

Abstract
RNA helicases have been shown to be important for the function of RNA molecules at several levels, although their putative involvement in microbial pathogenesis has remained elusive. We have previously shown that Listeria monocytogenes DExD-box RNA helicases are important for bacterial growth, motility, ribosomal maturation, and rRNA processing. We assessed the importance of the RNA helicase Lmo0866 (here named CshA) for expression of virulence traits. We observed a reduction in hemolytic activity in a strain lacking CshA compared to the wild type. This phenomenon was less evident in strains lacking other RNA helicases. The reduced hemolysis was accompanied by lower expression of major listerial virulence factors in the ΔcshA strain, mainly listeriolysin O, but also to some degree the actin polymerizing factor ActA. Reduced expression of these virulence factors in the strain lacking CshA did not, however, correlate with a decreased level of the virulence regulator PrfA. When combining the ΔcshA knockout with a mutation creating a constitutively active PrfA protein (PrfA*), the effect of the ΔcshA knockout on LLO expression was negated. These data suggest a role for the RNA helicase CshA in posttranslational activation of PrfA. Surprisingly, although the expression of several virulence factors was reduced, the ΔcshA strain did not demonstrate any reduced ability to infect nonphagocytic cells compared to the wild-type strain.
INTRODUCTION
RNA helicases play important roles in RNA metabolism (1–7). RNA helicases are ubiquitous enzymes found in all kingdoms of life (8, 9) and are subdivided into DExD and DExH types depending on catalytic site consensus sequence (10, 11). These enzymes have been found to unwind structured RNA and RNA duplexes by hydrolyzing ATP (12). In addition, RNA helicases have been suggested to be involved in translation elongation (13–15). Involvement of RNA helicases during RNA maturation and degradation has been shown in Escherichia coli (RhlB and RhlE), Bacillus subtilis (CshA, CshB, DeaD, and YfmL), and Staphylococcus aureus (CshA) where they also been shown to associate with RNA degradosome components (14, 16–23). One RNA helicase in Listeria monocytogenes, Lmo1722, was shown to be important for ribosome biogenesis and interacted with the 50S subunit through its C terminus (6). Not surprisingly, the absence of Lmo1722 and other listerial RNA helicases has the largest effect at low temperatures and under adverse stress conditions (6, 15, 24–27).
The human pathogen Listeria monocytogenes is the causative agent of listeriosis, which has a mortality rate of 20 to 30% in immunocompromised patients (28). Other high-risk groups are elderly, pregnant women and newborns (29). Even though listeriosis has a low incidence rate and may occur sporadically, outbreaks of listeriosis are increasing worldwide and therefore becoming a public health issue, especially as the average population age is increasing (30). Listeria monocytogenes is a food-borne pathogen and a major concern for the food industry due to its ability to grow at very low temperatures, such as refrigeration temperature (31, 32).
Only a few prokaryotic RNA helicases have previously been implicated in virulence. Two of these are DExH-box RNA helicases: an Escherichia coli strain deficient in HrpA showed altered stability of the mRNA daaE, which codes for a fimbriae subunit (33). The absence of HrpA or mutations in its ATPase or RNA-binding motif decreased mouse infectivity in the spirochete Borrelia burgdorferi (34). The DExD-box RNA helicase CshA of Staphylococcus aureus was shown to affect the stability of the agr mRNA, resulting in increased levels of RNAIII and elevated hemolysin production in the cshA mutant (14). In Pseudomonas aeruginosa, the DEAD box RNA helicase (DeaD) was required for efficient translation of the type III secretion regulator ExsA (35). Recently, the absence of different RNA helicases was shown to affect virulence factor expression in Listeria monocytogenes and a role for the RNA helicases during translation of LLO was suggested (15). mRNA regulation is also crucial for modifying the switch for Listeria monocytogenes to a pathogenic lifestyle. Expression of the majority of virulence factors important for host cell invasion (InlA and InlB), phagosomal escape (LLO, PlcA, and PlcB), cytosolic replication (UhpT), and direct cell-to-cell spread (ActA) are all controlled by the transcriptional regulator PrfA, which functions using complex posttranscriptional regulation (36–40).
In the present study, we show that a Listeria monocytogenes strain deficient in one DExD-box RNA helicase, Lmo0866 (here named CshA due to its homology to CshA in S. aureus and B. subtilis) (15), displays a significant decrease in hemolysis coupled with reduction in listeriolysin O (LLO), the pore-forming cytolysin. Also, the expression of ActA was reduced in the strain lacking CshA, although the effect was less evident. Interestingly, a reduction in virulence factor expression in the ΔcshA strain was not accompanied by decreased expression of PrfA, the transcriptional regulator activating virulence gene expression. The reduced expression of virulence factors observed in the ΔcshA strain could be rescued by introducing a constitutively active PrfA protein, indicating a role for CshA during PrfA posttranslational activation. Despite decreased virulence factor expression, the ΔcshA strain was able to infect nonphagocytic cells with an efficiency similar to that of the wild-type (WT) strain, suggesting a specific role for this RNA helicase in activating PrfA under certain environmental conditions.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Bacterial strains are listed in Table 1. Bacteria were grown aerobically at 37°C at 150 rpm (Innova 3100; New Brunswick Scientific) in brain heart infusion (BHI) medium (broth or agar; Fluka) supplemented with 0.15 M NaCl, adjusted to pH 5.5 or pH 7.6. Bacterial growth was monitored using an Ultrospec 2100pro (GE), and the bacteria were harvested at an optical density at 600 nm (OD600) of 0.9 before further analysis.
TABLE 1.
Bacterial strains used in this study
| Strain | Genotype | Source or reference |
|---|---|---|
| Escherichia coli DH5α | 64 | |
| Listeria monocytogenes | ||
| WT | Wild-type L. monocytogenes strain EGDe | 65 |
| Δlmo1722 mutant | EGDe with lmo1722 deletion | 6 |
| Δlmo1450 mutant | EGDe with lmo1450 deletion | 15 |
| Δlmo1246 mutant | EGDe with lmo1246 deletion | 15 |
| ΔcshA mutant | EGDe with cshA (lmo0866) deletion | 15 |
| Δ4 quadruple mutant | EGDe with lmo1722, lmo1246, cshA, and lmo1450 deletions | 15 |
| ΔprfA mutant | EGDe with prfA deletion | 48 |
| Δhly mutant | EGDe with hly deletion | 56 |
| ΔactA mutant | EGDe with actA deletion | 56 |
| EGDe::pIMK3 mutant | EGDe with plasmid vector pIMK3 | 15 |
| ΔcshA::pIMK3 mutant | EGDe with cshA deletion and carrying plasmid vector pIMK3 | 15 |
| ΔcshA::pcshA mutant | EGDe with cshA deletion and complemented with plasmid vector pIMK3 carrying cshA | 15 |
| KAR987 | EGDe strain with prfA(G145S) allele | This study |
| KAR990 | EGDe ΔcshA strain with prfA(G145S) allele | This study |
Genetic manipulations.
Single and quadruple knockout strains, as well as complemented strains, were constructed as described previously (6, 15). Construction of the PrfAG145S substitution mutant was performed as follows: 10 ng of plasmid pLis35 (41) was amplified by PCR with Phusion DNA polymerase (Thermo Scientific) using the primer pair PrfA-G145S-F and PrfA-G145-R (42) and digested overnight with 10 U of DpnI (Thermo Scientific) according to the manufacturer's instructions. E. coli strain DH5α was transformed with the resulting reaction mix and the sequence of the prfA gene carrying the G145S substitution was verified by sequencing the insert in the resulting plasmid, pKVA609. The prfA G145S allele DNA fragment was excised from pKVA609 using PstI endonuclease, blunt end treated using mung bean nuclease (New England BioLabs), and ligated into the SmaI site of the pMAD vector to construct the allelic replacement plasmid pKVA973. Replacement of the prfA allele with the prfA(G145S) allele (prfA*) was performed using the plasmid pKVA973 in the L. monocytogenes EGDe and ΔcshA strains as described previously (43).
Hemolytic analysis.
Human venous blood was acquired from healthy donors into Vacutainer citrate tubes (BD) containing 3.2% buffered sodium citrate solution. The plasma and buffy coat were removed by pipetting before use. One milliliter of red blood cells (RBCs) was suspended in 9 ml of 1× phosphate-buffered saline (PBS; pH 7.4) and centrifuged at 900 × g for 15 min. The supernatant was removed using a 10-ml pipette. The precipitated pellet of RBCs was resuspended and washed in 1× PBS three more times until the supernatant was visibly clear. The cells were then resuspended in 1× PBS. The hemolytic activity was assayed as described previously, with the following minor modifications (44). After growth to an OD600 of 0.9 (see above), 6 ml of bacterial culture was harvested and filtered through a Millex GV Durapore polyvinylidene difluoride membrane filter (0.22-μm pore size). Then, 1 ml of supernatant was used for protein preparation. Dithiothreitol (DTT; Sigma-Aldrich) was added at 2 mM concentration to fully reduce LLO and increase the fraction of active LLO. 100 μl of filtered suspension was added to each well, and an equivalent volume of 10% RBC solution was supplemented at a final 1:1 ratio into a 96-well, clear, round-bottom microtiter plate (Sarstedt), followed by incubation at 37°C for 3 h. Sodium dodecyl sulfate (SDS) at 0.1% (Sigma-Aldrich) was used as a positive control, and BHI was used as a negative control. The supernatant was transferred after centrifugation at 3,250 × g for 5 min into a new microtiter plate, and the absorbance was measured in a Tecan microplate reader Infinite 2000 PRO series at 541 nm.
Protein preparation. (i) Detection of LLO or secreted P60.
Bacteria were grown as described above, and the bacterial culture supernatant was harvested at an OD600 of 0.9 and filtered. Next, 1 ml of sterile-filtered supernatant was mixed with 10 μl of 2% sodium deoxycholate (Sigma-Aldrich) for 10 min at room temperature before precipitation using 250 μl of 50% trichloroacetic acid (Sigma-Aldrich) and left on ice for 1 h. After centrifugation, at 20,800 × g at 4°C for 30 min, the supernatant was removed, and the pellet was resuspended in 580 μl of 80% ice-cold acetone. The suspension was centrifuged at 20,800 × g at 4°C for 30 min before the pellet was dried and resuspended in 15 μl of 1× Laemmli sample buffer (62.5 mM Tris-HCl [pH 6.8], 2% [wt/vol] SDS, 10% glycerol, 5% β-mercaptoethanol, 0.001% bromophenol blue) (45).
(ii) Detection of ActA.
Surface proteins were extracted by boiling bacterial pellet suspended in 1× Laemmli sample buffer for 20 min. After centrifugation at 20,800 × g for 5 min, the supernatant was collected and used for SDS-PAGE separation (see below).
(iii) Detection of InlB and P60.
Bacteria were grown as described above and bacteria were harvested at an OD600 of 0.9 before 10 ml was centrifuged at 6,000 × g for 10 min. The pellet was dissolved in 500 μl of buffer A (200 mM KCl, 50 mM Tris-HCl [pH 8.0], 1 mM EDTA, 10% glycerol, 1 mM DTT). The sample was transferred to beater tubes with 0.4 g of glass beads. The bacteria were disrupted using a mini bead beater (Biospec products) for 1 min. After centrifugation (5 min, 4°C at 13,000 × g), the upper phase was harvested and used for SDS-PAGE separation.
(iv) Detection of PrfA.
Detection of PrfA was performed as described previously with minor modifications (46). Bacteria were grown as described above, and bacteria were harvested at an OD600 of 0.9 before 2 ml was centrifuged at 20,800 × g for 5 min. The pellet was washed in 500 μl of wash buffer (30 mM Tris-HCl [pH 8.0], 50 mM NaCl, 5 mM EDTA), suspended in 100% ice-cold acetone, and kept on ice for 10 min. The sample was centrifuged at 20,800 × g at 4°C for 30 min, and the pellet was dried. The pellet was resuspended in Mutanolysin mix (0.5 U/μl) with DNase (0.2 U/μl) and left on a 37°C heat block for 30 min before vortexing for 5 min. The sample was mixed with 4× Laemmli buffer and boiled at 95°C for 10 min. For surface protein detection, 50 μl of 4× Laemmli buffer was added to 1 ml of pelleted bacterial sample, followed by boiling for 5 min, and briefly vortexed before SDS-PAGE separation.
SDS-PAGE and Western blotting.
Protein samples were separated on SDS-polyacrylamide gels (Bio-Rad) before transfer onto polyvinylidene difluoride membranes using a wet transfer apparatus (Bio-Rad). Membranes were blocked in 5% dry milk at 4°C overnight. Primary antibodies were diluted (anti-PrfA, 1:3,000; anti-LLO, 1:2,000; anti-ActA, 1:4,000; anti-InlB, 1:3,000; anti-P60, 1:1,500; and anti-InlB, 1:10,000) before incubation at room temperature for 1 h or at 4°C overnight. Membranes were washed before incubation at room temperature with anti-rabbit-horseradish peroxidase secondary antibody, diluted 1:3,000. The expression levels of each protein (the intensity of chemiluminescence) were detected and measured using a LAS4000 image analyzer (Fuji).
Cell culture and infection.
J774 cells (J774A.1, ATCC TIB-67) and Caco-2 cells (ATCC HTB-37; LGC Standards) were grown in Dulbecco modified Eagle minimum essential medium (DMEM; Gibco) supplemented with 10% fetal calf serum at 37°C in the presence of 5% CO2. Caco-2 cells were additionally supplemented with nonessential amino acids and seeded on BD BioCoat collagen-coated 24-well plates (Corning, Tewksbury, MA). J774 cells were seeded on Thermo Scientific 24-well plates. Cells were grown for 24 h to confluence. Bacteria were grown as described above in BHI broth and harvested at an OD600 of 0.9, before centrifugation at 3,443 × g for 5 min. The bacteria were washed once in 1× PBS and diluted in DMEM before being used to infect cells at an MOI of ∼10. J774 cells and bacteria were centrifuged for 10 min at 800 × g, and the bacteria were allowed to infect for 1 h. Caco-2 cells and bacteria were not centrifuged prior to allowing bacteria to infect for 1 h. At 1 h postinfection, the cells were washed twice with DMEM, the Caco-2 cells were incubated for another hour or 8 h and J774 for 1 or 3 h with 150 μg of gentamicin/ml to kill extracellular bacteria and washed twice in PBS (Gibco). Internalized bacteria were released by lysing with 1% Triton X-100 (Sigma-Aldrich). Viable bacterial counts were determined by plating different cell concentrations on tryptic soy agar plates and growth for 24 h at 37°C prior to counting.
Immunohistochemistry.
Caco-2 cells were seeded on 12-mm glass coverslips (Thermo scientific) coated with collagen type 1, Rat Tail (BD Biosciences). J774 cells was seeded on glass slides without collagen in DMEM (Gibco) supplemented with 10% fetal calf serum at 37°C in the presence of 5% CO2. The cells were grown for 24 h to confluence and infected with bacteria that were grown as described above in BHI broth and harvested at an OD600 of 0.9. J774 cells were infected for 1 or 3 h, and Caco-2 cells were infected for 1 or 8 h. For intracellular visualization of bacterial infection, the cells were fixed using 4% paraformaldehyde and permeabilized using 0.5% Triton X-100 after infection. The cells were blocked with 0.5% bovine serum albumin (Sigma-Aldrich) for 1 h at 37°C, before incubation with primary anti-Listeria monocytogenes antibody (1:200; Becton Dickinson) for 1 h at 37°C. Secondary antibody goat anti-rabbit IgG(H&L) 488 Alexa Fluor (1:200; Agrisera) was added and allowed to incubate for 1 h at 37°C. DAPI (4′,6′-diamidino-2-phenylindole; Invitrogen) was added for 5 min. The coverslips were mounted with Mowiol (Calbiochem) on glass slides and left to dry at 37°C. Intracellular bacteria were visualized with a Nikon 90i Eclipse microscope using NIS-E AR software.
RNA isolation.
Bacteria were cultured as described above and grown to an OD600 of 0.9 before being mixed with 0.2 volumes of 5% phenol in 95% ethanol (47), and bacteria were harvested by centrifugation. Bacterial pellets were frozen in liquid nitrogen and stored at −80°C. RNA from L. monocytogenes was isolated using a modification of the guanidinium thiocyanate-phenol-chloroform extraction method (48). Briefly, total cellular RNA was isolated from L. monocytogenes by dissolving pelleted cultures in resuspension solution (10% glucose, 12.5 mM Tris [pH 7.6], 70 mM EDTA). Samples were immediately transferred to bead beater tubes with roughly 0.4 g of glass beads and 500 μl of acid phenol (pH 4.5). The bacteria were disrupted using a mini-bead beater (Biospec Products) for 75 s. After centrifugation (15 min at 20,800 × g), RNA was recovered by the addition of 1 ml of TRI-Reagent solution (Ambion) and 100 μl of chloroform, followed by centrifugation. Samples were thereafter subjected to two additional chloroform/isoamylalcohol (IAA) extractions. The aqueous phase was precipitated by adding isopropanol (0.7×), followed by incubation at −20°C for 20 min. To collect the pellet, the RNA samples were centrifuged for 25 min at 20,800 × g. The pellet was dissolved in 200 μl of RNase-free water. To remove the remaining DNA, samples were treated with 20 U of DNase I (Roche) for 45 min at 37°C. The reaction was terminated by the addition of phenol-chloroform/IAA (1:24:1, pH 6.6). Centrifuged samples were chloroform/IAA extracted and ethanol precipitated. The pellet was resuspended in 200 μl of RNase-free water, and the RNA concentration was measured using a NanoDrop spectrophotometer (ND-1000). The RNA integrity was determined by electrophoresis on a 1.2% agarose gel. Only RNA samples showing distinct nonprocessed precursors to rRNA were used in the following experiments.
Northern blotting.
Twenty micrograms of RNA was separated on a 1.2% agarose gel containing 1× HEPES buffer (10× HEPES buffer = 0.2 M HEPES, 50 mM sodium acetate, and 10 mM EDTA, adjusted to pH 7) and 7.3% formaldehyde. The gel was run in 1× HEPES buffer at 100 V for 4 h, and the RNA was transferred to a Hybond-N membrane (Amersham) by capillary transfer in 20× SSC buffer (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate). The membranes were hybridized at 60°C overnight with [α-32P]dATP-labeled DNA fragments amplified with corresponding primers (Table 2) using a Prime-a-Gene DNA labeling system (Promega). Membranes were washed (0.5% SDS–2× SSC at room temperature for 15 min, followed by 0.5% SDS–0.1× SSC at 60°C for 15 min), exposed to a phosphorimager cassette, and developed using a Storm imager (Molecular Dynamics) or a Typhoon FLA9500 (GE).
TABLE 2.
Oligonucleotides used in this study
| Oligonucleotide | Sequence 5′-3′ | Reference |
|---|---|---|
| tmRNA-U | CGGCACTTAATATCTACGAGC | |
| tmRNA-D | CCTCGTTATCAACGTCAAAGCC | |
| hly-U | GAAGCAAAGGATGCATCTGC | |
| hly-D | CCATCTTTGTAACCTTTTCTTGG | |
| PrfA-G145S-F | GGAAGCTTGGCTCTATTTGCTCTCAACTTTTAATCCTGACC | 42 |
| PrfA-G145S-R | GTCAGGATTAAAAGTTGAGAGCAAATAGAGCCAAGCTTCC | 42 |
| inlA-U | GCAATATTAGTATTGGCAGCG | |
| inlA-D | CTAGATCTGTTTGTGAGACCG |
Glutathione fluorometric assay.
The method was performed according to the assay kit (glutathione fluorometric assay kit (K264-100; BioVision) with minor modifications. Bacteria were grown as described above, and bacteria were harvested at an OD600 of 0.9 before 20 ml of culture was centrifuged at 6,000 × g for 10 min. The bacteria pellet was washed three times in 1× PBS and then dissolved in 200 μl of ice-cold glutathione assay buffer. The sample was transferred to beater tubes with roughly 0.2 g of glass beads. The bacteria were disrupted using a mini-bead beater (Biospec Products) for 1 min. After centrifugation (5 min, 4°C at 13,000 × g), a 60-μl sample was transferred to ice-cold tubes containing 20 μl of 6 N perchloric acid (PA). To precipitate PA and neutralize the samples, 20 μl of ice-cold 6 N KOH was added to a 40-μl sample before centrifugation for 2 min at 4°C (13,000 × g). Then, 10 μl of sample, together with 90 μl of assay buffer, was transferred to a 96-well plate before 10 μl of OPA probe (o-phthalaldehyde) was added, and the samples were incubated for 40 min. To determine the amount of glutathione, the samples were read on a fluorescence plate reader (Tecan Infinite M200) set at excitation/emission wavelengths of 340/420 nm and correlated to a standard curve.
RESULTS
Listeria monocytogenes lacking the DExD-box RNA helicase CshA has reduced hemolytic activity.
We have previously observed a role for the DExD-box RNA helicase Lmo1722 during growth, motility, and ribosomal maturation; the latter function presumably occurs through its interaction with the 50S subunit of the ribosome (6). In that study, we were, however, unable to detect any role for Lmo1722 during listerial pathogenesis. Also, in another study, we observed involvement of the RNA helicases in virulence factor expression (15). To further analyze a putative role for RNA helicases during infection, different mutant strains were tested for their hemolytic activity. L. monocytogenes hemolysis is primarily caused by the pore-forming cytolysin, listeriolysin O (LLO), which is most active at pH 5.5 (49–52), a condition the bacterium encounters in the phagosome (52, 53). Hemolytic activity was therefore assessed at pH 5.5, using human red blood cells in the presence of supernatants from Listeria strains with different genetic backgrounds (Fig. 1). Analyzing single-knockout mutants of listerial RNA helicases showed that the absence of Lmo1722, Lmo1450, or Lmo1246 only marginally affected hemolysis compared to the wild-type strain. In contrast, the absence of CshA dramatically decreased hemolysis to a level comparable to that of a strain lacking the master regulator of virulence, PrfA, or the pore-forming cytolysin, LLO (Fig. 1). The strain lacking all RNA helicases (Δ4) also displayed reduced hemolysis, albeit not to the level observed in the ΔcshA strain.
FIG 1.
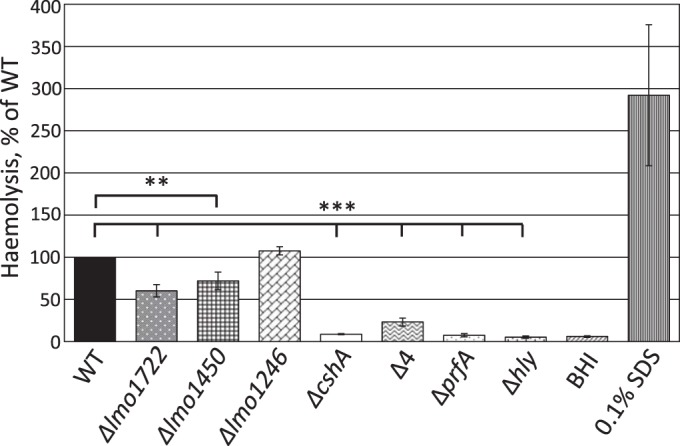
Hemolytic activity at pH 5.5. The indicated strains were grown at 37°C to an OD600 of 0.9 in pH-adjusted BHI (pH 5.5) before the supernatant was removed. Filtered supernatants were added at a 1:1 ratio to a 10% suspension of red blood cells, followed by incubation for 3 h at 37°C. The absorbance was measured at 541 nm. The figure shows the hemolytic activity of the indicated strains in percentages relative to the wild type (100%) with the standard deviations. All samples were compared to the wild type using a two-tailed Student t test (**, P < 0.01; ***, P < 0.001).
We next sought to determine whether CshA-dependent hemolytic activity was exclusively manifested at low pH or whether the effect could also be detected at a higher pH. The difference in hemolytic activity between wild-type and ΔcshA strains was less pronounced at pH 7.6 compared to pH 5.5, suggesting that CshA is more important for virulence factor expression at a lower pH (Fig. 2). The reduced hemolysis could be reestablished to wild-type levels in the ΔcshA strain expressing CshA from another location of the chromosome, clearly demonstrating that the reduced hemolysis was due to the RNA helicase itself, not to any downstream effect (see Fig. S1 in the supplemental material) (15). The ΔcshA strain grew ca. 20% slower compared to the wild type (Table 3), but the growth rate could be reestablished to wild-type levels in a ΔcshA strain expressing CshA in trans (Table 3).
FIG 2.
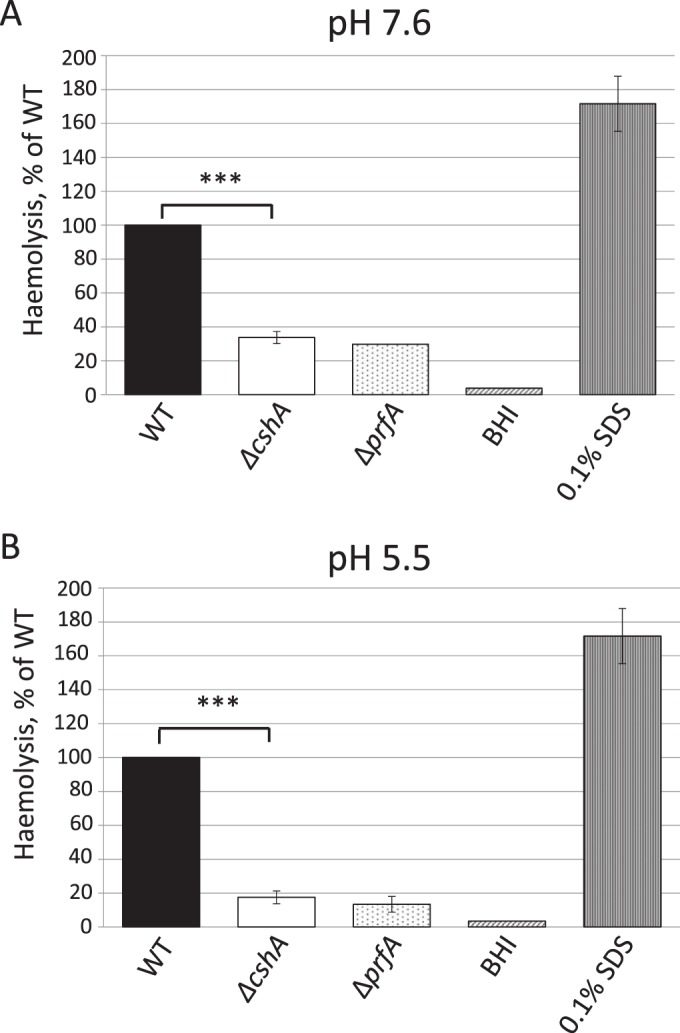
Hemolytic activity at different pH levels. The indicated strains were grown at 37°C to an OD600 of 0.9 in BHI of pH 7.6 (A) or pH 5.5 (B) before the supernatant was removed. Filtered supernatants were added to a 10% suspension of red blood cells at a 1:1 ratio and incubated for 3 h at 37°C. The absorbance was measured at 541 nm. The figure shows the hemolytic activity of the indicated strains relative to the wild type (100%) with the standard deviations. All samples were compared to the wild type using a two-tailed Student t test (***, P < 0.001).
TABLE 3.
Growth rate of the indicated bacterial strains at 37°C in BHI at pH 5.5
| Strain | Avg doubling time (min) ± SD |
|---|---|
| WT | 48 ± 1.0 |
| ΔcshA mutant | 59 ± 1.8 |
| Δhly mutant | 51 ± 1.3 |
| ΔprfA mutant | 48 ± 3.1 |
| WT::pIMK3 | 51 ± 1.3 |
| ΔcshA::pIMK3 mutant | 60 ± 1.1 |
| ΔcshA::pcshA mutant | 51 ± 1.1 |
Absence of CshA reduces the level of LLO and ActA but not of the transcriptional regulator PrfA.
Since the Listeria strain lacking CshA was not hemolytic (Fig. 1), we next examined whether the reduced hemolytic activity in this strain was accompanied by a reduction in LLO levels. The absence of CshA decreased LLO expression compared to the wild-type strain (Fig. 3A, upper panel, and Fig. 3B). Also, LLO expression could be reestablished in the ΔcshA strain expressing CshA in trans, in contrast to the ΔcshA strain carrying the vector construct. Since RNA helicases have been suggested to participate in various steps of posttranscriptional events (9), it could be hypothesized that CshA aids translation of the hly transcript (encoding LLO), an idea that has been suggested previously (15). To examine this, we first examined the level of hly transcript in the wild-type, ΔcshA, and ΔprfA strains (see Fig. S2A in the supplemental material). The results show that absence of CshA decreased hly expression to a level observed in a strain lacking the transcriptional regulator PrfA. Hence, our results suggest that CshA is important for transcriptional activation of hly rather than translation of the hly transcript. Most of the virulence factors in L. monocytogenes are controlled by the virulence transcriptional regulator PrfA (38, 39, 54, 55). To examine whether expression of other PrfA regulated genes were also altered in the ΔcshA strain, we analyzed the level of ActA, the factor responsible for intracellular actin-polymerization as well as for bacterial aggregation and stress perception (48, 56). The level of ActA was reduced in the strain lacking CshA compared to the wild-type strain, but the difference was not as pronounced as the reduction in LLO expression (Fig. 3A, middle panel, and Fig. 3B). ActA expression could be restored to wild-type levels in the strain expressing CshA in trans. We also examined whether expression of one adhesin, InlB was affected in a ΔcshA strain. The absence of CshA did not affect InlB expression, although the expression of the inlB gene (as well as the gene encoding internalin, inlA) appeared to be increased (see Fig. S3 in the supplemental material). The expression and activity of the transcriptional activator PrfA is controlled at the transcriptional, translational, and posttranslational levels (54, 55). Hypothetically, the reduced expression of virulence factors observed in the ΔcshA strain could be due to decreased levels of PrfA or, alternatively, the activity of PrfA could be lowered. To test these possibilities, the PrfA level was examined in various strains (Fig. 3, lower panel). We were unable to identify any differences in PrfA levels among the bacterial strains tested, suggesting that the activity of PrfA rather than its expression level was reduced in the ΔcshA strain compared to the wild-type strain.
FIG 3.
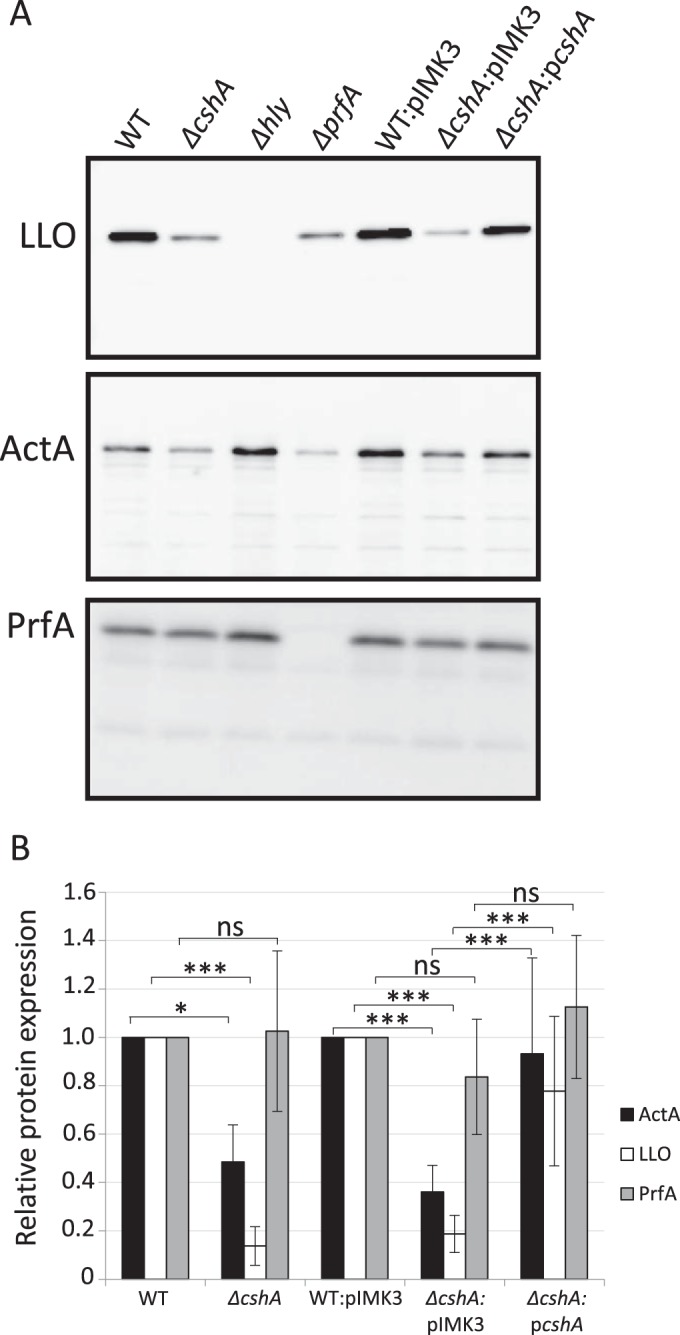
Virulence factor expression in different strain backgrounds. (A) The indicated strains were grown at 37°C in pH-adjusted BHI (pH 5.5) to an OD600 of 0.9 before protein extraction, SDS-PAGE separation, and Western blot analysis. In the upper panel, the expression levels of LLO were analyzed using LLO-specific antibodies in samples of protein precipitated by trichloroacetic acid from filtered culture supernatants. In the middle panel, the expression levels of ActA were examined by extraction of boiled bacterial cells in Laemmli buffer using ActA-specific antibodies. In the lower panel, the PrfA level was examined from whole-cell fractions using PrfA-specific antibodies. (B) The expression of LLO, ActA, and PrfA, respectively, was quantified from panel A, and the results are shown relative to the wild type (WT) for EGDe or ΔcshA or to the WT::pIMK3 strain for WT::pIMK3, ΔcshA::pIMK3, or ΔcshA::pcshA, respectively. WT and WT::pIMK3 were arbitrarily set to 1.0. Error bars show the standard deviations. Statistics show two-tailed Student t test determinations (*, P < 0.05; ***, P < 0.001).
A constitutively active PrfA protein overcomes the regulatory effect of CshA deficiency.
PrfA has been suggested to require posttranslational modification for activation. This activation has long remained elusive, but glutathione was recently discovered to directly bind to and activate PrfA (57). In light of this, we examined whether absence of CshA affected glutathione levels. No significant difference in the level of glutathione could be observed in a strain lacking CshA compared to the wild-type strain (Fig. 4A). To further examine the effect of CshA on PrfA activity, we made a constitutively active PrfA construct (PrfAG145S) expressed from its native site on the chromosome. The PrfAG145S protein has a stabilized helix-turn-helix motif that increases its DNA-binding affinity compared to the wild type (42). As a consequence, a strain carrying PrfAG145S continuously expresses PrfA-regulated genes. PrfAG145S has previously been used to examine whether amino acid substitutions at other positions would affect PrfA activity (58). We reasoned that if CshA affects any step in the activation of PrfA, this effect should be bypassed in a strain with a constitutively active PrfA protein. On the other hand, if the PrfA regulatory effect still can be observed in a ΔcshA deletion strain expressing PrfAG145S, it would indicate that CshA affects steps downstream of PrfA activation, possibly by influencing other factors required for virulence gene expression. Expressing the PrfAG145S protein increased LLO levels, compared to LLO levels in a PrfAWT background (Fig. 4B, upper panel, WT* versus WT, respectively, and Fig. 4C). In a PrfAG145S background, the LLO levels were not significantly different in strains with or without CshA (Fig. 4B, upper panel, WT* versus ΔcshA*, respectively, and Fig. 4C). This shows that a constitutively active PrfA protein can overcome the absence of CshA that is observed in the PrfAWT background (Fig. 3 and 4). In conclusion, our results suggest that CshA is important for PrfA activation, rather than acting downstream by affecting other putative regulatory factors.
FIG 4.
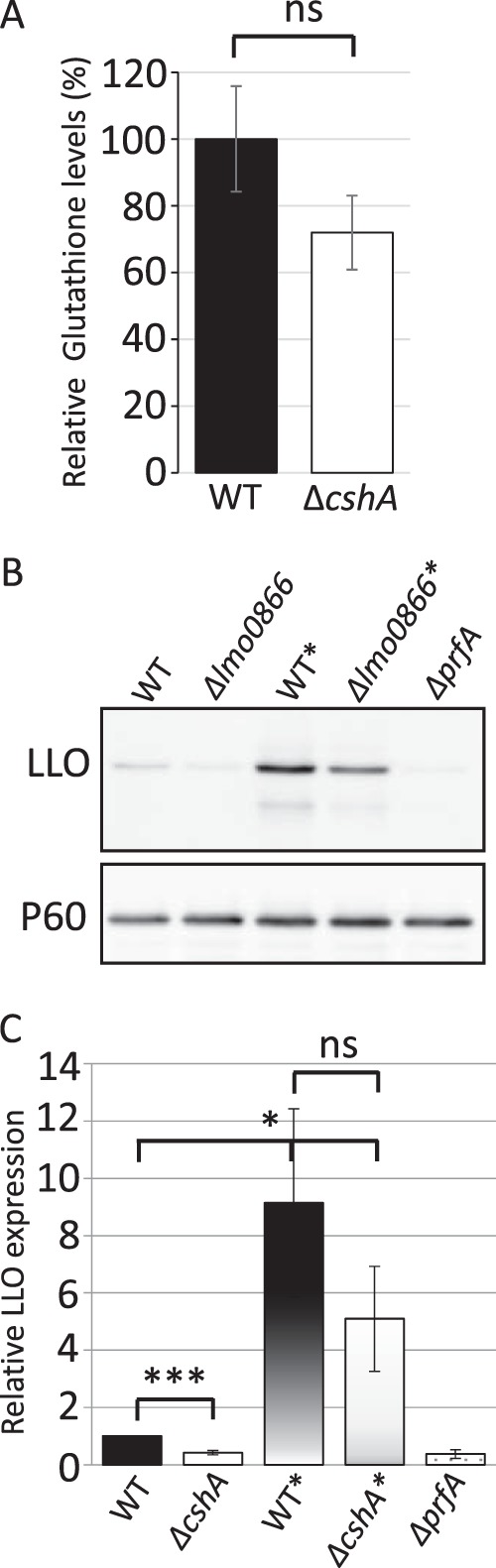
Glutathione levels and virulence factor expression in various strain backgrounds. (A) Wild-type or ΔcshA strains were grown at 37°C in pH-adjusted BHI (pH 5.5) to an OD600 of 0.9 before the intracellular glutathione concentration was determined. The glutathione concentration in the wild-type strain was arbitrarily set to 100% (n = 3). For statistics, the sample from the ΔcshA strain was compared to the WT using a two-tailed Student t test (ns, not significant). (B) The indicated strains were grown at 37°C in pH-adjusted BHI (pH 5.5) until an OD600 of ∼0.9 before protein extraction, SDS-PAGE separation, and Western blot analysis. The expression levels of LLO (upper panel) or P60 (control, lower panel) were analyzed, using LLO- or P60-specific antibodies, in samples of protein precipitated by trichloroacetic acid from filtered culture supernatant (n = 3). (C) LLO expression (from panel A) was quantified in the indicated strains and related to P60 expression. LLO expression in EGDe was arbitrarily set to 1. Error bars show the standard deviations. For statistics, all samples were compared to EGDe using a two-tailed Student t test (*, P < 0.05; ***, P < 0.001; ns, not significant).
The ΔcshA strain does not display reduced infection capacity.
Since the strain lacking CshA shows reduced expression of LLO and ActA, as well as lowered hemolytic activity, compared to the wild-type strain, it would be expected that infectivity of this strain is also reduced. To test this, we allowed the different strains to infect the nonphagocytic cell line Caco-2. Using both viable counts and microscopy, the absence of LLO or PrfA reduced the infection capacity at both 1 and 8 h postinfection, as has been shown by others (Fig. 5) (59–61). Surprisingly, despite displaying reduced virulence factor expression, the ΔcshA strain was equally infectious compared to the wild type (Fig. 5) at both 1 and 8 h postinfection. Using a phagocytic cell line, J774, a slight reduction in infectivity was observed for the ΔcshA strain compared to the wild-type strain, whereas the strain lacking PrfA showed a more pronounced reduction in infectivity at both 1 and 3 h postinfection (Fig. 6).
FIG 5.
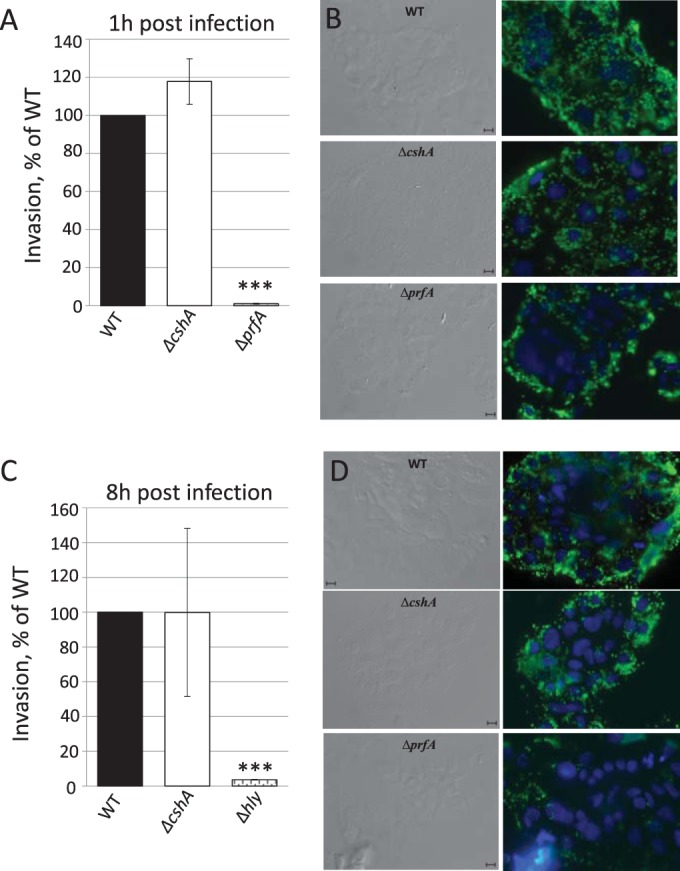
Infection assay. (A and C) Caco-2 cells were infected with wild-type, ΔprfA, Δhly, or ΔcshA strains for the indicated time points before cells were lysed, and bacteria were plated and counted. The infectivity of the ΔprfA, Δhly, and ΔcshA strains are shown relative to the wild-type strain (100%). Error bars show the standard deviations. All samples were compared to the wild-type using a two-tailed Student t test (***, P < 0.001). (B and D) Phase-contrast and fluorescence microscopy. The right panels show Caco-2 cells infected with the indicated strains for 1 h (B) or 8 h (D) and stained for Listeria (green) or cell nuclei (blue). The left panels show phase-contrast images of right panels. Bars, 10 μm.
FIG 6.
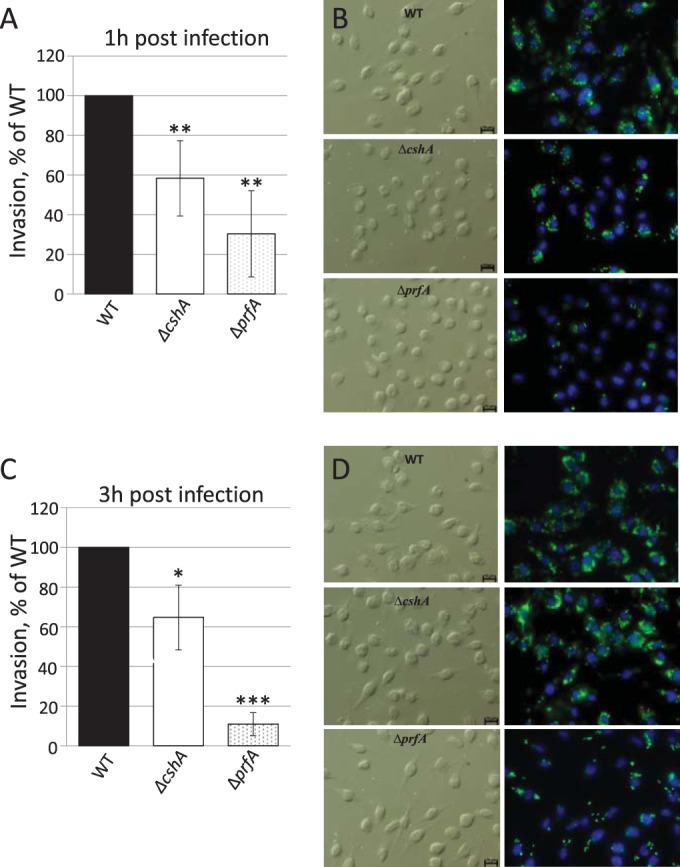
Infection assay. (A and C) J774 cells were infected with wild-type, ΔprfA, or ΔcshA strains for the indicated time points before cells were lysed, and bacteria were plated and counted. The infectivity of the ΔprfA and ΔcshA strains is shown relative to the wild-type strain (100%). Error bars show the standard deviations. All samples were compared to the wild type using a two-tailed Student t test (*, P < 0.05; **, P < 0.01; ***, P < 0.001). (B and D) Phase-contrast and fluorescence microscopy. The right panels show J774 cells infected with the indicated strains for 1 h (B) or 8 h (D) and stained for Listeria (green) or cell nuclei (blue). The left panels show phase-contrast images of right panels. Bars, 10 μm.
DISCUSSION
We analyzed here the importance of a DExD-box RNA helicase, CshA, during L. monocytogenes virulence factor expression and infection. Our data show that the absence of CshA reduces the expression of the most prominent virulence factor, LLO, of L. monocytogenes (Fig. 3). The absence of CshA also decreased ActA levels, although the effect was less pronounced (Fig. 3). The reduced virulence factor expression observed in the ΔcshA strain seems to be exerted at the transcriptional level, as suggested by Northern analysis (see Fig. S2 in the supplemental material). In line with reduced LLO expression, we observed lowered hemolytic activity in the strain lacking CshA compared to the wild-type strain (Fig. 1 and 2). However, the reduced expression of LLO and ActA were not due to lower expression of the transcriptional regulator PrfA (Fig. 3). The expression and activity of PrfA is controlled at several levels. At the transcriptional level, prfA expression is controlled by three different promoters: one constitutive, one stress regulated, and one PrfA regulated (55). At the posttranscriptional level, a thermosensor in its 5′-untranslated RNA allows translation of PrfA only at temperatures above 30°C (37). In addition, PrfA translation is also controlled by regulatory RNAs, and it requires an unstructured 5′ region of the coding RNA for efficient translation (36, 40). Full expression of PrfA is, however, not sufficient for maximal virulence; the protein requires a coactivator, recently suggested to be glutathione (57). We were, however, unable to observe any significant differences in glutathione levels between a wild-type strain and a ΔcshA strain (Fig. 4A). Using a constitutively active PrfA protein, we determined that CshA is important for PrfA activation rather than acting at steps downstream (Fig. 4B and C). Alternatively, CshA might only affect PrfA activity at noninducing conditions (i.e., in the absence of the putative cofactor). The mechanism by which CshA enhances PrfA activity remains to be elucidated, but it could involve regulation of glucose or iron levels—both have been shown to affect PrfA activity (62, 63). Also, the reason why the expression of LLO is more sensitive to variations in PrfA activity than ActA expression requires further studies.
Surprisingly, the reduced LLO and ActA expression in the ΔcshA strain at low pH was not accompanied by reduced infectivity of nonphagocytic Caco-2 cells (Fig. 5). A small but statistically significant reduction was observed when infecting phagocytic J774 cells with the ΔcshA strain compared to the wild type (Fig. 6). Why a strain lacking CshA shows a diverse capacity to infect different cell types is exciting and requires further study. It is nevertheless interesting that although strains lacking either CshA or PrfA affect virulence factor expression and hemolytic activity similarly, the impact of each of these proteins during infection is radically different: the strain lacking PrfA shows a dramatic reduction in infectivity, whereas the strain lacking CshA has infectivity similar to that of the wild type. This indicates that CshA has a much narrower function during pathogenesis, despite reducing the activity of PrfA. Whether this reflects a larger importance for CshA when Listeria is exposed to red blood cells than during intracellular phagosome escape needs to be clarified. Our study also suggests that maximal virulence factor expression is not needed for full infection, at least under the conditions studied here.
It is interesting that the CshA homolog in Staphylococcus aureus, CshASa, has also been shown to be important for hemolytic activity, although in a manner opposite that of CshALm (more hemolysis in the absence of CshASa) (14). CshA of both S. aureus and Bacillus subtilis is part of a protein complex, known as the degradosome, which is important for mRNA degradation (19, 20). The putative association of CshA with other degradosome components in L. monocytogenes has yet to be revealed.
Efficient translation of the transcriptional activator of type III secretion in P. aeruginosa, ExsA, was shown to require the RNA helicase DeaD (35). Although we also observed a role for an RNA helicase controlling a transcriptional activator (PrfA), the effect is not at the level of translation but rather at the level of activity (Fig. 3 and 4).
The different RNA helicases in L. monocytogenes are involved in overlapping but also disparate processes of the bacterium. We and others have recently characterized the physiological importance of these RNA helicases. The roles of the different listerial RNA helicases have been analyzed under various stress conditions (26, 27). By deleting all RNA helicases in different combinations, we have pinpointed the role of the individual enzymes (15). We identified the RNA helicase Lmo1450 as the most important regarding growth and ribosomal maturation; accordingly, the overexpression of Lmo1450 could partially compensate for the lack of all four RNA helicases. In another study, we showed that the RNA helicase Lmo1722 was associated with the 50S ribosomal subunit through its C terminus (6). In this study, we show that Lmo1450 and Lmo1722 play a minor role in virulence factor expression and hemolytic activity, whereas CshA appears to be more important during these processes. Further characterization of the interplay of these RNA helicases is required to fully appreciate their role during virulence, as well as under normal physiological conditions.
Supplementary Material
ACKNOWLEDGMENTS
J.J. was supported by Umeå University, Swedish Research Council grants K2011-56X-15144-08-6 and 621-2012-2451, and ERC starting grant 260764-RNAntibiotics.
We declare no conflict of interest.
We are grateful to P. Cossart for antibodies and T. Tiensuu for technical assistance.
Funding Statement
This study was supported by Umeå University.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00849-15.
REFERENCES
- 1.Py B, Higgins CF, Krisch HM, Carpousis AJ. 1996. A DEAD-box RNA helicase in the Escherichia coli RNA degradosome. Nature 381:169–172. doi: 10.1038/381169a0. [DOI] [PubMed] [Google Scholar]
- 2.Coburn GA, Miao X, Briant DJ, Mackie GA. 1999. Reconstitution of a minimal RNA degradosome demonstrates functional coordination between a 3′ exonuclease and a DEAD-box RNA helicase. Genes Dev 13:2594–2603. doi: 10.1101/gad.13.19.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klostermeier D. 2013. Lifelong companions: RNA helicases and their roles in RNA metabolism. RNA Biol 10:2–3. doi: 10.4161/rna.23500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaberdin VR, Blasi U. 2013. Bacterial helicases in posttranscriptional control. Biochim Biophys Acta 1829:878–883. doi: 10.1016/j.bbagrm.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 5.Portier C. 1975. Quaternary structure of Escherichia coli polynucleotide phosphorylase: new evidence for a trimeric structure. FEBS Lett 50:79–81. doi: 10.1016/0014-5793(75)81045-3. [DOI] [PubMed] [Google Scholar]
- 6.Netterling S, Vaitkevicius K, Nord S, Johansson J. 2012. A Listeria monocytogenes RNA helicase essential for growth and ribosomal maturation at low temperatures uses its C terminus for appropriate interaction with the ribosome. J Bacteriol 194:4377–4385. doi: 10.1128/JB.00348-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Redder P, Hausmann S, Khemici V, Yasrebi H, Linder P. 2015. Bacterial versatility requires DEAD-box RNA helicases. FEMS Microbiol Rev 39:392–412. doi: 10.1093/femsre/fuv011. [DOI] [PubMed] [Google Scholar]
- 8.Linder P, Fuller-Pace FV. 2013. Looking back on the birth of DEAD-box RNA helicases. Biochim Biophys Acta 1829:750–755. doi: 10.1016/j.bbagrm.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 9.Iost I, Bizebard T, Dreyfus M. 2013. Functions of DEAD-box proteins in bacteria: current knowledge and pending questions. Biochim Biophys Acta 1829:866–877. doi: 10.1016/j.bbagrm.2013.01.012. [DOI] [PubMed] [Google Scholar]
- 10.Jarmoskaite I, Russell R. 2011. DEAD-box proteins as RNA helicases and chaperones. Wires RNA 2:135–152. doi: 10.1002/wrna.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tanner NK, Linder P. 2001. DExD/H box RNA helicases: from generic motors to specific dissociation functions. Mol Cell 8:251–262. doi: 10.1016/S1097-2765(01)00329-X. [DOI] [PubMed] [Google Scholar]
- 12.Nagahama M, Yamazoe T, Hara Y, Tani K, Tsuji A, Tagaya M. 2006. The AAA-ATPase NVL2 is a component of pre-ribosomal particles that interacts with the DExD/H-box RNA helicase DOB1. Biochem Biophys Res Commun 346:1075–1082. doi: 10.1016/j.bbrc.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 13.Vakulskas CA, Pannuri A, Cortes-Selva D, Zere TR, Ahmer BM, Babitzke P, Romeo T. 2014. Global effects of the DEAD-box RNA helicase DeaD (CsdA) on gene expression over a broad range of temperatures. Mol Microbiol 92:945–958. doi: 10.1111/mmi.12606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oun S, Redder P, Didier JP, Francois P, Corvaglia AR, Buttazzoni E, Giraud C, Girard M, Schrenzel J, Linder P. 2013. The CshA DEAD-box RNA helicase is important for quorum sensing control in Staphylococcus aureus. RNA Biol 10:157–165. doi: 10.4161/rna.22899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bareclev C, Vaitkevicius K, Netterling S, Johansson J. 2014. DExD-box RNA-helicases in Listeria monocytogenes are important for growth, ribosomal maturation, rRNA processing, and virulence factor expression. RNA Biol 11:1457–1466. doi: 10.1080/15476286.2014.996099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Redder P, Linder P. 2012. DEAD-box RNA helicases in gram-positive RNA decay. Methods Enzymol 511:369–383. doi: 10.1016/B978-0-12-396546-2.00017-6. [DOI] [PubMed] [Google Scholar]
- 17.Dominguez-Malfavon L, Islas LD, Luisi BF, Garcia-Villegas R, Garcia-Mena J. 2013. The assembly and distribution in vivo of the Escherichia coli RNA degradosome. Biochimie 95:2034–2041. doi: 10.1016/j.biochi.2013.07.022. [DOI] [PubMed] [Google Scholar]
- 18.Lehnik-Habrink M, Rempeters L, Kovacs AT, Wrede C, Baierlein C, Krebber H, Kuipers OP, Stulke J. 2013. DEAD-Box RNA helicases in Bacillus subtilis have multiple functions and act independently from each other. J Bacteriol 195:534–544. doi: 10.1128/JB.01475-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roux CM, DeMuth JP, Dunman PM. 2011. Characterization of components of the Staphylococcus aureus mRNA degradosome holoenzyme-like complex. J Bacteriol 193:5520–5526. doi: 10.1128/JB.05485-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lehnik-Habrink M, Pfortner H, Rempeters L, Pietack N, Herzberg C, Stulke J. 2010. The RNA degradosome in Bacillus subtilis: identification of CshA as the major RNA helicase in the multiprotein complex. Mol Microbiol 77:958–971. doi: 10.1111/j.1365-2958.2010.07264.x. [DOI] [PubMed] [Google Scholar]
- 21.Giraud C, Hausmann S, Lemeille S, Prados J, Redder P, Linder P. 2015. The C-terminal region of the RNA helicase CshA is required for the interaction with the degradosome and turnover of bulk RNA in the opportunistic pathogen Staphylococcus aureus. RNA Biol 12:658–674. doi: 10.1080/15476286.2015.1035505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carpousis AJ, Khemici V, Poljak L. 2008. Assaying DEAD-box RNA helicases and their role in mRNA degradation in Escherichia coli. Methods Enzymol 447:183–197. doi: 10.1016/S0076-6879(08)02210-6. [DOI] [PubMed] [Google Scholar]
- 23.Liou GG, Chang HY, Lin CS, Lin-Chao S. 2002. DEAD box RhlB RNA helicase physically associates with exoribonuclease PNPase to degrade double-stranded RNA independent of the degradosome-assembling region of RNase E. J Biol Chem 277:41157–41162. doi: 10.1074/jbc.M206618200. [DOI] [PubMed] [Google Scholar]
- 24.Azizoglu RO, Kathariou S. 2010. Inactivation of a cold-induced putative RNA helicase gene of Listeria monocytogenes is accompanied by failure to grow at low temperatures but does not affect freeze-thaw tolerance. J Food Prot 73:1474–1479. [DOI] [PubMed] [Google Scholar]
- 25.Chan YC, Raengpradub S, Boor KJ, Wiedmann M. 2007. Microarray-based characterization of the Listeria monocytogenes cold regulon in log- and stationary-phase cells. Appl Environ Microbiol 73:6484–6498. doi: 10.1128/AEM.00897-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Markkula A, Lindstrom M, Johansson P, Bjorkroth J, Korkeala H. 2012. Roles of four putative DEAD-box RNA helicase genes in growth of Listeria monocytogenes EGD-e under heat, pH, osmotic, ethanol, and oxidative stress conditions. Appl Environ Microbiol 78:6875–6882. doi: 10.1128/AEM.01526-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Markkula A, Mattila M, Lindstrom M, Korkeala H. 2012. Genes encoding putative DEAD-box RNA helicases in Listeria monocytogenes EGD-e are needed for growth and motility at 3°C. Environ Microbiol 14:2223–2232. doi: 10.1111/j.1462-2920.2012.02761.x. [DOI] [PubMed] [Google Scholar]
- 28.Hernandez-Milian A, Payeras-Cifre A. 2014. What is new in listeriosis? Biomed Res Int 2014:358051. doi: 10.1155/2014/358051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lamont RF, Sobel J, Mazaki-Tovi S, Kusanovic JP, Vaisbuch E, Kim SK, Uldbjerg N, Romero R. 2011. Listeriosis in human pregnancy: a systematic review. J Perinatal Med 39:227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goulet V, Hebert M, Hedberg C, Laurent E, Vaillant V, De Valk H, Desenclos JC. 2012. Incidence of listeriosis and related mortality among groups at risk of acquiring listeriosis. Clin Infect Dis 54:652–660. doi: 10.1093/cid/cir902. [DOI] [PubMed] [Google Scholar]
- 31.Bae D, Crowley MR, Wang C. 2011. Transcriptome analysis of Listeria monocytogenes grown on a ready-to-eat meat matrix. J Food Prot 74:1104–1111. doi: 10.1016/j.jprot.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 32.Jemmi T, Stephan R. 2006. Listeria monocytogenes: food-borne pathogen and hygiene indicator. Rev Sci Technol 25:571–580. [PubMed] [Google Scholar]
- 33.Koo JT, Choe J, Moseley SL. 2004. HrpA, a DEAH-box RNA helicase, is involved in mRNA processing of a fimbrial operon in Escherichia coli. Mol Microbiol 52:1813–1826. doi: 10.1111/j.1365-2958.2004.04099.x. [DOI] [PubMed] [Google Scholar]
- 34.Salman-Dilgimen A, Hardy PO, Radolf JD, Caimano MJ, Chaconas G. 2013. HrpA, an RNA helicase involved in RNA processing, is required for mouse infectivity and tick transmission of the Lyme disease spirochete. PLoS Pathog 9:e1003841. doi: 10.1371/journal.ppat.1003841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Intile PJ, Balzer GJ, Wolfgang MC, Yahr TL. 2015. The RNA helicase DeaD stimulates ExsA translation to promote expression of the Pseudomonas aeruginosa type III secretion system. J Bacteriol 197:2664–2674. doi: 10.1128/JB.00231-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Loh E, Memarpour F, Vaitkevicius K, Kallipolitis BH, Johansson J, Sonden B. 2012. An unstructured 5′-coding region of the prfA mRNA is required for efficient translation. Nucleic Acids Res 40:1818–1827. doi: 10.1093/nar/gkr850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johansson J, Mandin P, Renzoni A, Chiaruttini C, Springer M, Cossart P. 2002. An RNA thermosensor controls expression of virulence genes in Listeria monocytogenes. Cell 110:551–561. doi: 10.1016/S0092-8674(02)00905-4. [DOI] [PubMed] [Google Scholar]
- 38.de las Heras A, Cain RJ, Bieleckal MK, Vazquez-Boland JA. 2011. Regulation of Listeria virulence: PrfA master and commander. Curr Opin Microbiol 14:118–127. doi: 10.1016/j.mib.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 39.Xayarath B, Freitag NE. 2012. Optimizing the balance between host and environmental survival skills: lessons learned from Listeria monocytogenes. Future Microbiol 7:839–852. doi: 10.2217/fmb.12.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Loh E, Dussurget O, Gripenland J, Vaitkevicius K, Tiensuu T, Mandin P, Repoila F, Buchrieser C, Cossart P, Johansson J. 2009. A trans-acting riboswitch controls expression of the virulence regulator PrfA in Listeria monocytogenes. Cell 139:770–779. doi: 10.1016/j.cell.2009.08.046. [DOI] [PubMed] [Google Scholar]
- 41.Mengaud J, Dramsi S, Gouin E, Vazquez-Boland JA, Milon G, Cossart P. 1991. Pleiotropic control of Listeria monocytogenes virulence factors by a gene that is autoregulated. Mol Microbiol 5:2273–2283. doi: 10.1111/j.1365-2958.1991.tb02158.x. [DOI] [PubMed] [Google Scholar]
- 42.Eiting M, Hageluken G, Schubert WD, Heinz DW. 2005. The mutation G145S in PrfA, a key virulence regulator of Listeria monocytogenes, increases DNA-binding affinity by stabilizing the HTH motif. Mol Microbiol 56:433–446. doi: 10.1111/j.1365-2958.2005.04561.x. [DOI] [PubMed] [Google Scholar]
- 43.Arnaud M, Chastanet A, Debarbouille M. 2004. New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, gram-positive bacteria. Appl Environ Microbiol 70:6887–6891. doi: 10.1128/AEM.70.11.6887-6891.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kingdon GC, Sword CP. 1970. Biochemical and immunological effects of Listeria monocytogenes hemolysin. Infect Immun 1:363–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 46.Kampfer P. 1995. An efficient method for preparation of extracts from Gram-positive bacteria for comparison of cellular protein patterns. J Microbiol Methods 21:55–60. doi: 10.1016/0167-7012(94)00033-4. [DOI] [Google Scholar]
- 47.Udekwu KI, Darfeuille F, Vogel J, Reimegard J, Holmqvist E, Wagner EG. 2005. Hfq-dependent regulation of OmpA synthesis is mediated by an antisense RNA. Genes Dev 19:2355–2366. doi: 10.1101/gad.354405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Toledo-Arana A, Dussurget O, Nikitas G, Sesto N, Guet-Revillet H, Balestrino D, Loh E, Gripenland J, Tiensuu T, Vaitkevicius K, Barthelemy M, Vergassola M, Nahori MA, Soubigou G, Regnault B, Coppee JY, Lecuit M, Johansson J, Cossart P. 2009. The Listeria transcriptional landscape from saprophytism to virulence. Nature 459:950–956. doi: 10.1038/nature08080. [DOI] [PubMed] [Google Scholar]
- 49.Dramsi S, Cossart P. 2002. Listeriolysin O: a genuine cytolysin optimized for an intracellular parasite. J Cell Biol 156:943–946. doi: 10.1083/jcb.200202121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kayal S, Charbit A. 2006. Listeriolysin O: a key protein of Listeria monocytogenes with multiple functions. FEMS Microbiol Rev 30:514–529. doi: 10.1111/j.1574-6976.2006.00021.x. [DOI] [PubMed] [Google Scholar]
- 51.Schnupf P, Portnoy DA. 2007. Listeriolysin O: a phagosome-specific lysin. Microbes Infect 9:1176–1187. doi: 10.1016/j.micinf.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 52.Glomski IJ, Gedde MM, Tsang AW, Swanson JA, Portnoy DA. 2002. The Listeria monocytogenes hemolysin has an acidic pH optimum to compartmentalize activity and prevent damage to infected host cells. J Cell Biol 156:1029–1038. doi: 10.1083/jcb.200201081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schuerch DW, Wilson-Kubalek EM, Tweten RK. 2005. Molecular basis of listeriolysin O pH dependence. Proc Natl Acad Sci U S A 102:12537–12542. doi: 10.1073/pnas.0500558102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scortti M, Monzo HJ, Lacharme-Lora L, Lewis DA, Vazquez-Boland JA. 2007. The PrfA virulence regulon. Microbes Infect 9:1196–1207. doi: 10.1016/j.micinf.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 55.Freitag NE, Port GC, Miner MD. 2009. Listeria monocytogenes: from saprophyte to intracellular pathogen. Nat Rev Microbiol 7:623–628. doi: 10.1038/nrmicro2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tiensuu T, Andersson C, Ryden P, Johansson J. 2013. Cycles of light and dark co-ordinate reversible colony differentiation in Listeria monocytogenes. Mol Microbiol 87:909–924. doi: 10.1111/mmi.12140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reniere ML, Whiteley AT, Hamilton KL, John SM, Lauer P, Brennan RG, Portnoy DA. 2015. Glutathione activates virulence gene expression of an intracellular pathogen. Nature 517:170–173. doi: 10.1038/nature14029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xayarath B, Volz KW, Smart JI, Freitag NE. 2011. Probing the role of protein surface charge in the activation of PrfA, the central regulator of Listeria monocytogenes pathogenesis. PLoS One 6:e23502. doi: 10.1371/journal.pone.0023502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guzman CA, Rohde M, Chakraborty T, Domann E, Hudel M, Wehland J, Timmis KN. 1995. Interaction of Listeria monocytogenes with mouse dendritic cells. Infect Immun 63:3665–3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gaillard JL, Berche P, Mounier J, Richard S, Sansonetti P. 1987. In vitro model of penetration and intracellular growth of Listeria monocytogenes in the human enterocyte-like cell line Caco-2. Infect Immun 55:2822–2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goebel W, Leimeister-Wachter M, Kuhn M, Domann E, Chakraborty T, Kohler S, Bubert A, Wuenscher M, Sokolovic Z. 1993. Listeria monocytogenes: a model system for studying the pathomechanisms of an intracellular microorganism. Zentralbl Bakteriol 278:334–347. doi: 10.1016/S0934-8840(11)80850-9. [DOI] [PubMed] [Google Scholar]
- 62.Milenbachs AA, Brown DP, Moors M, Youngman P. 1997. Carbon-source regulation of virulence gene expression in Listeria monocytogenes. Mol Microbiol 23:1075–1085. doi: 10.1046/j.1365-2958.1997.2711634.x. [DOI] [PubMed] [Google Scholar]
- 63.Bockmann R, Dickneite C, Middendorf B, Goebel W, Sokolovic Z. 1996. Specific binding of the Listeria monocytogenes transcriptional regulator PrfA to target sequences requires additional factor(s) and is influenced by iron. Mol Microbiol 22:643–653. doi: 10.1046/j.1365-2958.1996.d01-1722.x. [DOI] [PubMed] [Google Scholar]
- 64.Bethesda Research Laboratories. 1986. BRL pUC host: Escherichia coli DH5α competent cells. Focus 8:9. [Google Scholar]
- 65.Glaser P, Frangeul L, Buchrieser C, Rusniok C, Amend A, Baquero F, Berche P, Bloecker H, Brandt P, Chakraborty T, Charbit A, Chetouani F, Couve E, de Daruvar A, Dehoux P, Domann E, Dominguez-Bernal G, Duchaud E, Durant L, Dussurget O, Entian KD, Fsihi H, Garcia-del Portillo F, Garrido P, Gautier L, Goebel W, Gomez-Lopez N, Hain T, Hauf J, Jackson D, Jones LM, Kaerst U, Kreft J, Kuhn M, Kunst F, Kurapkat G, Madueno E, Maitournam A, Vicente JM, Ng E, Nedjari H, Nordsiek G, Novella S, de Pablos B, Perez-Diaz JC, Purcell R, Remmel B, Rose M, Schlueter T, Simoes N, Tierrez A, Vazquez-Boland JA, Voss H, Wehland J, Cossart P. 2001. Comparative genomics of Listeria species. Science 294:849–852. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.