y. pestis, Y. pestis Pla D206A, or trypsin or incubation alone for 2 h at 37°C. Prdx6 activity is calculated as the percentage of H2O2 removed, based on the ratio of the amount of H2O2 removed in the presence to that removed in the absence of Prdx6. (B) Phospholipase A2 activity of Prdx6 following incubation with Y. pestis, Y. pestis Pla D206A, or the inhibitor MJ33 or incubation alone. One representative experiment of 2 biological replicates is shown, and error bars represent the SEM (*, P < 0.05; **, P < 0.01; ***, P < 0.001 by one-way ANOVA with Bonferroni's multiple-comparison test).
Incubation of Y. pestis with BALF and iTRAQ analysis.
All procedures involving animals were carried out in compliance with protocols approved by the Institutional Animal Care and Use Committee of Northwestern University. Mouse bronchoalveolar lavage fluid (BALF) was collected from uninfected, female C57BL/6 mice using 1 ml phosphate-buffered saline (PBS) for each lavage for a total of two lavages per animal as described previously (43). Samples were pooled and centrifuged at 300 × g for 10 min to separate cells and cell debris; supernatants were passed through a 0.22-μm filter. The protein content of the BALF (supernatant) was measured with Bradford reagent (Bio-Rad). Y. pestis strains grown overnight at 37°C in BHI were diluted to an optical density at 620 nm (OD620) of 0.1 into 1 ml of filter-sterilized cell-free BALF (diluted to a concentration of 100 μg/ml total protein with PBS). Three independent 1-ml assay mixtures of either BALF alone, BALF plus Y. pestis, or BALF plus Y. pestis Pla D206A were incubated in a rotor drum at 37°C for 6 h. Following incubation, the 3 replicates for each condition were pooled and centrifuged at 6,000 × g for 10 min at 4°C to pellet the bacteria. The supernatants were removed and precipitated with 20% trichloroacetic acid (TCA). The resulting pellets were analyzed by 4-plex isobaric tag for relative and absolute quantitation (iTRAQ) by the W. M. Keck Foundation Biotechnology Resource Laboratory at Yale University. Samples were prepared and analysis was performed as previously described (45). The samples were labeled with iTRAQ tags as follows: 114, BALF only; 115, Y. pestis plus BALF; 116, Y. pestis Pla D206A plus BALF; and 117, Y. pestis plus BALF. Mass spectrometric (MS) analysis was performed on an AB Sciex TripleTOF 5600 mass spectrometer with AB Sciex ProteinPilot software used for protein identification and quantitation (46). Amino acid sequences were further analyzed with BLAST and analysis tools available through the ExPASy Molecular Biology Server (http://www.expasy.ch/) and UniProt (http://www.uniprot.org/).
Plasminogen activation assay, cleavage of Prdx6, and immunoblot analyses.
The plasminogen-activating ability of Y. pestis strains was assessed as described previously (35, 47). Briefly, strains were cultured for 6 h at 37°C in either BHI or BALF before being diluted to 4 × 106 CFU in 20 mM HEPES buffer and combined with purified human Glu-plasminogen (Hematologic Technologies; 4 μg) and the chromogenic substrate D-AFK-ANSNH-iC4H9-2HBr (SN5; Hematologic Technologies; 50 μM) in a total volume of 200 μl and incubated at 37°C for 3 h. The absorbance at 460 nm was measured every 10 to 11 min in a Molecular Devices SpectraMax M5 fluorescence microplate reader.
The cleavage of Prdx6 by Y. pestis or E. coli strains producing Pla was assessed in a manner similar to that previously described for FasL (43). Briefly, strains were cultured in BHI for 6 h at 37°C before being diluted to 4 × 106 CFU or 8 × 106 CFU in 20 mM HEPES and combined with 0.1 μg recombinant His-tagged human Prdx6 (Novus Biologicals) and incubated at 37°C. For experiments with Pla-producing E. coli, pla expression was induced by the addition of isopropyl-β-d-thiogalactopyranoside (IPTG) (1 mM) for 14 to 16 h at 37°C before being diluted to 8 × 106 CFU. Following incubation for the indicated times, bacteria were removed by centrifugation and proteins contained within the supernatant were mixed with reducing sample buffer, separated by SDS-PAGE, and transferred to nitrocellulose for analysis by immunoblotting with an anti-Prdx6 antibody (1:1,000 dilution) (Abcam; catalog no. 59543). Densitometry of all blots was measured using ImageJ.
Peroxidase and phospholipase assays.
The peroxidase activity of Prdx6 was assessed by quantifying the removal of H2O2 by the peroxidase activity assay (48). Recombinant human Prdx6 (0.2 μg) or mock samples (PBS only) were digested in triplicate with trypsin (0.1 μg) at 37°C overnight or incubated with Y. pestis strains (4 × 106 CFU) as described above. Supernatants (5 μl) were then mixed with 45 μl reaction buffer (1 mM dithiothreitol [DTT], 0.03× PBS, 0.5% glycerol) and 5 μl of 5 mM H2O2. Following incubation at room temperature for 20 min, 20 μl of 26% TCA was added to stop the reaction. Remaining peroxide levels were quantified by measuring the A475 on a Molecular Devices SpectraMax M5 fluorescence microplate reader after the addition of 30 μl formation solution (10 mM ferrous ammonium sulfate and 2.5 M potassium thiocyanate). Prdx6 activity is presented as percentage of H2O2 removed, based on the ratio of the amount of H2O2 removed in the presence to that removed in the absence of Prdx6.
The phospholipase A2 (PLA2) activity of Prdx6 was assessed using a cytosolic PLA2 (cPLA2) assay kit (Cayman Chemical, Ann Arbor, MI). Following the incubation of recombinant human Prdx6 with the indicated Y. pestis strains for 2 h as described above or with the PLA2 inhibitor MJ33 (1 mM) in duplicate, 10 μl of the supernatant was assayed as described by the manufacturer. Phospholipase A2 activity was quantified by measuring the A405 on a Molecular Devices SpectraMax M5 fluorescence microplate reader after the addition of 10 μl of 25 mM 5,5′-dithiobis(2-dinitrobenzoic acid) (DTNB) to stop enzyme catalysis and develop the reaction.
Identification of Pla cleavage site(s) of Prdx6.
Recombinant human Prdx6 (0.5 μg) was incubated in the presence or absence of Y. pestis (4 × 106 CFU) at 37°C for 2 h in 20 mM HEPES. Following incubation, bacteria were removed by centrifugation and proteins contained within the supernatant were mixed with reducing sample buffer and separated by SDS-PAGE. To determine the cleavage site(s) of Prdx6, cleaved fragments were cut from the gel, digested with trypsin, and analyzed by matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS). The peptides were analyzed with the Mascot search engine (www.matrixscience.com). MS and peptide analysis were performed by the Northwestern University proteomics core.
Animal infections.
Wild-type C57BL/6 mice were bred at Northwestern University. Breeder stocks of C57BL/6 prdx6−/− mice (49) were generously supplied by Aaron Fisher (University of Pennsylvania). All mice were maintained in a specific-pathogen-free facility at Northwestern University. Mice were sex and age matched at 6 to 8 weeks for all experiments. Mice were anesthetized with ketamine and xylazine and infected by the intranasal (i.n.) route with Y. pestis strains diluted in PBS at a dose of 1 × 104 CFU or 1 × 108 CFU as previously described (35, 43). The inoculating dose was confirmed by plating on BHI agar. Mice were monitored twice daily for the length of the experiment. Mice were euthanized by intraperitoneal (i.p.) injection of pentobarbital sodium followed by organ removal or cervical dislocation. All animal infections were performed at least twice, and the data were combined. To determine bacterial burden, mice were inoculated i.n. with Y. pestis strains and euthanized at 48 h postinfection. Organs were subsequently removed and weighed, homogenized in sterile PBS, serially diluted, and plated onto BHI agar. Following incubation at 26°C for 2 to 3 days, the CFU per organ were enumerated.
Quantification of total Prdx6 in BALF.
C57BL/6 mice were inoculated via the i.n. route with PBS (mock), Y. pestis, or Y. pestis Δpla, and after 48 h, BALF was collected. Prdx6 abundance in BALF was assessed both by immunoblotting and by the Prdx6 human enzyme-linked immunosorbent assay (ELISA) kit Quantikine ELISA (Abcam). For ELISA, 100-μl aliquots of BALF were analyzed in duplicate as described by the manufacturer. The absorbance at 450 nm was measured in a Molecular Devices SpectraMax M5 microplate reader. For immunoblot analysis, 50-μl aliquots of BALF were separated by SDS-PAGE and transferred to nitrocellulose for analysis with an anti-Prdx6 antibody. Densitometry of all blots was measured using ImageJ.
Histopathology.
Mice were inoculated i.n. with PBS (mock) or the indicated Y. pestis strains as described above, and at 48 h postinfection, mice were euthanized and lungs were inflated with 1 ml of 10% neutral buffered formalin via tracheal cannulation. Lungs were removed, fixed in 10% formalin, and subsequently embedded in paraffin. Two 5-micrometer sections, 200 μm apart per lung, were stained with hematoxylin and eosin (H&E) for examination. Tissue embedding, sectioning, and staining with H&E were performed by the Northwestern University Mouse Histology and Phenotyping Laboratory. Slides were imaged using a Zeiss Axioskop/Nuance camera.
Innate immune cell analysis.
Mice were infected via the i.n. route with PBS (mock) or the indicated Y. pestis strains as described above, and at 48 h postinfection, mice were euthanized and BALF was collected using 1 ml PBS for each lavage for a total of 5 lavages per animal as described previously (43). Samples were centrifuged at 300 × g for 10 min to separate cells and cell debris; the supernatant from the first wash was saved for cytokine analyses (see below). Cells were washed once each in PBS and twice in flow buffer (2% fetal bovine serum in PBS). For ex vivo cell surface marker detection, cells were stained with antibodies for CD45 (BioLegend; clone 30-F11), CD11b (BioLegend; clone M1/70), CD11c (BD Biosciences; clone HL3), Ly6G (BioLegend; clone IA8), F4/80 (eBioscience; clone BM8), and aqua Live/Dead fixable stain (Invitrogen) for 30 min at 4°C. All antibodies were used at 1:100 dilutions in flow buffer, while Live/Dead cell stain was used at 1:1,000. An anti-CD16/32 FcBlock antibody was included to minimize nonspecific staining (eBioscience). Cells were washed with flow buffer and fixed with 2% paraformaldehyde. Samples were analyzed using a BD FACSCanto II flow cytometer and FlowJo software (TreeStar). Identification of specific cell populations was performed as previously described (50) based on the following markers: neutrophils, CD45+ F4/80− Ly6G+ CD11b+; alveolar macrophages, CD45+ F4/80+ Ly6G− CD11b− CD11c+; CD11b-high macrophages, CD45+ F4/80+ Ly6G− CD11bhi; monocytes, CD45+ Ly6G− F4/80− CD11b+ CD11c−; and both CD11b-high and -low dendritic cells, CD45+ Ly6G− F4/80− CD11c+.
Cytokine analysis.
The levels of tumor necrosis factor (TNF), gamma interferon (IFN-γ), monocyte chemoattractant protein 1 (MCP-1), interleukin-6 (IL-6), and IL-10 were quantitatively established from the supernatants of collected BALF (see above) using the cytometric bead array technique (BD cytometric bead array mouse inflammation kit; BD Biosciences) as specified by the manufacturer (43). Prior to analysis, supernatants were passed through a 0.22-μm filter and plated for sterilization. Data were analyzed using the BD cytometric bead array software.
Albumin quantification.
The albumin in the supernatants of collected BALF from mock-infected (PBS) or Y. pestis-infected (see above) mice at 48 h postinoculation was quantitatively measured by the mouse albumin ELISA as described in the manufacturer's instructions (Immunology Consultants Laboratory, Inc.).
Statistics.
In all cases, statistical means are graphed and error bars represent standard errors of the means (SEM) for combined experiments. For bacterial burden comparisons, the Mann-Whitney U test was performed; for all other experiments, Student’s two-tailed unpaired t test or one-way analysis of variance (ANOVA), as indicated, was performed using GraphPad Prism 5. P values are indicated as follows: *, P ≤ 0.05; **, P ≤ 0.01; and ***, P ≤ 0.001.
RESULTS
Identification of Pla substrates within mouse BALF.
Previous work has shown that the protease activity of Pla is necessary for the rapid proliferation of Y. pestis within the small airways of the lungs during pneumonic plague (34, 35). As Y. pestis infection is generally extracellular in nature, it is therefore likely that Pla cleaves host targets within the alveolar space to alter innate immune responses within the lungs and to allow for rapid bacterial replication. Thus, the lung-air interface represents an ideal location to discover and examine previously unknown Pla substrates in order to further elucidate the mechanisms by which Y. pestis causes pneumonic plague.
To do so, we employed a shotgun proteomic approach using isobaric tag for relative and absolute quantitation (iTRAQ) to identify putative Pla substrates within cell-free BALF isolated from uninfected C57BL/6 mice. We incubated BALF alone or with wild-type Y. pestis or a strain of Y. pestis in which the native allele of pla is replaced with a protease-inactive mutant (Pla D206A) (35) for 6 h at 37°C. We subsequently separated the cells from the BALF by centrifugation and confirmed that Pla was still active in the wild-type-treated sample after 6 h by plg activation assay (see Fig. S1 in the supplemental material). The cell-free supernatants from each sample were then normalized for protein level, and protein abundance was analyzed by iTRAQ. A total of 212 different (mouse) proteins were identified from these cell-free BALF samples, of which the abundance of 23 proteins changed significantly (1.25-fold compared to BALF alone) in the presence of Y. pestis expressing the wild-type form of Pla compared to untreated controls (see Table S2). Compared to Y. pestis Pla D206A, the abundances of 14 of these proteins were significantly reduced, demonstrating a specific effect by the protease activity of Pla (see Table S2). The abundances of the previously described Pla targets complement component C3 and plg (51) were significantly reduced only when incubated with wild-type Y. pestis, providing internal validity to our substrate discovery analysis. The 14 identified putative Pla substrates within the BALF were analyzed and ranked based upon their biological function and processes, as well as the relative fold change compared to BALF alone or with the proteolytically inactive Pla D206A variant (see Table S2). Based upon these criteria, the physiological role, and the observed altered levels within the lungs during respiratory disease, Prdx6 was chosen as a newly identified Pla substrate to pursue from this substrate discovery screen. Immunoblot analysis of the same materials as those used for iTRAQ analysis confirmed a significant reduction in Prdx6 within the BALF but only when incubated with Y. pestis producing wild-type Pla (Fig. 1).
Validation of Prdx6 as a Pla substrate.
Analysis of mouse Prdx6 shows a high degree of identity at the amino acid level with Prdx6 encoded by genes in other mammals, including human Prdx6 (90% identity/93% similarity) and rat Prdx6 (93% identity/94% similarity). Therefore, to validate Prdx6 as a Pla substrate, we incubated recombinant full-length human Prdx6 with wild-type Y. pestis, Y. pestis Δpla, or Y. pestis carrying the Pla D206A variant. Immunoblot analysis shows a time-dependent cleavage of Prdx6, with the appearance of four distinct cleavage products only when incubated with bacteria producing wild-type Pla but not when incubated with the isogenic Δpla mutant or the D206A variant of Pla (Fig. 2A). To show that Pla is sufficient for Prdx6 cleavage, we performed a similar assay with a strain of E. coli that produces either the wild type or the D206A variant of Pla and found that only the wild-type pla-expressing strain is able to cleave Prdx6 (Fig. 2, compare panels A and B).
FIG 2.

Cleavage of Prdx6 requires catalytically active Pla. (A) Immunoblot analysis of recombinant human Prdx6 following incubation with Y. pestis, Y. pestis Pla D206A, or Y. pestis Δpla at 37°C for the indicated times. (B) Immunoblot analysis of recombinant human Prdx6 following incubation with E. coli producing Pla or Pla D206A or E. coli carrying the corresponding empty expression vector at 37°C for the indicated times. (C) Amino acid sequence of human Prdx6. Black arrowheads indicate identified Pla cleavage sites. The lipase and peroxidase motifs are boxed, and residues for the active site for peroxidase activity (C47, H39, and R132) or the catalytic triad for lipase activity (S32, D140, and H26) are in bold. Lowercase letters in panels A and B represent Pla-cleaved Prxd6 products. Numbers to the left of the blots indicate molecular masses in kilodaltons. Data are representative of at least 3 independent experiments.
To define the Pla cleavage site(s) within Prdx6, isolated peptides corresponding to bands a, b, c, and d in Fig. 2A were analyzed by MALDI-TOF MS analysis. This analysis identified two cleavage sites, one between amino acids K173 and R174 and a second between amino acids K201 and L202 (Fig. 2C). We were unable to identify a third cleavage site that would correspond with band b, although we cannot rule out the possibility that this is a result of the partial digestion product of the generated band a. Thus, these data demonstrate that Pla directly cleaves Prdx6 into 4 discrete peptides within the C-terminal domain of the protein.
Cleavage by Pla disrupts both peroxidase and phospholipase A2 activities of Prdx6.
Our results indicate that through the activity of Pla, Y. pestis is able to cleave both human and mouse Prdx6. The major physiological function associated with Prdx6 is its peroxidase activity (12). Therefore, to assess the consequences of the Pla-dependent cleavage on Prdx6 function, we measured the peroxidase activity of Prdx6 after incubation with Y. pestis producing either native Pla or the protease-inactive (D206A) variant of Pla. Incubation of Prdx6 with Y. pestis producing wild-type Pla results in significantly reduced peroxidase activity compared to untreated Prdx6 or Prdx6 incubated with Y. pestis producing the D206A variant of Pla, but not to the same extent as treatment with trypsin (Fig. 3A). While incubation of Prdx6 with Y. pestis D206A did significantly reduce the peroxidase activity of Prdx6 compared to Prdx6 alone, it was to only a modest extent, demonstrating that the protease activity of Pla is the primary mechanism by which Y. pestis disrupts Prdx6 function (Fig. 3A).
In addition to its peroxidase activity, and unlike the other peroxiredoxins, Prdx6 has also been demonstrated to have phospholipase A2 activity (12). We therefore assessed the consequences of the Pla-dependent cleavage of Prdx6 on its ability to hydrolyze arachidonic acid. Prdx6 was incubated alone or with wild-type Y. pestis, Y. pestis carrying Pla D206A, or the phospholipase A2 inhibitor MJ33 (52). We found that wild-type Y. pestis significantly reduced the phospholipase A2 activity of Prdx6 to levels similar to those of MJ33, and while bacteria carrying the protease-inactive variant of Pla also significantly affected the activity of Prdx6 compared to Prdx6 alone, again it was only minimal (Fig. 3B). Taken together, these data indicate that Y. pestis disrupts both the peroxidase and phospholipase A2 activities of Prdx6 primarily through the protease activity of Pla.
Y. pestis affects Prdx6 abundance in the lungs during primary pneumonic plague.
As Pla is required for the full virulence of Y. pestis during pneumonic plague (34, 35), we set out to determine if (i) the total levels of Prdx6 are altered in the lungs of mice during respiratory infection with Y. pestis and (ii) if Pla cleaves Prdx6 in vivo. As the Δpla mutant of Y. pestis is attenuated during respiratory infections (35), to compare the effects of Pla on Prdx6 levels when bacterial loads are normalized between the wild-type and Δpla strains, we can increase the inoculum of the Δpla mutant (“input CFU”) so that the CFU in the lungs at 48 h (“output CFU”) is equivalent between the wild-type and mutant strains, as we have done previously (43, 50). To do so, we infected C57BL/6 mice via the i.n. route with 104 CFU of wild-type Y. pestis or 108 CFU of Δpla Y. pestis, doses of each strain that result in approximately 108 CFU in the lungs by 48 h (43), or 104 CFU of the Δpla mutant (to compare the effects of equivalent input CFU on Prdx6 levels), or mice were mock infected with PBS. After 48 h, BALF was collected and analyzed by ELISA to quantify the total levels of Prdx6 present in the lung airspace. We found that the abundance of total Prdx6 within the BALF of mice is significantly increased in mice infected with 108 CFU of Δpla Y. pestis compared to both mock-infected mice and mice infected with 104 CFU of Δpla Y. pestis, indicating that increased numbers of bacteria in the lungs result in greater abundance of Prdx6 within the airspace (Fig. 4A). In contrast, however, we determined that, in mice infected with wild-type Y. pestis, the levels of Prdx6 are significantly reduced compared to those found in mice with an equivalent number of Y. pestis Δpla bacteria (108 output CFU), suggesting that the presence of Pla reduces the abundance of Prdx6 in the airspace (Fig. 4A).
FIG 4.

Pla-dependent changes in Prdx6 levels within the lung during Y. pestis respiratory infection. Abundance of extracellular Prdx6 recovered by BAL of C57BL/6 mouse lungs 48 h post-inoculation with PBS (mock), 104 CFU of Y. pestis, 104 CFU of Y. pestis Δpla, or 108 CFU of Y. pestis Δpla as measured by ELISA (A) or immunoblotting (B). Data from mock- or wild-type Y. pestis-infected C57BL/6 prdx6−/− mice are shown as controls in panel B. The input CFU and output CFU are included below the graph and blot to denote the number of bacteria inoculated and present at the time of assessment. The density of each band relative to BALF is indicated beneath the immunoblot. Data in panel A are combined from 2 independent experiments (n = 10), and error bars represent the SEM. Data in panel B are representative of 3 independent experiments (*, P < 0.05; ns, not significant by one-way ANOVA with Bonferroni's multiple-comparison test).
Therefore, to determine if we could detect the cleavage of Prdx6 by Pla from samples isolated from infected mice, we analyzed the same BALF by immunoblotting with an antibody to Prdx6. Consistent with our ELISA data, there is a significant increase in Prdx6 abundance in the airspace of mice infected with 108 CFU of Δpla Y. pestis compared to either mock-infected or wild-type Y. pestis-infected mice as determined by ImageJ densitometry (Fig. 4B). Similarly to our ELISA data, we observed a reduction in the abundance of Prdx6 present in the airspace of wild-type Y. pestis-infected mice compared to a dose-matched infection with Δpla Y. pestis (108 output CFU) at 48 h (Fig. 4B). In contrast to the in vitro cleavage of Prdx6 by Pla (Fig. 2), however, we were unable to detect cleavage products by immunoblotting of Prdx6 present in the BALF of wild-type Y. pestis-infected mice. In total, these data demonstrate that although pulmonary infection with high doses of Y. pestis Δpla results in an increase in Prdx6 levels within the airspace, the presence of Pla reduces these levels to ones approaching those for uninfected mice, suggesting that Prdx6 is a Pla substrate during pneumonic plague.
The cleavage or absence of Prdx6 does not influence the outgrowth or dissemination of Y. pestis following respiratory infection.
As Pla is necessary for the outgrowth of Y. pestis in the lungs during pneumonic plague, we hypothesized that if the cleavage of Prdx6 by Pla contributes to the virulence of Y. pestis, then infections of mice lacking Prdx6 with the Δpla strain would result in an increased bacterial burden or dissemination compared to wild-type mice. To test this, wild-type or prdx6−/− C57BL/6 mice were infected via the i.n. route with equivalent doses (104 CFU) of either wild-type or Δpla Y. pestis, and after 48 h, the bacteria in the lungs and spleens were enumerated. We found no difference in bacterial outgrowth in either the lungs or the spleens of Prdx6-deficient mice compared to wild-type mice infected with wild-type Y. pestis or between the same sets of mice infected with the Δpla mutant (Fig. 5), suggesting that Prdx6 has little impact on the outgrowth or dissemination of Y. pestis during pneumonic plague.
FIG 5.
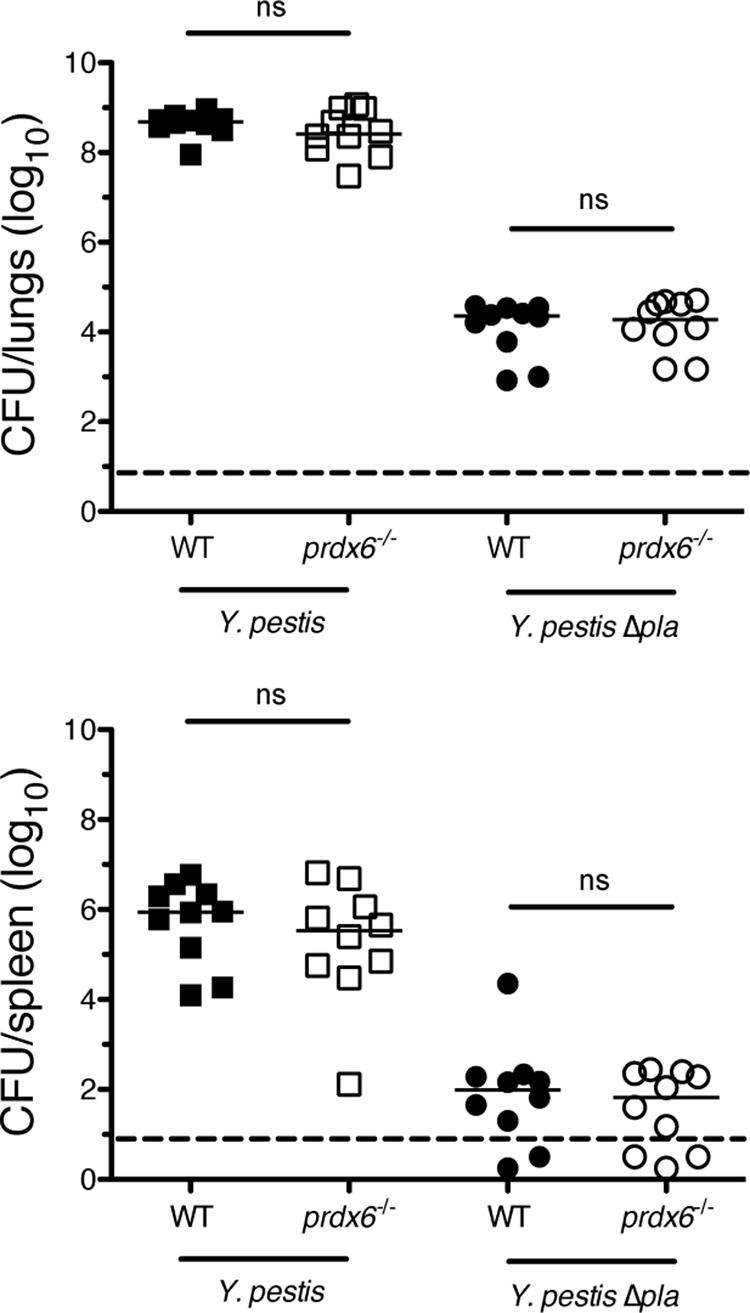
Absence of Prdx6 does not alter bacterial burden during Y. pestis respiratory infection. Bacterial burden within lungs (A) and spleens (B) of C57BL/6 or C57BL/6 prdx6−/− mice infected via the i.n. route with 104 CFU of Y. pestis or Y. pestis Δpla after 48 h. Each point represents the number of bacteria recovered from a single mouse. The median CFU is denoted by a solid line, and the dashed line indicates the limit of detection. Data are combined from two independent experiments (n = 10 for each group; ns, not significant by Mann-Whitney U test). WT, wild type.
Prdx6-deficient mice are not altered in the host response to pulmonary Y. pestis infection.
While we found that the absence of Prdx6 does not affect bacterial numbers following respiratory infection, it is possible that Prdx6 may influence the innate immune response to Y. pestis in the lungs (11, 53). Therefore, to test the impact of Prdx6 deficiency on the host response, we first examined the pulmonary pathology of Prdx6-deficient mouse lungs at 48 h postinfection following i.n. infection with equivalent doses of wild-type or Δpla Y. pestis. Analysis of pulmonary histopathology by H&E staining revealed no qualitative difference in the size of the pulmonary lesions either between Prdx6-deficient and wild-type mice following infection with wild-type Y. pestis or between the same sets of mice following infection with Δpla Y. pestis (Fig. 6A, insets, arrows). Closer examination of these individual lesions reveals similarity in cellular architecture between Prdx6-deficient and wild-type mice, with the Δpla Y. pestis infection resulting in small compact foci comprised of PMN and with greater hematoxylin staining than that of wild-type Y. pestis, which shows more diffuse infiltration of PMN (Fig. 6A).
FIG 6.

Absence of Prdx6 does not alter the host immune response during Y. pestis respiratory infection. (A) Sections of formalin-fixed lungs stained with H&E from C57BL/6 or C57BL/6 prdx6−/− mice inoculated with PBS (mock), 104 CFU of Y. pestis, or 104 CFU of Y. pestis Δpla after 48 h. Representative images of inflammatory lesions are shown (arrows; n = 3). Bars, 100 μm (inset images) and 50 μm (larger images). (B) Enumeration of total immune cells present in the BALF of C57BL/6 or C57BL/6 prdx6−/− mice inoculated with PBS (mock), 104 CFU of Y. pestis, or 104 CFU of Y. pestis Δpla after 48 h. (C) Abundance of indicated inflammatory cytokines present in supernatants of the same BALF samples as those collected for panel B. Data are combined from 2 independent experiments (n = 10 for each group), and error bars represent the SEM (ns, not significant by one-way ANOVA with Bonferroni's multiple-comparison test).
To assess this more quantitatively, we then performed flow cytometry to enumerate the numbers and types of immune cells infiltrating into the lung airspace in the presence or absence of Prdx6. Prdx6-deficient and wild-type mice infected with wild-type Y. pestis exhibit a similar and significant recruitment of immune cells into the lungs compared to mock and Δpla Y. pestis infections (Fig. 6B). Using the percentages (data not shown) and the total number of immune cells recruited, the absolute numbers of neutrophils, alveolar macrophages, CD11b-high macrophages, monocytes, and both CD11b-high and -low dendritic cells were determined. We found no significant difference in the numbers of any immune cell type measured between wild-type and Prdx6-deficient mice when infected with Y. pestis or between the same sets of mice infected with Δpla Y. pestis (see Fig. S2 in the supplemental material).
We also investigated the effects of Prdx6 deficiency on the production of proinflammatory cytokines in response to pulmonary Y. pestis infection. Examination of the pulmonary inflammatory cytokine response by cytometric bead array showed no differences between wild-type and Prdx6-deficient mice when infected with Y. pestis or between the same sets of mice infected with Δpla Y. pestis (Fig. 6C).
The loss of Prdx6 does not impact lung damage during Y. pestis infection.
Pneumonic plague results in severe damage to the lung-bloodstream barrier, primarily due to the effects of the resulting immune response (43, 54). As Prdx6 is known to affect lung homeostasis and pulmonary surfactant turnover (10, 12), we investigated the effects of Prdx6 deficiency on lung injury during respiratory Y. pestis infection by assessing serum albumin levels in the airspace by ELISA. We observed no difference in albumin levels between wild-type and Prdx6-deficient mice when infected with Y. pestis or between the same sets of mice infected with Δpla Y. pestis (Fig. 7). Taken together, these data demonstrate that Prdx6 does not play a measurable role in the host response or protect against tissue damage during Y. pestis infection of the lungs.
FIG 7.
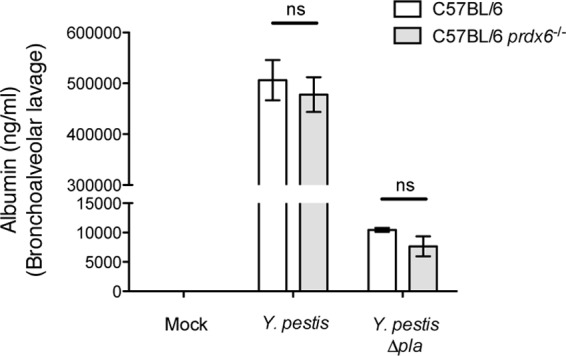
Absence of Prdx6 does not alter lung damage during Y. pestis respiratory infection. Abundance of albumin present in supernatants of the same BALF samples as those collected for the experiment in Fig. 6 as measured by ELISA. Data are combined from 2 independent experiments (n = 10 for each group), and error bars represent the SEM (ns, not significant by one-way ANOVA with Bonferroni's multiple-comparison test).
DISCUSSION
The Pla protease of Y. pestis has been shown to be a critical virulence factor in the emergence of the plague bacillus as a respiratory pathogen (34, 35). However, the means by which Pla manipulates lung function and innate immunity are largely unknown, and host substrates of Pla are just beginning to be elucidated. In this study, we sought to discover previously unknown Pla substrates within the pulmonary environment in order to assess their roles in the development of pneumonic plague. We identified 14 host substrates whose abundances were altered in a Pla-dependent manner within the cell-free BALF isolated from uninfected mice, 8 of which have been characterized as having a role in immune system processes or proteolysis. In addition, the abundances of 9 additional BALF proteins were reduced in a Pla-independent manner, suggesting that Y. pestis may cleave these proteins via other mechanisms, or that they may adhere to the surface of Y. pestis bacilli, thus being removed from solution. The use of uninfected BALF for these experiments restricts our analysis to proteins that are normally present in the airspace; it is likely that there are additional potential substrates released into the airway in response to infection that would not be identified in this screen. It is of interest that the abundances of SP-A and SP-D, which comprise the majority of surfactant protein content (7), were unaltered within the BALF, suggesting that the impact of Y. pestis on pulmonary and immune function may occur independently of these collectins.
We identified Prdx6, an abundant protein found within pulmonary surfactant (6), as a Pla substrate that Y. pestis encounters in the lungs of mice during pneumonic plague. In the absence of Pla, extracellular levels of Prdx6 increase in the lungs following infection with a high dose of Y. pestis, which is consistent with observations of other severe respiratory pathogens such as K. pneumoniae and F. tularensis, neither of which harbors a bacterial plasminogen activator (25, 26). While it is possible that the host response to a high dose of the Δpla mutant delivered to the lungs may differ from that of wild-type Y. pestis given at lower doses, our results are consistent with a Pla-dependent cleavage of Prdx6 in vivo at 48 h, while pla is maximally expressed. Indeed, the snapshot of the infection at 48 h allows us to make the best examination and comparison of Pla-dependent processes within the lungs, as this is when pla is upregulated by Y. pestis during pneumonic plague and when the proinflammatory response is fully induced in response to the infection (35, 55). While there is a Pla-dependent decrease in extracellular Prdx6 protein levels in the lungs after 48 h, we were unable to detect Prdx6 cleavage products in vivo, as is observed following cleavage of Prdx6 by Pla in vitro. This may be due to the cleavage peptides of Prdx6 being further degraded and removed by the host, or they may be present at low levels below our limit of detection. Alternatively, it is possible that the antibody that we used for our immunoblot analyses, which was raised in rats, is able to detect cleavage products of human Prdx6 but not mouse Prdx6. Nonetheless, the Pla-dependent decrease in the full-length peptide both in vivo and ex vivo strongly suggests that cleavage of Prdx6 occurs in the lung airspace during pneumonic plague.
The cleavage of Prdx6 by Pla generates four distinct peptides; two of the three Pla cleavage sites within Prdx6 are located near the C terminus of the protein. The nature of these cleavage sites is consistent with other Pla substrate cleavages, typically located between basic amino acids such as lysine (56). While our analyses of Prdx6 function following cleavage by Pla showed significant reductions in both the peroxidase and phospholipase A2 activity, in neither case did we observe a complete elimination of either activity. Our data suggest that Pla may not completely cleave Prdx6, as evidenced by the residual full-length band present on the immunoblot, which is still present after incubation for 8 h with wild-type Y. pestis (data not shown). Alternatively, the C-terminal cleavage of Pla on Prdx6 may explain the remaining activity following proteolysis, as the active serine-32 (phospholipase) and cysteine-45 (peroxidase) residues for these biochemical activities are located near the N terminus of the protein (57). Indeed, one or more of the cleaved peptides may retain these activities. Therefore, based on these results, it is plausible that Prdx6 activities are not completely inactivated within the lung during the development of pneumonic plague.
Although recent work has indicated that the phospholipase activity of Prdx6 is involved in mediating the proinflammatory response through the regulation of TNF-induced apoptosis via arachidonic acid release and interleukin-1β production (53), we did not observe any change in the influx of immune cells or the production of proinflammatory cytokines (TNF, IFN-γ, MCP-1, or IL-6) in the lungs in the absence of Prdx6. While the loss of Prdx6 results in increased ROS production during ALI/ARD, during infection with Y. pestis, the bacterial Yop-Ysc type III secretion system might otherwise serve as a mechanism to suppress ROS generation within the host, even in the absence of Prdx6. Based on gene expression data collected from rat buboes and infected macrophages, it has been shown that Y. pestis encounters ROS during the initial stage of infection but not later (58). Therefore, it remains possible that the cleavage of Prdx6 may affect the infection in ways not assessed here or that the impact may be masked by the activities of other Y. pestis virulence factors.
Indeed, we found that the loss of Prdx6 and Pla has no effect on the disease progression of Y. pestis in the lungs or bacterial dissemination from the lungs into deeper tissues. These results suggest that Prdx6 is not a protective factor within the pulmonary compartment against an acute respiratory pathogen such as Y. pestis, at least as measured by the assays described here. Although it has yet to be determined if Prdx6 contributes to the host response during other bacterial respiratory infections such as those with K. pneumoniae and F. tularensis, it may be possible that Prdx6 is an important host response only to noninfectious lung injury (such as ALI/ARD), rather than to respiratory pathogens.
The lack of a conserved consensus motif for Pla cleavage suggests that, while selective, Pla could act upon several different and/or overlapping substrates, depending on the route of infection. Based upon the data presented here and collected by our group and others previously, we propose classifying substrates of Pla into three categories: group I substrates that are cleaved both in vitro and in vivo and contribute to bacterial virulence and the host response (these substrates would be represented by the apoptotic molecule FasL [43]); group II substrates such as Prdx6 that are cleaved in vitro and in vivo but do not play a detectable role in bacterial virulence or the host response; and finally, group III substrates that are cleaved in vitro but not in vivo and thus have a limited role during disease progression; A2AP would be a representative example of this class of substrate (50). Therefore, while the identification of new host substrates of Pla in vitro can provide new insights into the mechanisms by which Y. pestis causes disease in mammals, it is important to consider that any individual host substrate may not necessarily play a role in vivo, at least at our current level of investigation. Regardless of the effects of Pla on Prdx6 function in vivo, the study presented here expands our understanding of the role of Prdx6 during bacterial respiratory infections and suggests that in some cases, this host factor may have little overall impact on disease progression and innate immunity.
Supplementary Material
ACKNOWLEDGMENTS
iTRAQ analysis was performed by the W. M. Keck Foundation Biotechnology Resource Laboratory at Yale University. This work was supported by the Northwestern University Interdepartmental Immunobiology Flow Cytometry Core Facility. Histopathology was performed by the Northwestern University Mouse Histology and Phenotyping Laboratory, supported by NCI CA060553. Imaging was performed at the Northwestern University Center for Advanced Microscopy, supported by NCI CCSG P30 CA060553 awarded to the Robert H. Lurie Comprehensive Cancer Center.
We thank Jeremy Ritzert, Adam Caulfield, and Dhaval Nanavati for technical assistance. We are grateful to Aron Fisher, University of Pennsylvania, for providing the prdx6−/− mice.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01168-15.
REFERENCES
- 1.Kingma PS, Whitsett JA. 2006. In defense of the lung: surfactant protein A and surfactant protein D. Curr Opin Pharmacol 6:277–283. doi: 10.1016/j.coph.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 2.Eisele NA, Anderson DM. 2011. Host defense and the airway epithelium: frontline responses that protect against bacterial invasion and pneumonia. J Pathog 2011:249802. doi: 10.4061/2011/249802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Glasser JR, Mallampalli RK. 2012. Surfactant and its role in the pathobiology of pulmonary infection. Microbes Infect 14:17–25. doi: 10.1016/j.micinf.2011.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bedard K, Krause KH. 2007. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 5.Wattiez R, Falmagne P. 2005. Proteomics of bronchoalveolar lavage fluid. J Chromatogr B Analyt Technol Biomed Life Sci 815:169–178. doi: 10.1016/j.jchromb.2004.10.029. [DOI] [PubMed] [Google Scholar]
- 6.Gharib SA, Nguyen E, Altemeier WA, Shaffer SA, Doneanu CE, Goodlett DR, Schnapp LM. 2010. Of mice and men: comparative proteomics of bronchoalveolar fluid. Eur Respir J 35:1388–1395. doi: 10.1183/09031936.00089409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nayak A, Dodagatta-Marri E, Tsolaki AG, Kishore U. 2012. An insight into the diverse roles of surfactant proteins, SP-A and SP-D in innate and adaptive immunity. Front Immunol 3:131. doi: 10.3389/fimmu.2012.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Laube DM, Yim S, Ryan LK, Kisich KO, Diamond G. 2006. Antimicrobial peptides in the airway. Curr Top Microbiol Immunol 306:153–182. [DOI] [PubMed] [Google Scholar]
- 9.Salzano S, Checconi P, Hanschmann EM, Lillig CH, Bowler LD, Chan P, Vaudry D, Mengozzi M, Coppo L, Sacre S, Atkuri KR, Sahaf B, Herzenberg LA, Herzenberg LA, Mullen L, Ghezzi P. 2014. Linkage of inflammation and oxidative stress via release of glutathionylated peroxiredoxin-2, which acts as a danger signal. Proc Natl Acad Sci U S A 111:12157–12162. doi: 10.1073/pnas.1401712111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y, Feinstein SI, Fisher AB. 2008. Peroxiredoxin 6 as an antioxidant enzyme: protection of lung alveolar epithelial type II cells from H2O2-induced oxidative stress. J Cell Biochem 104:1274–1285. doi: 10.1002/jcb.21703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang D, Song Y, Wang X, Sun J, Ben Y, An X, Tong L, Bi J, Wang X, Bai C. 2011. Deletion of peroxiredoxin 6 potentiates lipopolysaccharide-induced acute lung injury in mice. Crit Care Med 39:756–764. doi: 10.1097/CCM.0b013e318206befd. [DOI] [PubMed] [Google Scholar]
- 12.Fisher AB. 2011. Peroxiredoxin 6: a bifunctional enzyme with glutathione peroxidase and phospholipase A(2) activities. Antioxid Redox Signal 15:831–844. doi: 10.1089/ars.2010.3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diet A, Abbas K, Bouton C, Guillon B, Tomasello F, Fourquet S, Toledano MB, Drapier JC. 2007. Regulation of peroxiredoxins by nitric oxide in immunostimulated macrophages. J Biol Chem 282:36199–36205. doi: 10.1074/jbc.M706420200. [DOI] [PubMed] [Google Scholar]
- 14.Power JH, Nicholas TE. 1999. Immunohistochemical localization and characterization of a rat Clara cell 26-kDa protein (CC26) with similarities to glutathione peroxidase and phospholipase A2. Exp Lung Res 25:379–392. doi: 10.1080/019021499270141. [DOI] [PubMed] [Google Scholar]
- 15.Kinnula VL, Lehtonen S, Kaarteenaho-Wiik R, Lakari E, Paakko P, Kang SW, Rhee SG, Soini Y. 2002. Cell specific expression of peroxiredoxins in human lung and pulmonary sarcoidosis. Thorax 57:157–164. doi: 10.1136/thorax.57.2.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stripp BR, Reynolds SD, Boe IM, Lund J, Power JH, Coppens JT, Wong V, Reynolds PR, Plopper CG. 2002. Clara cell secretory protein deficiency alters clara cell secretory apparatus and the protein composition of airway lining fluid. Am J Respir Cell Mol Biol 27:170–178. doi: 10.1165/ajrcmb.27.2.200200270c. [DOI] [PubMed] [Google Scholar]
- 17.Guo Y, Ma SF, Grigoryev D, Van Eyk J, Garcia JG. 2005. 1-DE MS and 2-D LC-MS analysis of the mouse bronchoalveolar lavage proteome. Proteomics 5:4608–4624. doi: 10.1002/pmic.200500052. [DOI] [PubMed] [Google Scholar]
- 18.Wu YZ, Manevich Y, Baldwin JL, Dodia C, Yu K, Feinstein SI, Fisher AB. 2006. Interaction of surfactant protein A with peroxiredoxin 6 regulates phospholipase A2 activity. J Biol Chem 281:7515–7525. doi: 10.1074/jbc.M504525200. [DOI] [PubMed] [Google Scholar]
- 19.Schremmer B, Manevich Y, Feinstein SI, Fisher AB. 2007. Peroxiredoxins in the lung with emphasis on peroxiredoxin VI. Subcell Biochem 44:317–344. doi: 10.1007/978-1-4020-6051-9_15. [DOI] [PubMed] [Google Scholar]
- 20.Chatterjee S, Feinstein SI, Dodia C, Sorokina E, Lien YC, Nguyen S, Debolt K, Speicher D, Fisher AB. 2011. Peroxiredoxin 6 phosphorylation and subsequent phospholipase A2 activity are required for agonist-mediated activation of NADPH oxidase in mouse pulmonary microvascular endothelium and alveolar macrophages. J Biol Chem 286:11696–11706. doi: 10.1074/jbc.M110.206623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ellison MA, Thurman GW, Ambruso DR. 2012. Phox activity of differentiated PLB-985 cells is enhanced, in an agonist specific manner, by the PLA2 activity of Prdx6-PLA2. Eur J Immunol 42:1609–1617. doi: 10.1002/eji.201142157. [DOI] [PubMed] [Google Scholar]
- 22.Bast A, Erttmann SF, Walther R, Steinmetz I. 2010. Influence of iNOS and COX on peroxiredoxin gene expression in primary macrophages. Free Radic Biol Med 49:1881–1891. doi: 10.1016/j.freeradbiomed.2010.09.015. [DOI] [PubMed] [Google Scholar]
- 23.Wang X, Phelan SA, Forsman-Semb K, Taylor EF, Petros C, Brown A, Lerner CP, Paigen B. 2003. Mice with targeted mutation of peroxiredoxin 6 develop normally but are susceptible to oxidative stress. J Biol Chem 278:25179–25190. doi: 10.1074/jbc.M302706200. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Phelan SA, Manevich Y, Feinstein SI, Fisher AB. 2006. Transgenic mice overexpressing peroxiredoxin 6 show increased resistance to lung injury in hyperoxia. Am J Respir Cell Mol Biol 34:481–486. doi: 10.1165/rcmb.2005-0333OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ali M, Umstead TM, Haque R, Mikerov AN, Freeman WM, Floros J, Phelps DS. 2010. Differences in the BAL proteome after Klebsiella pneumoniae infection in wild type and SP-A−/− mice. Proteome Sci 8:34. doi: 10.1186/1477-5956-8-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Varnum SM, Webb-Robertson BJ, Pounds JG, Moore RJ, Smith RD, Frevert CW, Skerrett SJ, Wunschel D. 2012. Proteomic analysis of bronchoalveolar lavage fluid proteins from mice infected with Francisella tularensis ssp. novicida. J Proteome Res 11:3690–3703. doi: 10.1021/pr3001767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamada Y, Limmon GV, Zheng D, Li N, Li L, Yin L, Chow VT, Chen J, Engelward BP. 2012. Major shifts in the spatio-temporal distribution of lung antioxidant enzymes during influenza pneumonia. PLoS One 7:e31494. doi: 10.1371/journal.pone.0031494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bertherat EG. 2015. Plague in Madagascar: overview of the 2014-2015 epidemic season. Wkly Epidemiol Rec 90:250–252. [PubMed] [Google Scholar]
- 29.Kwit N, Nelson C, Kugeler K, Petersen J, Plante L, Yaglom H, Kramer V, Schwartz B, House J, Colton L, Feldpausch A, Drenzek C, Baumbach J, DiMenna M, Fisher E, Debess E, Buttke D, Weinburke M, Percy C, Schriefer M, Gage K, Mead P. 2015. Human plague—United States, 2015. MMWR Morb Mortal Wkly Rep 64:918–919. doi: 10.15585/mmwr.mm6433a6. [DOI] [PubMed] [Google Scholar]
- 30.Lathem WW, Crosby SD, Miller VL, Goldman WE. 2005. Progression of primary pneumonic plague: a mouse model of infection, pathology, and bacterial transcriptional activity. Proc Natl Acad Sci U S A 102:17786–17791. doi: 10.1073/pnas.0506840102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Navarro L, Alto NM, Dixon JE. 2005. Functions of the Yersinia effector proteins in inhibiting host immune responses. Curr Opin Microbiol 8:21–27. doi: 10.1016/j.mib.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 32.Price PA, Jin J, Goldman WE. 2012. Pulmonary infection by Yersinia pestis rapidly establishes a permissive environment for microbial proliferation. Proc Natl Acad Sci U S A 109:3083–3088. doi: 10.1073/pnas.1112729109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sebbane F, Lemaitre N, Sturdevant DE, Rebeil R, Virtaneva K, Porcella SF, Hinnebusch BJ. 2006. Adaptive response of Yersinia pestis to extracellular effectors of innate immunity during bubonic plague. Proc Natl Acad Sci U S A 103:11766–11771. doi: 10.1073/pnas.0601182103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zimbler DL, Schroeder JA, Eddy JL, Lathem WW. 2015. Early emergence of Yersinia pestis as a severe respiratory pathogen. Nat Commun 6:7487. doi: 10.1038/ncomms8487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lathem WW, Price PA, Miller VL, Goldman WE. 2007. A plasminogen-activating protease specifically controls the development of primary pneumonic plague. Science 315:509–513. doi: 10.1126/science.1137195. [DOI] [PubMed] [Google Scholar]
- 36.Zhang SS, Park CG, Zhang P, Bartra SS, Plano GV, Klena JD, Skurnik M, Hinnebusch BJ, Chen T. 2008. Plasminogen activator Pla of Yersinia pestis utilizes murine DEC-205 (CD205) as a receptor to promote dissemination. J Biol Chem 283:31511–31521. doi: 10.1074/jbc.M804646200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suomalainen M, Haiko J, Ramu P, Lobo L, Kukkonen M, Westerlund-Wikstrom B, Virkola R, Lahteenmaki K, Korhonen TK. 2007. Using every trick in the book: the Pla surface protease of Yersinia pestis. Adv Exp Med Biol 603:268–278. doi: 10.1007/978-0-387-72124-8_24. [DOI] [PubMed] [Google Scholar]
- 38.Degen JL, Bugge TH, Goguen JD. 2007. Fibrin and fibrinolysis in infection and host defense. J Thromb Haemost 5(Suppl 1):24–31. doi: 10.1111/j.1538-7836.2007.02519.x. [DOI] [PubMed] [Google Scholar]
- 39.Kukkonen M, Lahteenmaki K, Suomalainen M, Kalkkinen N, Emody L, Lang H, Korhonen TK. 2001. Protein regions important for plasminogen activation and inactivation of alpha2-antiplasmin in the surface protease Pla of Yersinia pestis. Mol Microbiol 40:1097–1111. doi: 10.1046/j.1365-2958.2001.02451.x. [DOI] [PubMed] [Google Scholar]
- 40.Kukkonen M, Suomalainen M, Kyllonen P, Lahteenmaki K, Lang H, Virkola R, Helander IM, Holst O, Korhonen TK. 2004. Lack of O-antigen is essential for plasminogen activation by Yersinia pestis and Salmonella enterica. Mol Microbiol 51:215–225. [DOI] [PubMed] [Google Scholar]
- 41.Haiko J, Laakkonen L, Juuti K, Kalkkinen N, Korhonen TK. 2010. The omptins of Yersinia pestis and Salmonella enterica cleave the reactive center loop of plasminogen activator inhibitor 1. J Bacteriol 192:4553–4561. doi: 10.1128/JB.00458-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sodeinde OA, Subrahmanyam YV, Stark K, Quan T, Bao Y, Goguen JD. 1992. A surface protease and the invasive character of plague. Science 258:1004–1007. doi: 10.1126/science.1439793. [DOI] [PubMed] [Google Scholar]
- 43.Caulfield AJ, Walker ME, Gielda LM, Lathem WW. 2014. The Pla protease of Yersinia pestis degrades fas ligand to manipulate host cell death and inflammation. Cell Host Microbe 15:424–434. doi: 10.1016/j.chom.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Finegold MJ. 1969. Pneumonic plague in monkeys. An electron microscopic study. Am J Pathol 54:167–185. [PMC free article] [PubMed] [Google Scholar]
- 45.Cabral MP, Soares NC, Aranda J, Parreira JR, Rumbo C, Poza M, Valle J, Calamia V, Lasa I, Bou G. 2011. Proteomic and functional analyses reveal a unique lifestyle for Acinetobacter baumannii biofilms and a key role for histidine metabolism. J Proteome Res 10:3399–3417. doi: 10.1021/pr101299j. [DOI] [PubMed] [Google Scholar]
- 46.Shifman MA, Li Y, Colangelo CM, Stone KL, Wu TL, Cheung KH, Miller PL, Williams KR. 2007. YPED: a web-accessible database system for protein expression analysis. J Proteome Res 6:4019–4024. doi: 10.1021/pr070325f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eddy JL, Gielda LM, Caulfield AJ, Rangel SM, Lathem WW. 2014. Production of outer membrane vesicles by the plague pathogen Yersinia pestis. PLoS One 9:e107002. doi: 10.1371/journal.pone.0107002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ambruso DR. 2013. Peroxiredoxin-6 and NADPH oxidase activity. Methods Enzymol 527:145–167. doi: 10.1016/B978-0-12-405882-8.00008-8. [DOI] [PubMed] [Google Scholar]
- 49.Mo Y, Feinstein SI, Manevich Y, Zhang Q, Lu L, Ho YS, Fisher AB. 2003. 1-Cys peroxiredoxin knock-out mice express mRNA but not protein for a highly related intronless gene. FEBS Lett 555:192–198. doi: 10.1016/S0014-5793(03)01199-2. [DOI] [PubMed] [Google Scholar]
- 50.Eddy JL, Schroeder JA, Zimbler DL, Bellows LE, Lathem WW. 2015. Impact of the Pla protease substrate α2-antiplasmin on the progression of primary pneumonic plague. Infect Immun 83:4837–4847. doi: 10.1128/IAI.01086-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Caulfield AJ, Lathem WW. 2012. Substrates of the plasminogen activator protease of Yersinia pestis. Adv Exp Med Biol 954:253–260. doi: 10.1007/978-1-4614-3561-7_32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Benipal B, Feinstein SI, Chatterjee S, Dodia C, Fisher AB. 2015. Inhibition of the phospholipase A2 activity of peroxiredoxin 6 prevents lung damage with exposure to hyperoxia. Redox Biol 4:321–327. doi: 10.1016/j.redox.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim SY, Chun E, Lee K-Y. 2011. Phospholipase A2 of peroxiredoxin 6 has a critical role in tumor necrosis factor-induced apoptosis. Cell Death Differ 18:1573–1583. doi: 10.1038/cdd.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pechous RD, Sivaraman V, Price PA, Stasulli NM, Goldman WE. 2013. Early host cell targets of Yersinia pestis during primary pneumonic plague. PLoS Pathog 9:e1003679. doi: 10.1371/journal.ppat.1003679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lathem WW, Schroeder JA, Bellows LE, Ritzert JT, Koo JT, Price PA, Caulfield AJ, Goldman WE. 2014. Posttranscriptional regulation of the Yersinia pestis cyclic AMP receptor protein Crp and impact on virulence. mBio 5:e01038-13. doi: 10.1128/mBio.01038-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Agarkov A, Chauhan S, Lory PJ, Gilbertson SR, Motin VL. 2008. Substrate specificity and screening of the integral membrane protease Pla. Bioorg Med Chem Lett 18:427–431. doi: 10.1016/j.bmcl.2007.09.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen JW, Dodia C, Feinstein SI, Jain MK, Fisher AB. 2000. 1-Cys peroxiredoxin, a bifunctional enzyme with glutathione peroxidase and phospholipase A2 activities. J Biol Chem 275:28421–28427. doi: 10.1074/jbc.M005073200. [DOI] [PubMed] [Google Scholar]
- 58.Pradel E, Lemaitre N, Merchez M, Ricard I, Reboul A, Dewitte A, Sebbane F. 2014. New insights into how Yersinia pestis adapts to its mammalian host during bubonic plague. PLoS Pathog 10:e1004029. doi: 10.1371/journal.ppat.1004029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.