

Abstract
Streptococcus agalactiae causes both symptomatic cystitis and asymptomatic bacteriuria (ABU); however, growth characteristics of S. agalactiae in human urine have not previously been reported. Here, we describe a phenotype of robust growth in human urine observed in ABU-causing S. agalactiae (ABSA) that was not seen among uropathogenic S. agalactiae (UPSA) strains isolated from patients with acute cystitis. In direct competition assays using pooled human urine inoculated with equal numbers of a prototype ABSA strain, designated ABSA 1014, and any one of several UPSA strains, measurement of the percentage of each strain recovered over time showed a markedly superior fitness of ABSA 1014 for urine growth. Comparative phenotype profiling of ABSA 1014 and UPSA strain 807, isolated from a patient with acute cystitis, using metabolic arrays of >2,500 substrates and conditions revealed unique and specific l-malic acid catabolism in ABSA 1014 that was absent in UPSA 807. Whole-genome sequencing also revealed divergence in malic enzyme-encoding genes between the strains predicted to impact the activity of the malate metabolic pathway. Comparative growth assays in urine comparing wild-type ABSA and gene-deficient mutants that were functionally inactivated for the malic enzyme metabolic pathway by targeted disruption of the maeE or maeK gene in ABSA demonstrated attenuated growth of the mutants in normal human urine as well as synthetic human urine containing malic acid. We conclude that some S. agalactiae strains can grow in human urine, and this relates in part to malic acid metabolism, which may affect the persistence or progression of S. agalactiae ABU.
INTRODUCTION
Streptococcus agalactiae is a leading cause of infection in newborns, pregnant women, and older persons with chronic medical illness (1). This organism also causes symptomatic cystitis (2) and asymptomatic bacteriuria (ABU) (3, 4). These infections have been associated with diverse patient groups, including pregnant and nonpregnant women and elderly individuals (2, 5–9). Bacteriuria due to S. agalactiae in pregnant women is important due to the risk for vertical transmission of the bacterium to the newborn that can lead to life-threatening infections such as central nervous system invasion and meningitis (10–13). S. agalactiae bacteriuria of any count during pregnancy is routinely treated with antibiotics to minimize the risk of vertical transmission to the neonate (14–17). S. agalactiae bacteriuria has an incidence of between 1.0 and 3.5% in individuals with suspected urinary tract infection (UTI) (4, 18–21). S. agalactiae bacteriuria loads average between 50,000 and 70,000 CFU ml−1 in infected individuals (3), although these loads are dynamic and have been reported to change dramatically over just a few hours (2). The significance of S. agalactiae bacteriuria in other patient populations and the cost-benefit of universal treatment for this condition in pregnancy remain unclear (22, 23). Finally, it is unknown whether S. agalactiae bacteriuria might act as a reservoir for persistence or contribute to chronic ongoing infection in the host.
An important fitness trait that contributes to the persistence of bacteria such as ABU-causing Escherichia coli in the urinary tract is the ability of the bacteria to grow in human urine. This enables bacteria such as E. coli to persist in the host by regrowth of nonvoided organisms in residual urine and offers a competitive advantage independent of urothelial cell binding and inflammation (24, 25). In addition to E. coli, other organisms have been reported to grow in urine, including staphylococci (26) and enterococci (27). To achieve this, the bacteria must endure the low pH of urine, high urea levels, nutrient limitation, nitrite in mildly acidified urine, and exposure to antimicrobial proteins (e.g., Tamm-Horsfall protein) and peptides (e.g., cathelicidin). Studies of the metabolic basis underlying growth in human urine for ABU-causing E. coli have shown links to transport and degradation pathways for galacturonate, glucuronide, and galactonate (24), as well as to synthesis of guanine, arginine, and glutamine (28). Antioxidant defense mechanisms have also been identified as important for urine growth of E. coli (29). Russo et al. reported that synthesis of guanine-dependent products was critical for this phenotype (30). Arginine metabolism was also shown to contribute to growth of E. coli (31). In addition, tolerance to high levels of d-serine, or requirement for this compound, has been linked to survival of Staphylococcus saprophyticus (26) and E. coli in urine (28), respectively. Iron acquisition by E. coli (32) and Enterococcus faecalis (27) is also important for this phenotype.
Uropathogenic S. agalactiae (UPSA) strains, isolated from individuals with acute cystitis and pyelonephritis, were shown in two prior studies to be incapable of human-urine growth (33, 34). These organisms adhered to urothelial cells and induced inflammatory responses but had no capacity for urine growth, findings consistent with data published in a prior study of S. agalactiae and other diverse bacteria (35). The potential of ABU-causing S. agalactiae (ABSA) to grow in human urine has not previously been investigated. In this study, we report a novel fitness trait of robust growth of ABSA in human urine, which we did not observe in multiple UPSA clinical strains analyzed. These findings show that some S. agalactiae strains are capable of robust urine growth. We show that this is related, at least in part, to malic acid metabolism.
MATERIALS AND METHODS
Bacterial strains.
The ABSA strain used for the initial experiments in this study was cultured from urine obtained by catheter from an asymptomatic 26-year-old pregnant woman undergoing routine screening. Repeat urine cultures grew pure S. agalactiae at 50,000 CFU ml−1, which continued over a 5-week period, and the woman was diagnosed with ABU. The strain was designated ABSA 1014. A UPSA isolate, designated UPSA 807, was used for the initial comparative growth studies. UPSA 807 was cultured from a clean-catch voided urine sample obtained from a 59-year-old woman who presented with frequency, urgency, hematuria, pyuria, and bacteriuria of 80,000 CFU ml−1. Urinalysis was consistent with a clinical diagnosis of acute uncomplicated cystitis. S. agalactiae isolates were routinely identified and serotyped using latex agglutination, as previously described (2). Details of other isolates used for comparative assays in subsequent studies are provided in Table 1. This study had ethical approval from the University of Alabama committee on human experimentation (X070619009) and the Griffith University human ethics committee (MSC/06/08/HREC), and all work was undertaken in accordance with the Helsinki Declaration.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristic(s)a | Reference |
|---|---|---|
| Strains | ||
| ABSA 1014 | 26; F; asymptomatic; 50,000b; II; 28 (19); G | This study |
| ABSA 729 | 69; F; asymptomatic; 30,000b; NTc; 452 (24); G | This study |
| ABSA 834 | 57; F; asymptomatic; 50,000b; III; 182 (19); G | This study |
| UPSA 807 | 59; F; Fr/U, GH, P; 80,000; V; 1 (1); G, C, T | This study |
| UPSA 714 | 89; F; Dys, GH, P; 50,000; III; 17 (17); G, T | This study |
| UPSA 058 | 77; M; Dys, Fr/U, GH, P; 100,000; Ia; 23 (23); G, T | This study |
| UPSA 008 | 57; F; Dys, GH; 100,000; V; 1 (1); G, C | This study |
| UPSA 872 | 69; F; Dys, GH, P; 50,000; Ia; 1 (1); G, T | This study |
| GU2136 | E. coli DH5α containing pDI102 | This study |
| GU2293 | ABSA 834 containing pDI102 | This study |
| GU2296 | ABSA 834ΔmaeE (maeE mutant, Cmr) | This study |
| GU2320 | GU2296 with pDI110 (maeE complementation plasmid) | This study |
| GU2356 | E. coli DH5α containing pDI108 | This study |
| GU2318 | E. coli DH5α containing pDI110 | This study |
| GU2436 | E. coli DH5α containing pGU2436 | This study |
| GU2446 | ABSA 834ΔmaeK (maeK mutant, Cmr) | This study |
| GU2468 | GU2446 with pDI111 (maeK complementation plasmid) | This study |
| GU2321 | WT UPSA 807 with pDI111 (maeK complementation plasmid) | This study |
| GU2319 | E. coli DH5α containing pDI111 | This study |
| GU2060 | E. coli DH5α containing pLZ12 | This study |
| GU2032 | E. coli DH5α containing pHY304 | |
| GU2298 | E. coli DH5α containing pMSP3545 | This study |
| GU2356 | E. coli DH5α containing pMSP3545-aad9 | This study |
| Plasmids | ||
| pHY304 | ori(Ts), temperature-sensitive shuttle vector, Emr | 42 |
| pHY304-aad9 | ori(Ts), pHY304 derivative, Sptr | This study |
| pLZ12 | E. coli-Streptococcus shuttle vector, Cmr | 44 |
| pDI102 | pHY304-aad9 derivative, maeE::Cmr construct, Sptr Cmr | This study |
| pGU2436 | pHY304-aad9 derivative, maeK::Cmr construct, Sptr Cmr | This study |
| pMSP3545 | Emr, Gram-positive bacterial expression vector | 47 |
| pDI108 | pMSP3545 derivative, Emr cassette replaced with Sptr | This study |
| pDI110 | maeE from ABSA 834 cloned into pDI108 using BglII, XhoI sites | This study |
| pDI111 | maeK from ABSA 834 cloned into pDI108 using BglII, XhoI sites | This study |
For ABSA and UPSA strains, the characteristics are those of the patient from whom the strain was isolated and are as follows: age (years), gender (F, female; M, male), clinical presentation (Fr/U, frequency/urgency; GH, gross hematuria; P, pyuria; Dys, dysuria), and bacteriuria count (CFU per milliliter). The characteristics of the strain are as follows: serotype, sequence type (clonal complex), and antibiotic resistance (G, gentamicin; C, clindamycin; T, tetracycline).
Bacteriuria count was equal or higher in repeat urine samples.
NT, nontypeable.
Human-urine growth assays and culture conditions.
For growth assays, we used pooled urine collected from six healthy female and male volunteers who had no recent history of UTI or antibiotic usage (preceding month). Urine was combined in equal volumes, filter sterilized (0.45 μm), stored at 4°C, and used within 48 h. Growth of S. agalactiae in urine and Todd-Hewitt broth (THB) was quantified using colony counts and turbidity measurements. Duplicate cultures were grown in 200-μl volumes in 96-well microtiter plates that were inoculated with approximately 5 × 103 CFU, which were prepared by washing with sterile phosphate-buffered saline (PBS; pH 7.4) and back-diluting 1/100,000 from an overnight culture. Cultures were grown at 37°C with shaking at 200 rpm. Colony counts were performed using Todd-Hewitt agar (THA), with antibiotic selection as indicated, and optical density at 600 nm (OD600) was measured to monitor culture turbidity. Growth assays were repeated at least three to five times in independent experiments using different batches of pooled urine. Based on the results generated from initial growth assays, we also performed direct competition assays using mixed cultures to determine whether ABSA 1014 could outcompete different UPSA strains in human urine. These assays were based on the differential susceptibilities of ABSA 1014 and (several) UPSA isolates to clindamycin and tetracycline, respectively, as listed in Table 1. For competition assays, ABSA 1014 and any one of several UPSA isolates were mixed at a 1:1 ratio and incubated in pooled human urine for 72 h. The absolute numbers of each strain were determined by plating onto selective THA (5 μg ml−1 clindamycin or 20 μg ml−1 tetracycline; permitting growth of only UPSA strain) and nonselective THA (permitting growth of both ABSA and UPSA) to enable the calculation of the relative proportions of each strain over time.
To examine the growth of bacteria in a defined rich medium, we used C medium. For this, approximately 5 × 105 cells were inoculated in 0.1-ml aliquots and incubated at 37°C. Culture density was monitored using a POLARstar Omega plate reader (BMG Labtech) with shaking at 500 rpm, and OD600 was measured every hour. Bacterial growth (CFU per milliliter) was quantified after 20 h of culture using colony counts. To examine the growth of bacteria in a chemically defined minimal medium, we used M9 medium (36), with minor modifications. Briefly, the following (presterilized) ingredients were added to 700 ml of autoclaved water: 200 ml of M9 salts, 2 ml of 1 M MgSO4, 100 μl of 1 M CaCl2, 0.025% (vol/vol) yeast extract (from a 10% filter-sterilized stock solution), and 0.1% (vol/vol) Casamino Acids (from a 20% filter-sterilized stock solution), with exclusion of 0.4% glucose, which was replaced with 40 mM malic acid as a carbon source where necessary.
PM arrays.
A comprehensive comparison of the metabolic profiles of ABSA 1014 and UPSA 807 was performed using phenotype metabolic (PM) array fingerprinting (Biolog Inc., Hayward, CA). These arrays tested the growth of both strains by evaluating >2,500 substrates and physiologic exposures, including carbon, nitrogen, and phosphorus utilization; biosynthetic pathway and nutrient stimulation; osmotic, ionic, and pH responses; and chemical sensitivity. A complete list of the test conditions (of PM1-20 arrays) is available at http://www.biolog.com. Additional PM arrays were also performed on ABSA 834 and ABSA 729. The cultures in the array plates were incubated at 37°C for 24 to 48 h, and the data were collected using an OmniLog V1.5 module. We performed pairwise comparisons using two independent experiments for the analysis with biological duplicates incorporated into each independent experiment.
DNA isolation, genome sequencing, and bioinformatic analysis.
To investigate the genetic basis for the metabolic differences between ABSA 1014 and UPSA 807, we performed whole-genome sequencing followed by comparative genomic analysis. Genomic DNA was isolated from overnight cultures of S. agalactiae grown in THB, centrifuged (8,000 × g, 10 min), and resuspended in 395 μl DNA isolation buffer (50 mM Tris-HCl, 0.145 M NaCl, pH 7.5) with 5 μl mutanolysin (10 U μl−1). The cells were incubated for 90 min at 37°C with shaking, followed by the addition of 154 μl DNA isolation buffer, 30 μl 10% SDS, 6 μl proteinase K (20 mg ml−1), and 10 μl RNase A (20 mg ml−1). Following 60 min of incubation at 37°C with shaking, 100 μl of 5 M NaCl was added and mixed. The remaining protocol for DNA isolation used the standard cetyltrimethylammonium bromide (CTAB) method (36). Genomic DNA from ABSA 1014, UPSA 807, and six other strains listed in Table 1 was used to generate 100-bp paired-end reads using the Illumina HiSeq2000 platform. Reads were then assembled using Velvet (37), and contigs of ≥200 bp were annotated using prokka (http://www.vicbioinformatics.com/software.prokka.shtml). Genome comparisons of syntenic loci were performed using the Artemis Comparison Tool (ACT) (38), and comparative genome images were generated using Easyfig (39). In silico antibiotic and virulence gene profiles were generated using BLAST (40) and SeqFindR (http://github.com/mscook/SeqFindR) using both assembled and mapping consensus modes to allow for read data to be used to identify query sequences that might not have assembled properly, such as repeat-containing sequences. The genomic contexts of tetM, ermA, and ermB were determined using ACT (38).
Targeted disruption of the ME metabolic pathway in ABSA.
To disrupt the malic enzyme (ME) pathway in ABSA 834, mutations were made in the maeE (SAG1919) and maeK (SAG1921) genes, encoding the ME pathway enzyme and sensor kinase, respectively. ABSA 834 was used because ABSA 1014 was not readily transformable, and the two strains showed equivalent urine growth phenotypes. Deletion of maeE and maeK was performed as described elsewhere (41) with modifications. We noted that ABSA 834 was resistant to erythromycin, so a spectinomycin (Spt)-resistant derivative of the temperature-sensitive allelic exchange vector pHY304 (42) was constructed by introducing the Spt resistance gene aad9 from pUCSpec (43) using primers listed in Table S1 in the supplemental material and termed pHY304-aad9. Briefly, constructs containing ∼500 bp of sequence flanking the sites for deletion were fused to a chloramphenicol (Cm) acetyltransferase (cat) cassette, amplified from pLZ12 (44), by overlapping extension PCR as described previously (45) using primers listed in Table S1. The upstream reverse and downstream forward primers for the flanking regions incorporated 5′ overhangs complementary to cat to facilitate three-way gene splicing by overlap extension (SOEing) PCR using equal amounts of the three amplicons. Primers were designed based on the S. agalactiae 2603V/R genome. Reaction mixtures contained 50 ng of DNA, 0.2 mM deoxynucleoside triphosphates (dNTPs), 1.5 to 4.0 mM MgCl2, 200 nmol of each primer, and 1 unit of Phusion DNA polymerase (New England BioLabs). Initial denaturation for 2 min at 98°C was followed by 35 cycles of 10 s at 95°C, 50 s at 65°C, and 30 s/kb at 72°C, with a final extension for 4 min at 72°C. The constructs for mutation of maeE and maeK were generated by introducing gel-purified PCR products (QIAquick gel extraction kit; Qiagen) into pHY304-aad9 using restriction sites listed in Table S1 and designated pDI102 and pGU2436, respectively. E. coli DH5α transformants were screened on LB agar with Spt (100 μg ml−1) and chloramphenicol (Cm; 20 μg ml−1) at 28°C and confirmed by restriction digest, PCR, and sequencing. Following electroporation into ABSA 834, as previously described (45, 46), allelic exchange, and selection of Spts, Cmr (37°C, THA supplemented with yeast extract [THY] with Cm; 10 μg ml−1) double crossover mutants, defective in maeE (strain GU2296) and maeK (strain GU2446), were obtained using methods essentially the same as described elsewhere (41). Mutations were verified by sequencing PCR products spanning the mutated region (AGRF, Brisbane, Australia). To ensure that no secondary mutations were present, both mutants were complemented in trans using wild-type (WT) maeE and maeK from ABSA 834, cloned into an Spt derivative of pMSP3545 (47), pDI108, to generate pDI110 (maeE) and pDI111 (maeK) complementation constructs. These plasmids were transformed into the respective mutant strains to generate strains GU2320 (maeE-deficient GU2296 complemented with pDI110) and GU2468 (maeK-deficient GU2446 complemented with pDI111).
Measurement of malate in urine.
The concentration of malate in some normal human-urine (NHU) samples and synthetic human urine (SHU) was determined using a commercial l-malic acid assay kit (manual format) (Megazyme International, Bray, Ireland), according to the manufacturer's instructions.
qRT-PCR expression analysis of the malic enzyme metabolic pathway in ABSA and UPSA.
For quantitative real-time reverse transcription-PCR (qRT-PCR) experiments, assays were performed using bacteria grown in SHU to minimize variability between different batches of fresh human urine. For RNA extractions, 12.5-ml cultures of mid-log-phase ABSA 834 and UPSA 807 were grown in SHU with or without 40 mM malic acid to cell densities of 105 to 107 CFU ml−1. Cultures were added to 5 ml of ice-cold 95% ethanol and 5% (vol/vol) phenol solution and incubated on ice for 30 min to stabilize the RNA and prevent degradation. Cells were then pelleted (10,000 × g, 10 min) and stored at −80°C until use. Cell pellets were resuspended in Tris-EDTA (TE) buffer containing 100 U mutanolysin (Sigma) and 30 mg ml−1 lysozyme (Sigma) and incubated for 1 h at 37°C prior to RNA isolation using the SV Total RNA isolation kit (Promega), according to the manufacturer's specifications. Trace DNA contamination was removed using Turbo DNA-free DNase (Life Technologies) and confirmed by standard PCR using RNA samples as the template. Nucleic acids were quantified spectrophotometrically, and the integrity of RNA samples was analyzed using an Experion automated electrophoresis platform (Bio-Rad) using RNA StdSens chips, according to the manufacturer's specifications. Total RNA (0.5 to 1 μg) was reverse transcribed to cDNA using the Superscript III first-strand synthesis system (Life Technologies) with random hexamers in a final volume of 20 μl, according to the manufacturer's specifications. cDNA was diluted 1:5 with PCR-grade H2O prior to qRT-PCR. Primers were designed using Primer3Plus software (48) (see Table S1 in the supplemental material) to amplify products between 80 and 100 bp, with a melting temperature (Tm) of approximately 60°C, and used at a final concentration of 0.4 μM. The primer binding regions of the target genes maeK, maeE, maeP, maeR, and rpoB had identical nucleotide sequences in ABSA 834 and UPSA 807 (data not shown).
Real-time quantification of transcripts was done using the Quantifast SYBR green kit (Qiagen) and a LightCycler 480 II real-time PCR detection system (Roche), according to the manufacturer's specifications. All reactions were performed in triplicate, and transcript amounts were quantified from three RNA preparations that were isolated from independent biological replicates. Standard curves and amplification efficiencies were determined using serially diluted (10-fold) genomic DNA. Expression values were normalized using the rpoB gene of S. agalactiae, which encodes RNA polymerase β subunit (SAG0160). Relative fold change values were calculated using amplification efficiencies, as described previously (49). All experiments conformed to the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines (50). For experiments comparing pH levels, C medium (pH 7.5) was used as a background medium as described previously (51). C medium containing either 100 mM morpholineethanesulfonic acid (MES) or 40 mM malic acid was adjusted to pH 6.0 and filter sterilized prior to use.
Murine model of UTI.
Female C57BL/6 mice (8 to 10 weeks) were purchased from the Animal Resources Centre (Canning Vale, WA, Australia). The murine model of UTI based on transurethral inoculation (52) was used for this study. Briefly, an inoculum of 40 μl, containing 5 × 108 CFU of bacteria in PBS, was instilled into the bladder using a 1-ml syringe attached to a catheter. Groups of 8 to 10 mice were euthanized after 24 h, and bladders were collected and homogenized in sterile PBS for colony counts on THB agar. The experiment was repeated independently. All procedures were approved by and conducted within the guidelines of the Griffith University Animal Research Ethics Committee (approval no. MSC/14/08/AEC).
Statistical analysis.
Bacterial growth was compared using analysis of area under the curve (AUC) and Student's t test. The mean numbers of bacterial CFU per milliliter were compared using an independent-sample t test. All statistical analyses were carried out using IBM SPSS statistics software (version 20.0) and GraphPad Prism software package 5.0. P values of <0.05 were considered significant.
RESULTS
Growth of ABSA and UPSA in human urine.
Initial examination of two different UPSA strains from patients with cystitis (33) and other S. agalactiae isolates from a prior study (34) showed that these organisms grew very poorly in human urine, a finding that was consistent with another previous study (35). Based on these data, we tested and exposed a novel phenotype of efficient growth in human urine for ABSA 1014 that was not observed in several UPSA strains, including UPSA 807 (Fig. 1A). The average generation time of ABSA 1014 was 170 min (calculated between 0 and 36 h), compared to UPSA 807 and other UPSA strains that grew poorly, with generation times of >600 min (two-sample two-tailed t test, P < 0.001). Subsequent analyses of several other UPSA and ABSA strains that were isolated from different patients with acute UTIs and individuals with ABU, respectively, showed similar urine growth patterns (Fig. 1B). Thus, these data establish that multiple ABSA strains can grow robustly in human urine, in contrast to UPSA strains that grow poorly in urine.
FIG 1.

(A) Robust growth of ABSA 1014 in urine contrasts with poor growth of UPSA strains 807, 714, and 058. (B) High-growth phenotypes of additional ABSA strains 729 and 834 in human urine compared to UPSA strains 008 and 872. (C) Direct competition assays revealed markedly superior fitness of ABSA 1014 for growth in urine versus UPSA 807, 714, 008, and 872; *, OD600 of ABSA 1014-UPSA 807 coculture shown; results were similar to those for other cocultures. (D) Robust growth of all strains in THB showed no general growth defect of UPSA strains. Data are pooled results of at least three to five independent assays and show mean values ± standard errors of the means.
The rapid doubling time of ABSA 1014 in human urine compared to UPSA indicated that this strain might outcompete UPSA strains in direct competition assays. In urine that was inoculated with equal numbers of ABSA 1014 and UPSA 807 organisms, or any one of several other UPSA strains (mixed 1:1 at start of assay), the percentage of each strain recovered over time showed the markedly superior fitness of ABSA 1014 for human-urine growth compared to the UPSA strains (Fig. 1C). In these assays, ABSA 1014 constituted 50% of the mixed population at t = 0 h but >90% of mixed cultures after 36 h (P < 0.001, paired two-tailed t test). The superior growth phenotype of ABSA 1014 was urine specific because all of the strains had similar log-phase growth rates in THB, as illustrated in Fig. 1D. Thus, ABSA 1014 outcompetes multiple UPSA strains for rapid growth in human urine through a urine-specific growth trait evidenced by an absence of any general growth defect in UPSA strains detectable using standard THB laboratory medium.
Metabolic profiling of ABSA 1014 and UPSA 807 using PM arrays.
We next undertook a comprehensive comparison of the core physiological properties of ABSA 1014 and UPSA 807 using PM arrays to gain insight into the metabolic potential of these strains. The arrays uncovered only a few metabolic differences between the strains; in fact, there were a total of only four gained phenotypes in ABSA 1014. These were efficient utilization of d,l-malic acid and l-malic acid and resistance to sodium arsenate and cadmium chloride. In addition, there were several phenotypes that were absent in ABSA 1014 compared to UPSA 807, including resistance to macrolides and lincomycin. A complete list of these metabolic profiles is provided in Table 2, which represents all of the metabolic phenotype differences that were observed between the two strains. Subsequent array profiling of two additional ABSA strains, 834 and 729, showed that these strains also exhibited phenotypes of efficient utilization of d,l-malic acid and l-malic acid compared to UPSA 807 (data not shown). A gene encoding a putative malate oxidoreductase has been annotated in the S. agalactiae 2603V/R reference genome; however, the ME pathway has not been characterized in S. agalactiae to date. Malic acid metabolism is typically associated with an operon comprising genes encoding a malate oxidoreductase enzyme (maeE), a permease/transporter (maeP), a transcriptional regulator (maeR), and/or an accessory membrane-anchored sensor kinase (maeK) (53). Bacterial MEs catalyze the oxidative decarboxylation of malate to pyruvate and CO2 (54), which underpins malolactic fermentation. While there is some evidence that MEs contribute to energy generation and bacterial survival at low pH, a role for MEs in bacterial growth in urine has not previously been investigated. Therefore, we examined the ME pathway in ABSA 1014 and UPSA 807 using a combination of genome sequencing, bioinformatics, and functional analyses.
TABLE 2.
Metabolic profile of ABSA 1014 compared to UPSA 807, derived from PM array, and genotype relating to phenotype (substrate or molecule), determined using draft genome assemblies
| Metabolic phenotype gained in ABSA 1014 | Activity level (OmniLog units) | Substrate or molecule | Phenotype relating to substrate or molecule | Genotype relating to substrate or molecule |
|---|---|---|---|---|
| Carbon utilization | 114 | d,l-Malic acid | Carbon source, carboxylic acid | maeK frameshift in UPSA 807 |
| 83 | l-Malic acid | Carbon source, carboxylic acid | maeK frameshift in UPSA 807 | |
| Resistance | 99 | Sodium arsenate | Toxic anion, phosphate analog | ICEsde3396 in ABSA 1014 |
| 305 | Cadmium chloride | Toxic cation | ICEsde3396 in ABSA 1014 | |
| Sensitivity | −148 | Poly-l-lysine | Membrane, detergent, cationic | NDa |
| −207 | Alexidine | Membrane, e− transport, biguanide | ND | |
| −253 | Lincomycin | Protein synthesis, lincosamide | Tn6002 in UPSA 807 | |
| −550 | Oleandomycin | Protein synthesis, macrolide | Tn6002 in UPSA 807 | |
| −465 | Troleandomycin | Protein synthesis, macrolide | Tn6002 in UPSA 807 | |
| −207 | Spiramycin | Protein synthesis, macrolide | Tn6002 in UPSA 807 |
ND, not determined.
Draft genome assembly and analysis of mae genes.
The draft assembled genomes of ABSA 1014 and UPSA 807 showed the presence of the mae gene cluster in both strains (Fig. 2). The mae gene clusters from ABSA 1014 and UPSA 807 shared identical structural organization with the homologous mae cluster from Lactobacillus casei BL23 (55). Notably, however, a frameshift mutation in a poly(A) sequence at the 5′ end of maeK of UPSA 807 was identified (Fig. 2). This mutation would lead to the production of a truncated nonfunctional protein (using an alternative downstream ATG start codon) that lacks the first 19 amino acids corresponding to part of the signal peptide of this membrane protein. The mutation in UPSA 807 (Fig. 2) was confirmed by PCR using primers uppS-F and galE-R (see Table S1 in the supplemental material) and sequencing of the entire mae gene cluster from ABSA 1014 and UPSA 807 (accession numbers KU061060 to KU061067). Further sequence comparisons showed the presence of homologous gene clusters in all complete genomes of S. agalactiae available in the databases. We observed equivalent structural organizations in all strains but noted several other unique disruptions, in particular in all non-human-associated strains (SA20-06, ILRI112, ILRI005, 2-22, and 09mas018883). The details of these disruptions, and their presence in the S. agalactiae strains available in the databases, are illustrated in Fig. 3.
FIG 2.

Organization of the mae locus in ABSA 1014 and UPSA 807. In UPSA 807, maeK contains a single-base-pair deletion in the homopolymeric tract at position +12 at the proximal 5′ end. A potential alternative start codon can be found downstream at position +52.
FIG 3.

Mutations in mae genes in ABSA and UPSA strains in this study (asterisks with accession numbers indicate data from Bioproject identifier PRJEB2837) and in the complete genomes of S. agalactiae strains available in GenBank.
Growth of UPSA 807 in urine following maeK complementation in trans.
To ascertain whether UPSA could grow in human urine in the presence of a fully functioning mae gene cluster, we cloned functional maeK from ABSA 834 into plasmid pDI108, derived from pMSP3545 (47), and transformed this into UPSA 807. Growth experiments in pooled NHU containing 40 mM malic acid demonstrated that the growth of WT UPSA 807 was significantly increased following genetic complementation with maeK (Fig. 4; area under the curve [AUC] analysis, 36 h to 72 h, P < 0.01). The two strains grew similarly in THB with equivalent growth curves over the same time course (data not shown).
FIG 4.
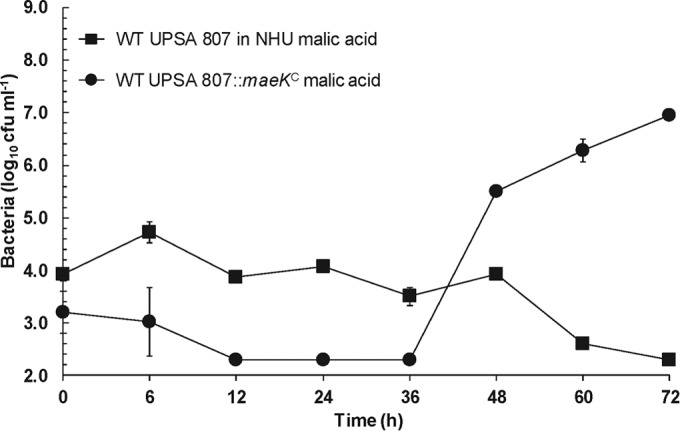
Growth of WT UPSA 807 in pooled normal human urine (NHU) containing 40 mM malic acid and comparison with a maeK genetically complemented strain. Genetic complementation with maeK in trans significantly increased the ability of UPSA 807 to grow in 5-ml NHU cultures containing 40 mM malic acid (area under the curve [AUC] analysis, 36 h to 72 h, P < 0.01). Data are pooled results of at least three to five independent assays and show mean values ± standard errors of the means.
Growth of ABSA and UPSA in synthetic and normal urine containing defined malate levels.
We retrospectively measured the concentrations of malate and creatinine in several of the normal human-urine samples used for growth assays. The malate concentrations ranged between ≤0.2 mM and 8 mM (limit of detection of assay, 0.2 mM), and creatinine ranged between 28 and 211 mg dl−1 (ratio of malic acid to creatinine ranged between 0 and 1,056 μg mg−1). Therefore, we analyzed whether the growth of ABSA and UPSA strains would differ in a synthetic medium that contained a defined amount of malate. For this, we used SHU as defined in previous studies (56–58) supplemented with 40 mM malic acid, a concentration consistent with prior studies that have used 30 to 50 mM malic acid in vitro (51, 53–55, 59–62). We observed that the growth of ABSA 834 and ABSA 729 was significantly higher than the growth of UPSA 058 and UPSA 714 in SHU (Fig. 5A), irrespective of the strain pair comparison and regardless of the presence of 40 mM malic acid (Fig. 5B). Notably, however, chemical supplementation with 40 mM malic acid significantly increased the growth of both ABSA strains and UPSA 058 and UPSA 714 (Fig. 5B, AUC analysis, 24 h to 72 h, P < 0.01); these strains have an intact mae gene cluster (UPSA 058) and a single frameshift mutation that is predicted to alter only the last three amino acids at the C terminus of the protein (and unlikely to disrupt function) (UPSA 714), respectively (Fig. 3).
FIG 5.
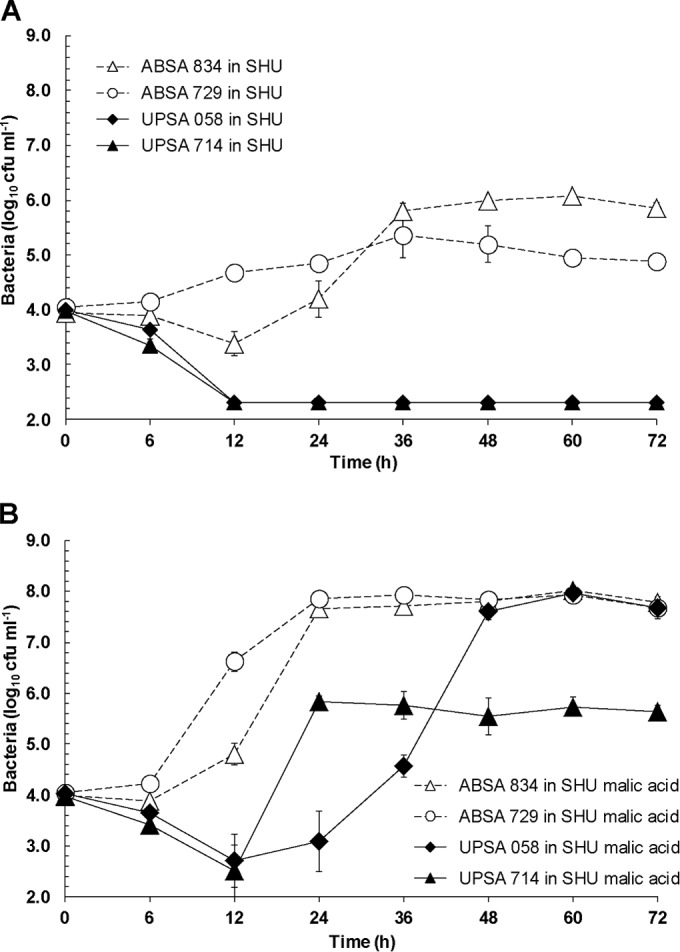
Growth of WT ABSA 834, ABSA 729, UPSA 058, and UPSA 714 in 5-ml cultures of synthetic human urine (SHU) (A) and SHU containing 40 mM malic acid (B). Growth of the ABSA strains was significantly higher than that of UPSA strains (AUC analysis, 24 h to 72 h, P < 0.01). Chemical supplementation with 40 mM malic acid significantly increased the growth of UPSA 058 and UPSA 714 compared to growth conditions without malic acid (AUC analysis, 24 h to 72 h, P < 0.01; comparisons were UPSA 058 without and with malic acid and UPSA 714 without and with malic acid). Data are pooled results of at least three to five independent assays and show mean values ± standard errors of the means.
We also compared the growth rates of WT ABSA 834 in two defined concentrations of malic acid, 8 mM and 40 mM, in NHU to explore any dose-dependent effects and contrasted these effects with the urine growth curves of ABSA 729, UPSA 807, and UPSA 714 based on the mae background of these strains (Fig. 3). These results showed that the growth of ABSA strains and UPSA 714 (harboring a predicted functional ME pathway) was significantly increased at 24 h in the presence of the higher concentration of malic acid (40 mM) (see Fig. S1 in the supplemental material; Student's t test, P < 0.01). The growth of UPSA 807 (harboring a predicted nonfunctional ME pathway) was not augmented by the presence of either concentration of malic acid (see Fig. S1). Thus, growth of ABSA in synthetic and normal urine increases with larger amounts of malic acid, and an intact ME pathway is required for this dose-dependent effect on growth.
In experiments using M9 minimal medium in which glucose was replaced with 40 mM malic acid as a carbon source, we observed significantly higher growth of ABSA 834 and 729 and UPSA 714 and 058 (both harboring predicted functional ME pathways) than of UPSA 807. ABSA 834 was unable to grow in M9 minimal medium in the absence of malic acid, but supplementation with 40 mM malic acid enabled growth to a cell density of 109 CFU ml−1. Other ABSA and UPSA strains with intact mae gene clusters showed similar enhanced growth in M9 medium supplemented with malic acid compared to M9 medium alone (see Fig. S2 in the supplemental material). These results demonstrate that the presence of malic acid as a carbon source can support robust growth of S. agalactiae in a minimal medium.
Mutation in maeK or maeE attenuates the growth of ABSA 834.
We inactivated the ME pathway in ABSA 834 by generating an isogenic maeK-deficient mutant and performed comparative urine growth assays. We used ABSA 834 because we found that ABSA 1014 was not easily transformable, and ABSA 834 exhibited urine growth properties equivalent to those of ABSA 1014. WT ABSA 834 grew significantly better than its maeK-deficient mutant in SHU and pooled NHU containing 40 mM malic acid (AUC analysis, 12 h to 72 h, P < 0.01) (Fig. 6). Complementation of the maeK-deficient mutant in trans using plasmid pDI111 (Table 1) restored the growth of the mutant in SHU (Fig. 6A) but only partially in NHU (Fig. 6B) (AUC analysis, 24 h to 48 h, P < 0.05). WT ABSA 834 also grew significantly better than the maeE- and maeK-deficient mutants in NHU in the absence of supplemental malic acid; however, growth rates of WT ABSA 834 did not differ between aerobic and microaerobic conditions or between shaking and static conditions (data not shown).
FIG 6.
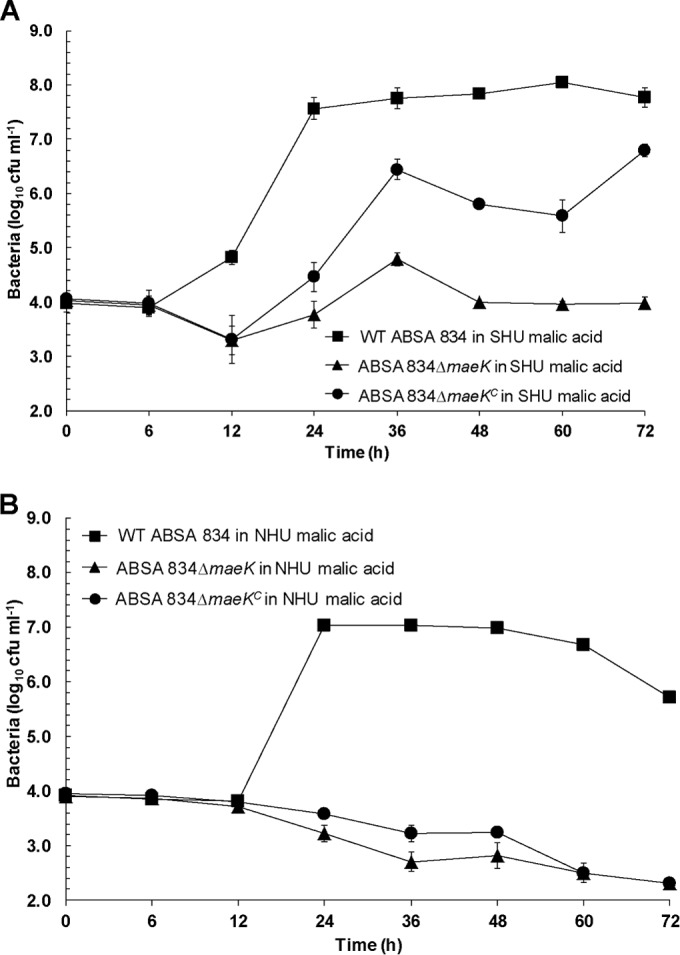
Growth of WT ABSA 834, ABSA 834ΔmaeK, and ABSA 834ΔmaeKC (superscripted “C” indicates complemented mutant strain) in synthetic human urine (SHU) (A) and pooled normal human urine (NHU) (B), both containing 40 mM malic acid (5-ml cultures). Growth of the WT was significantly higher than that of the ABSA 834ΔmaeK mutant (AUC analysis, 12 h to 72 h, P < 0.01). Complementation in trans using plasmid pDI111 partially restored the growth of the mutant in SHU (AUC analysis, 12 h to 72 h, P < 0.01). Data are pooled results of at least three to five independent assays and show mean values ± standard errors of the means.
To define the requirement for maeE for urine growth, we generated a maeE-deficient mutant in ABSA 834. The results of growth assays comparing the WT ABSA 834 and its ABSA 834ΔmaeE mutant derivative in SHU and pooled NHU, both containing 40 mM malic acid, are shown in Fig. 7. The ABSA 834ΔmaeE mutant was significantly attenuated for growth in both SHU and NHU compared to WT ABSA 834. Complementation of the maeE-deficient mutant in trans using plasmid pDI110 (Table 1) significantly restored the growth of the mutant in SHU (Fig. 7A) (AUC analysis, 24 h to 72 h, P < 0.01) and NHU (Fig. 7B) (AUC analysis, 24 h to 72 h, P < 0.01). In these experiments, measurement of the amount of l-malic acid that was present in the growth medium at the start of the assay (t = 0 h) and at 72 h showed a 65 to 70% reduction in malic acid concentration after 72 h of growth of WT ABSA 834 but no significant reduction in malic acid concentration in cultures of the ABSA 834ΔmaeE mutant. Together, these data show that functional maeK and maeE are required for optimal growth of ABSA in urine in the presence of malate.
FIG 7.
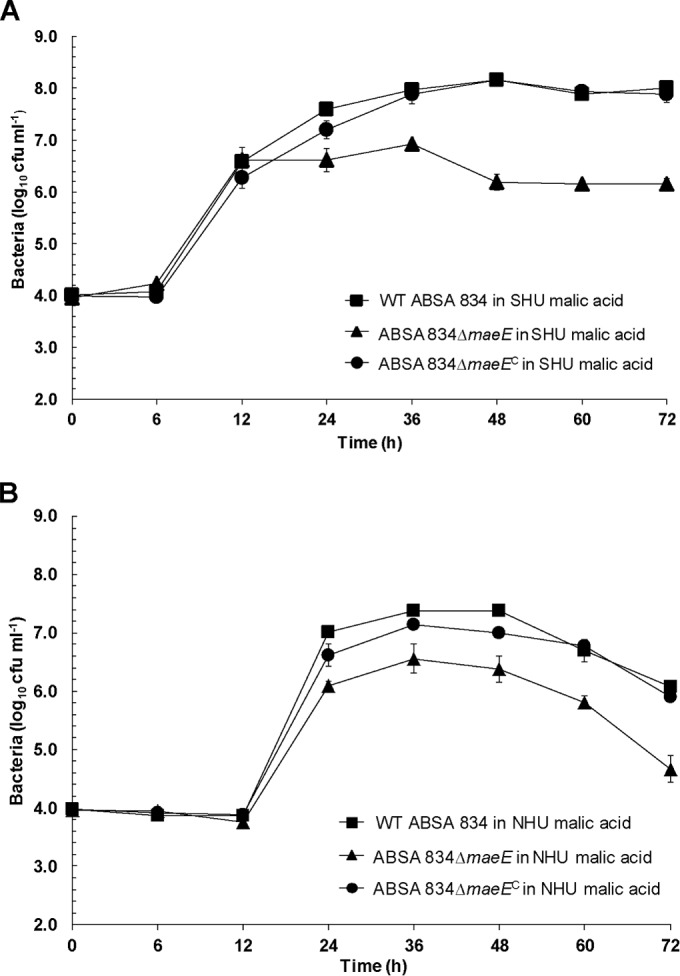
Growth of WT ABSA 834, ABSA 834ΔmaeE, and ABSA 834ΔmaeEC (superscripted “C” indicates complemented mutant strain) in synthetic human urine (SHU) (A) and pooled normal human urine (NHU) (B), both containing 40 mM malic acid (5-ml cultures). Growth of the WT was significantly higher than that of the ABSA 834ΔmaeE mutant (AUC analysis, 24 h to 72 h, P < 0.01). Complementation in trans using plasmid pDI110 partially restored the growth of the mutant (AUC analysis, 24 h to 72 h, P < 0.01). Data are pooled results of at least three to five independent assays and show mean values ± standard errors of the means.
Expression of mae genes in ABSA and UPSA during growth in medium containing malate.
The transcription of all four genes that comprise the mae gene cluster (maeK, maeE, maeP, and maeR) was examined by qRT-PCR in ABSA 834 and UPSA 807 grown in SHU with and without 40 mM malic acid. In these experiments, ABSA 834 significantly upregulated the transcription of maeE and maeP (>10-fold) in response to malic acid during growth in SHU (Fig. 8). Strikingly, and in contrast to ABSA 834, the transcription of these genes in UPSA 807 was unaffected by the presence of malic acid during growth in SHU (Fig. 8). We did not detect any significant changes (>2-fold) in the expression of either maeK or maeR in either ABSA 834 or UPSA 807 when comparing relative transcript abundances following growth in SHU in the presence or absence of malic acid. However, the absolute transcript levels of maeK and maeE did differ between ABSA 834 and UPSA 807 following growth in SHU in the presence of 40 mM malic acid (see Fig. S3 in the supplemental material). Together, these data show that there is a significant mae transcriptional response in ABSA 834, but not in UPSA 807, during growth in SHU containing malic acid.
FIG 8.
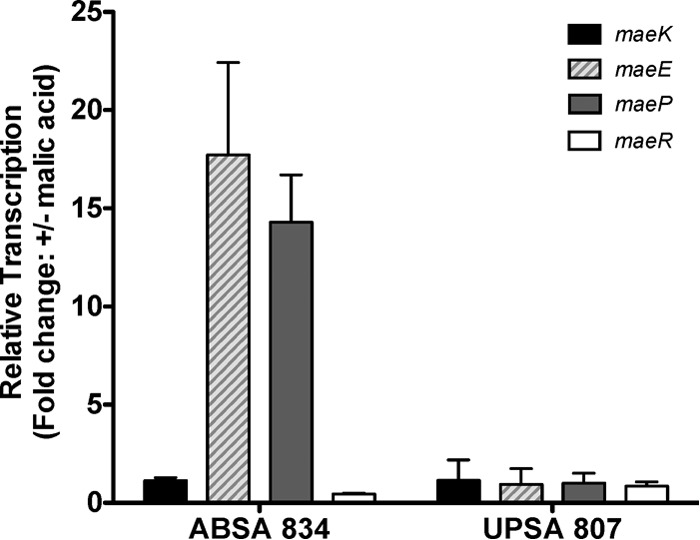
Relative expression of maeK, maeE, maeP, and maeR in ABSA 834 and UPSA 807 following growth in 12.5-ml cultures of synthetic human urine (SHU). qRT-PCR data show fold change relative to the reference rpoB in the presence and absence of 40 mM malic acid. Error bars represent standard deviations between three independent biological replicates.
We also examined whether acidic pH is sufficient to induce mae gene transcription in ABSA 834, independently of malic acid. We found that a shift in pH of 1 unit (6.5 to 5.5) induced transcription of maeE and maeP in SHU in the presence of malic acid (see Fig. S4A in the supplemental material), indicating an additional level of induction linked to pH. Further, using nutrient-rich C medium supplemented with either malic acid or MES, in an approach similar to a previous study (51), we compared transcription levels of maeE and maeP at pH 7.5 and pH 6.0. We observed that the mae genes were induced independently of malic acid as a major carbon source in the growth medium. In these assays (culture densities, 5 × 108 to 2 × 109 CFU ml−1 after 20 h), there were approximately 32- and 14-fold increases in maeE and maeP, respectively, in the presence of malic acid at pH 6.0 (see Fig. S4B), consistent with our experiments described for SHU. In addition, there were approximately 11- and 5-fold increases in maeE and maeP, respectively, in medium containing MES at pH 6.0, which was devoid of malic acid. Thus, consistent with a previous study (51), we observed that acidic conditions, in the absence of malic acid, are sufficient to induce the transcription of the mae genes in ABSA.
ABSA and UPSA colonization in vivo using a murine model of UTI.
We initially analyzed colonization of ABSA 1014 and UPSA 807 in mice to compare the relative fitnesses of these strains in vivo. There was a significant difference in the recovery of ABSA 1014 and UPSA 807 from the bladder of mice at 24 h following infection. ABSA 1014 was recovered in significantly higher numbers than UPSA 807 (Fig. 9A) (1.70 × 105 CFU of ABSA 1014 versus 5.37 × 104 CFU of UPSA 807, P = 0.004, Mann-Whitney U test). We also compared the colonization levels of ABSA 834 and its isogenic maeE-deficient mutant to study the effect of a nonfunctional ME pathway on in vivo colonization. There was a significant difference in the recoveries of WT ABSA 834 and the ABSA 834ΔmaeE mutant from the bladder at 24 h following infection, with 65% fewer mutant bacteria recovered (Fig. 9B) (3.47 × 105 CFU of WT ABSA versus 1.23 × 105 CFU of ABSA 834ΔmaeE, P = 0.042, independent-sample t test). Thus, ABSA 1014 colonizes the urinary tract of mice significantly better than UPSA 807 following experimental infection, and a functional ME pathway in ABSA 834 contributes significantly to this phenotype.
FIG 9.
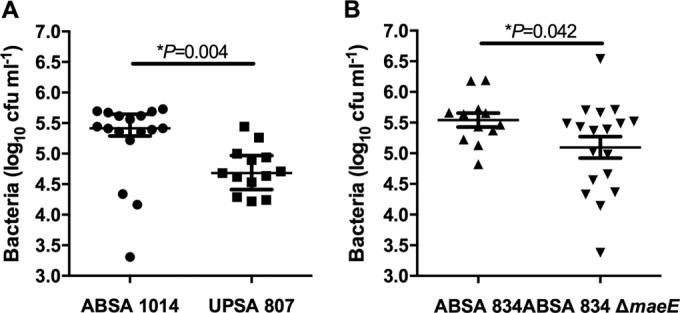
(A) Colonization in the murine UTI model demonstrates significantly higher recovery of ABSA 1014 than of UPSA 807 (*, P = 0.004, Mann-Whitney U test, based on non-normally distributed data according to Shapiro-Wilk test; bars represent medians and show interquartile ranges). (B) Significantly higher recovery of WT ABSA 834 than of ABSA 834ΔmaeE is shown (*, P = 0.042, independent-sample t test, normally distributed data according to Shapiro-Wilk test; bars represent means ± standard errors of the means). Data are presented as pooled results of two independent experiments each containing 8 to 10 mice per group. The limit of detection of the assay was 50 CFU ml−1 for bladder homogenates.
DISCUSSION
The principal aim of this study was to determine whether ABSA might be capable of urine growth, which is a trait of some ABU-causing E. coli strains (24, 25, 32). The central findings of this study are that (i) ABSA can grow efficiently in human urine, in contrast to UPSA strains tested to date that grow poorly; (ii) ABSA 1014 (and 834 and 729) can utilize malic acid in contrast to UPSA 807; (iii) malic acid utilization in ABSA correlates with an intact mae locus; and (iv) in ABSA, a functionally intact ME pathway is required for robust growth in human urine and affords activity for malic acid catabolism that takes place during urine growth. More broadly, the literature shows that ABU-causing E. coli strains bind poorly to urothelial cells and fail to stimulate a strong proinflammatory response during persistent infection (24, 25, 32, 63–65). In contrast, uropathogenic bacteria adhere to urothelial cells and induce inflammation (34, 66, 67). Prior studies showed that UPSA can adhere to urothelial cells efficiently; however, UPSA grew poorly in urine (33, 34), consistent with a report by Stamey and Mihara (35). In this study, the finding that ABSA can utilize urine for robust growth sheds light on how this organism may interact with the host during UTI, and this could influence the persistence of S. agalactiae ABU in some individuals. This hypothesis and the ability of ABSA strains to adhere to urothelial cells will be areas for future investigation.
In defining the phenotypic ABSA metabolome, and comparing this with genetic and mutational analyses, this study provides key insight into the mechanisms underlying urine growth for S. agalactiae. The inability of UPSA 807 to grow in urine correlated with a metabolic deficiency of malic acid catabolism, as revealed using PM arrays. Genome-sequence comparisons with ABSA 1014 highlighted the predicted nonfunctional ME pathway in UPSA 807. Crucial point mutations predicted to impact the functional integrity of the ME pathway in S. agalactiae were confirmed in multiple UPSA strains by comparative analyses. In addition, the targeted generation of a maeE-deficient ABSA mutant showed that inactivation of the ME pathway significantly restricts the growth in an otherwise “robust” strain in NHU and SHU containing malic acid; thus, a functionally active ME pathway contributes to the robust urine growth phenotype in ABSA. Genetic complementation of UPSA 807 with functional maeK in trans conferred an ability of this strain to grow in human urine containing malic acid. To our knowledge, this is the first description of a role for a bacterial ME pathway in human-urine utilization. Interestingly, the presence of a functionally intact ME pathway in two UPSA strains that grew poorly in urine (UPSA 714 and 058) implies that ME divergence is not the sole metabolic distinction between all ABSA and UPSA strains underlying differential urine growth. In other words, growth differences in all ABSA and UPSA strains are not solely explained by differences in malic acid metabolism, and we predict that these differences are likely to be influenced by other, unknown factors. Urine growth differences between some strains, for example, could be influenced by factors other than differences in malic acid metabolism, such as resistance to d-serine (26), metabolism of guanine (30), and iron acquisition (32). More broadly, chronic infection by uropathogenic E. coli (UPEC) is compounded by multiple virulence mechanisms, including adhesion, invasion of uroepithelial cells, evasion of host immune responses, intracellular survival, and replication in host cells, as reviewed elsewhere (67–69). Thus, the impact of virulence on persistence of UPSA in the urinary tract, independent of growth in urine, represents an area of future research for this uropathogen. Other studies using transcriptional profiling would also be of interest to explore the full gamut of gene activation pathways at play during ABSA growth in urine, beyond malic acid metabolism.
l-Malic acid is a common organic acid in fruits and vegetables, is synthesized in the kidney, and can be present in human urine (http://www.hmdb.ca/metabolites/HMDB00156) along with pyruvic acid (70), from which it can be derived. Its concentration in urine is dramatically affected by dietary intake (71). In bacteria, a metabolic signature of l-malic acid utilization is often related to functional expression of the ME pathway, as characterized in Lactobacillus casei (55) and Enterococcus faecalis (54); however, bacteria can metabolize malic acid through different pathways; in E. coli and Bacillus subtilis, this is controlled by a two-component system (72, 73); in lactic acid bacteria, including L. casei (55) and Streptococcus mutans (60, 61), l-malic acid is converted to l-lactate by malolactic enzyme. E. faecalis (54, 74), Streptococcus bovis (75), and L. casei (55) metabolize l-malic acid to pyruvate using the ME pathway. Oral streptococci degrade malic acid to create an alkaline environment that may protect against acid damage and oxidative and starvation stress (60–62). Utilization of malic acid via the ME pathway may therefore enable an organism to survive and grow at a lower pH (55). In our study, growth of ABSA in the acidic-pH environment of human urine (pH 5.5 to 6.2) required an intact ME pathway. A recent study on group A streptococcus (51) identified malate degradation as a link between pH and virulence. Our findings of increased transcriptional activity of the maeE and maeP genes in ABSA in response to malate are also consistent with the response of group A streptococcus (51).
Uropathogenic bacteria, including E. coli and enterococci, can utilize human urine for growth (27, 28, 35). Urinary components, including d-serine, guanine, and small peptides, can influence the growth of these organisms in urine (76, 77). Prior to the current study, the compounds in urine that S. agalactiae may use to grow have not been described. The finding that ABSA can metabolize l-malic acid in urine is interesting; this compound is a widespread organic acid found at high levels in currants, rhubarb, green apples, and grape musts (61, 78); pumpkin fruits (79); and wine (71) and is naturally excreted in human urine (70, 71, 80); its concentration varies dramatically depending on dietary intake. Other variable components in human urine include glucose (51, 53, 55, 70) and fructose, which may inhibit metabolic pathways, including the ME pathway, as reported in Streptococcus lactis (81), E. faecalis (74), and L. casei (55). Considering the various levels of these components in human urine, we used a standardized SHU medium to compare the growth rates of WT and mutant ABSA under controlled growth conditions. Synthetic medium to model human urine has been used in many studies to examine bacterial growth, although the compounds that bacteria utilize for growth remain undefined in most cases. The activity of the ME pathway also depends on divalent cofactors such as Mn2+ or Mg2+ (82). The requirement for cofactors in ABSA ME metabolism in urine remains to be defined.
The findings of the current study can also be compared more broadly to the literature in terms of implications for pathogenesis and infection in UTIs. Recent data from Kline et al. showed that the presence of S. agalactiae in the urinary tract in mice could predispose the host to other uropathogenic bacteria such as E. coli (83). Our findings suggest that ABSA strains that colonize urine more efficiently may have the potential to modify the progression of UTIs due to other causal organisms in some individuals. Furthermore, S. agalactiae has the ability to modulate host immune responses in the bladder, and studies to define potential differences between ABSA and UPSA in terms of immune modulation will now be important. More broadly, there may be potential dietary implications of the findings reported here, for example, for women during pregnancy. Establishing the ME pathway and the presence of malic acid as important for growth of some S. agalactiae strains in human urine points to a need for more studies on the effect of diet on urinary malate and the potential link between diet, malate, and ABU. Whether other bacterial species can utilize malic acid for urine growth is another interesting question to be addressed.
Recent studies on the genomics and virulence of S. agalactiae have identified a range of disease/host-associated genetic traits (84–87) and mechanisms of virulence (88–90). In this sense, we hypothesize that there is likely to be a spectrum of genotypes, phenotypes, and virulence strategies used by UPSA and ABSA to colonize and cause infection in the urinary tract; this would parallel knowledge for UPEC (67). While our data provide initial insight into the relationship between UPSA and ABSA, there are no data to support a concept of mutual exclusivity. Other findings in this study, such as the unique disruptions in the mae gene cluster in non-human-associated S. agalactiae strains, highlight the need for future investigation of mae genes in different host environments. Finally, the discovery of a novel phenotype of robust growth in human urine by ABSA, and the ability of ABSA to utilize malic acid in urine, has implications for our overall understanding of the mechanisms used by this organism to colonize the urinary tract and provides the basis for further studies.
Supplementary Material
ACKNOWLEDGMENTS
We thank the staff at the UAB University Hospital, Department of Pathology, for assistance in collecting samples; Janice King and Yvette Hale for technical assistance; and Flora Gathof for administrative assistance. We also thank David Briles for helpful discussions and laboratory support.
Funding Statement
This work was supported by the Wellcome Trust and Griffith University.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00938-15.
REFERENCES
- 1.Edwards MS, Baker CJ. 2005. Group B streptococcal infections in elderly adults. Clin Infect Dis 41:839–847. doi: 10.1086/432804. [DOI] [PubMed] [Google Scholar]
- 2.Ulett KB, Benjamin WH Jr, Zhuo F, Xiao M, Kong F, Gilbert GL, Schembri MA, Ulett GC. 2009. Diversity of group B streptococcus serotypes causing urinary tract infection in adults. J Clin Microbiol 47:2055–2060. doi: 10.1128/JCM.00154-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tan CK, Ulett KB, Steele M, Benjamin WH Jr, Ulett GC. 2012. Prognostic value of semi-quantitative bacteruria counts in the diagnosis of group B streptococcus urinary tract infection: a 4-year retrospective study in adult patients. BMC Infect Dis 12:273. doi: 10.1186/1471-2334-12-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McKenna DS, Matson S, Northern I. 2003. Maternal group B streptococcal (GBS) genital tract colonization at term in women who have asymptomatic GBS bacteriuria. Infect Dis Obstet Gynecol 11:203–207. doi: 10.1080/10647440300025522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Falagas ME, Rosmarakis ES, Avramopoulos I, Vakalis N. 2006. Streptococcus agalactiae infections in non-pregnant adults: single center experience of a growing clinical problem. Med Sci Monit 12:CR447–CR451. [PubMed] [Google Scholar]
- 6.Anderson BL, Simhan HN, Simons KM, Wiesenfeld HC. 2007. Untreated asymptomatic group B streptococcal bacteriuria early in pregnancy and chorioamnionitis at delivery. Am J Obstet Gynecol 196:524.e1–5. [DOI] [PubMed] [Google Scholar]
- 7.Hernaiz C, Anton N, Alos JI, Orden B, Orellana MA, Colomina J, Redondo J, Gomez-Garces JL. 2004. Clinical significance of Streptococcus agalactiae isolation from urine samples of outpatients from health care centers. Enferm Infecc Microbiol Clin 22:89–91. doi: 10.1016/S0213-005X(04)73040-2. [DOI] [PubMed] [Google Scholar]
- 8.Persson K, Christensen KK, Christensen P, Forsgren A, Jorgensen C, Persson PH. 1985. Asymptomatic bacteriuria during pregnancy with special reference to group B streptococci. Scand J Infect Dis 17:195–199. doi: 10.3109/inf.1985.17.issue-2.11. [DOI] [PubMed] [Google Scholar]
- 9.Munoz P, Coque T, Rodriguez Creixems M, Bernaldo de Quiros JC, Moreno S, Bouza E. 1992. Group B streptococcus: a cause of urinary tract infection in nonpregnant adults. Clin Infect Dis 14:492–496. doi: 10.1093/clinids/14.2.492. [DOI] [PubMed] [Google Scholar]
- 10.Maisey HC, Doran KS, Nizet V. 2008. Recent advances in understanding the molecular basis of group B streptococcus virulence. Expert Rev Mol Med 10:e27. doi: 10.1017/S1462399408000811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rajagopal L. 2009. Understanding the regulation of group B streptococcal virulence factors. Future Microbiol 4:201–221. doi: 10.2217/17460913.4.2.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pettersson K. 2007. Perinatal infection with group B streptococci. Semin Fetal Neonatal Med 12:193–197. doi: 10.1016/j.siny.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 13.Gibbs RS, Schrag S, Schuchat A. 2004. Perinatal infections due to group B streptococci. Obstet Gynecol 104:1062–1076. doi: 10.1097/01.AOG.0000144128.03913.c2. [DOI] [PubMed] [Google Scholar]
- 14.Ipe DS, Sundac L, Benjamin WH Jr, Moore KH, Ulett GC. 2013. Asymptomatic bacteriuria: prevalence rates of causal microorganisms, etiology of infection in different patient populations, and recent advances in molecular detection. FEMS Microbiol Lett 346:1–10. doi: 10.1111/1574-6968.12204. [DOI] [PubMed] [Google Scholar]
- 15.Lumbiganon P, Laopaiboon M, Thinkhamrop J. 2010. Screening and treating asymptomatic bacteriuria in pregnancy. Curr Opin Obstet Gynecol 22:95–99. doi: 10.1097/GCO.0b013e3283374adf. [DOI] [PubMed] [Google Scholar]
- 16.Schnarr J, Smaill F. 2008. Asymptomatic bacteriuria and symptomatic urinary tract infections in pregnancy. Eur J Clin Invest 38(Suppl 2):50–57. doi: 10.1111/j.1365-2362.2008.02009.x. [DOI] [PubMed] [Google Scholar]
- 17.Verani JR, McGee L, Schrag SJ. 2010. Prevention of perinatal group B streptococcal disease–revised guidelines from CDC, 2010. MMWR Recomm Rep 59(RR-10):1–36. [PubMed] [Google Scholar]
- 18.Aungst M, King J, Steele A, Gordon M. 2004. Low colony counts of asymptomatic group B streptococcus bacteriuria: a survey of practice patterns. Am J Perinatol 21:403–407. doi: 10.1055/s-2004-835310. [DOI] [PubMed] [Google Scholar]
- 19.Baker CJ. 1997. Group B streptococcal infections. Clin Perinatol 24:59–70. [PubMed] [Google Scholar]
- 20.Le J, Briggs GG, McKeown A, Bustillo G. 2004. Urinary tract infections during pregnancy. Ann Pharmacother 38:1692–1701. doi: 10.1345/aph.1D630. [DOI] [PubMed] [Google Scholar]
- 21.Whitney CG, Daly S, Limpongsanurak S, Festin MR, Thinn KK, Chipato T, Lumbiganon P, Sauvarin J, Andrews W, Tolosa JE. 2004. The international infections in pregnancy study: group B streptococcal colonization in pregnant women. J Matern Fetal Neonatal Med 15:267–274. doi: 10.1080/14767050410001668617. [DOI] [PubMed] [Google Scholar]
- 22.Allen VM, Yudin MH, Bouchard C, Boucher M, Caddy S, Castillo E, Money DM, Murphy KE, Ogilvie G, Paquet C, van Schalkwyk J, Senikas V. 2012. Management of group B streptococcal bacteriuria in pregnancy. J Obstet Gynaecol Can 34:482–486. [DOI] [PubMed] [Google Scholar]
- 23.Kessous R, Weintraub AY, Sergienko R, Lazer T, Press F, Wiznitzer A, Sheiner E. 2012. Bacteruria with group-B streptococcus: is it a risk factor for adverse pregnancy outcomes? J Matern Fetal Neonatal Med 25:1983–1986. doi: 10.3109/14767058.2012.671872. [DOI] [PubMed] [Google Scholar]
- 24.Roos V, Ulett GC, Schembri MA, Klemm P. 2006. The asymptomatic bacteriuria Escherichia coli strain 83972 outcompetes uropathogenic E. coli strains in human urine. Infect Immun 74:615–624. doi: 10.1128/IAI.74.1.615-624.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klemm P, Roos V, Ulett GC, Svanborg C, Schembri MA. 2006. Molecular characterization of the Escherichia coli asymptomatic bacteriuria strain 83972: the taming of a pathogen. Infect Immun 74:781–785. doi: 10.1128/IAI.74.1.781-785.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sakinc T, Michalski N, Kleine B, Gatermann SG. 2009. The uropathogenic species Staphylococcus saprophyticus tolerates a high concentration of d-serine. FEMS Microbiol Lett 299:60–64. doi: 10.1111/j.1574-6968.2009.01731.x. [DOI] [PubMed] [Google Scholar]
- 27.Vebo HC, Solheim M, Snipen L, Nes IF, Brede DA. 2010. Comparative genomic analysis of pathogenic and probiotic Enterococcus faecalis isolates, and their transcriptional responses to growth in human urine. PLoS One 5:e12489. doi: 10.1371/journal.pone.0012489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hull RA, Hull SI. 1997. Nutritional requirements for growth of uropathogenic Escherichia coli in human urine. Infect Immun 65:1960–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aubron C, Glodt J, Matar C, Huet O, Borderie D, Dobrindt U, Duranteau J, Denamur E, Conti M, Bouvet O. 2012. Variation in endogenous oxidative stress in Escherichia coli natural isolates during growth in urine. BMC Microbiol 12:120. doi: 10.1186/1471-2180-12-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Russo TA, Jodush ST, Brown JJ, Johnson JR. 1996. Identification of two previously unrecognized genes (guaA and argC) important for uropathogenesis. Mol Microbiol 22:217–229. doi: 10.1046/j.1365-2958.1996.00096.x. [DOI] [PubMed] [Google Scholar]
- 31.Vejborg RM, de Evgrafov MR, Phan MD, Totsika M, Schembri MA, Hancock V. 2012. Identification of genes important for growth of asymptomatic bacteriuria Escherichia coli in urine. Infect Immun 80:3179–3188. doi: 10.1128/IAI.00473-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watts RE, Totsika M, Challinor VL, Mabbett AN, Ulett GC, De Voss JJ, Schembri MA. 2012. Contribution of siderophore systems to growth and urinary tract colonization of asymptomatic bacteriuria Escherichia coli. Infect Immun 80:333–344. doi: 10.1128/IAI.05594-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ulett GC, Webb RI, Ulett KB, Cui X, Benjamin WH, Crowley M, Schembri MA. 2010. Group B streptococcus (GBS) urinary tract infection involves binding of GBS to bladder uroepithelium and potent but GBS-specific induction of interleukin 1alpha. J Infect Dis 201:866–870. doi: 10.1086/650696. [DOI] [PubMed] [Google Scholar]
- 34.Tan CK, Carey AJ, Cui X, Webb RI, Ipe D, Crowley M, Cripps AW, Benjamin WH Jr, Ulett KB, Schembri MA, Ulett GC. 2012. Genome-wide mapping of cystitis due to Streptococcus agalactiae and Escherichia coli in mice identifies a unique bladder transcriptome that signifies pathogen-specific antimicrobial defense against urinary tract infection. Infect Immun 80:3145–3160. doi: 10.1128/IAI.00023-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stamey TA, Mihara G. 1980. Observations on the growth of urethral and vaginal bacteria in sterile urine. J Urol 124:461–463. [DOI] [PubMed] [Google Scholar]
- 36.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 37.Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carver T, Berriman M, Tivey A, Patel C, Bohme U, Barrell BG, Parkhill J, Rajandream MA. 2008. Artemis and ACT: viewing, annotating and comparing sequences stored in a relational database. Bioinformatics 24:2672–2676. doi: 10.1093/bioinformatics/btn529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sullivan MJ, Petty NK, Beatson SA. 2011. Easyfig: a genome comparison visualizer. Bioinformatics 27:1009–1010. doi: 10.1093/bioinformatics/btr039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ulett GC, Bohnsack JF, Armstrong J, Adderson EE. 2003. Beta-hemolysin-independent induction of apoptosis of macrophages infected with serotype III group B streptococcus. J Infect Dis 188:1049–1053. doi: 10.1086/378202. [DOI] [PubMed] [Google Scholar]
- 42.Pritzlaff CA, Chang JC, Kuo SP, Tamura GS, Rubens CE, Nizet V. 2001. Genetic basis for the beta-haemolytic/cytolytic activity of group B streptococcus. Mol Microbiol 39:236–247. doi: 10.1046/j.1365-2958.2001.02211.x. [DOI] [PubMed] [Google Scholar]
- 43.Husmann LK, Yung DL, Hollingshead SK, Scott JR. 1997. Role of putative virulence factors of Streptococcus pyogenes in mouse models of long-term throat colonization and pneumonia. Infect Immun 65:1422–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perez-Casal J, Caparon MG, Scott JR. 1991. Mry, a trans-acting positive regulator of the M protein gene of Streptococcus pyogenes with similarity to the receptor proteins of two-component regulatory systems. J Bacteriol 173:2617–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yim HH, Rubens CE. 1998. Site-specific homologous recombination mutagenesis in group B streptococci. Methods Cell Sci 20:13–20. doi: 10.1023/A:1009810002276. [DOI] [Google Scholar]
- 46.Framson PE, Nittayajarn A, Merry J, Youngman P, Rubens CE. 1997. New genetic techniques for group B streptococci: high-efficiency transformation, maintenance of temperature-sensitive pWV01 plasmids, and mutagenesis with Tn917. Appl Environ Microbiol 63:3539–3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bryan EM, Bae T, Kleerebezem M, Dunny GM. 2000. Improved vectors for nisin-controlled expression in gram-positive bacteria. Plasmid 44:183–190. doi: 10.1006/plas.2000.1484. [DOI] [PubMed] [Google Scholar]
- 48.Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JA. 2007. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res 35:W71–W74. doi: 10.1093/nar/gkm306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT. 2009. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55:611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- 51.Paluscio E, Caparon MG. 2015. Streptococcus pyogenes malate degradation pathway links pH regulation and virulence. Infect Immun 83:1162–1171. doi: 10.1128/IAI.02814-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carey AJ, Tan CK, Ipe DS, Sullivan MJ, Cripps AW, Schembri MA, Ulett GC. 26 May 2015. Urinary tract infection of mice to model human disease: practicalities, implications and limitations. Crit Rev Microbiol doi: 10.3109/1040841X.2015.1028885. [DOI] [PubMed] [Google Scholar]
- 53.Mortera P, Espariz M, Suarez C, Repizo G, Deutscher J, Alarcon S, Blancato V, Magni C. 2012. Fine-tuned transcriptional regulation of malate operons in Enterococcus faecalis. Appl Environ Microbiol 78:1936–1945. doi: 10.1128/AEM.07280-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Espariz M, Repizo G, Blancato V, Mortera P, Alarcon S, Magni C. 2011. Identification of malic and soluble oxaloacetate decarboxylase enzymes in Enterococcus faecalis. FEBS J 278:2140–2151. doi: 10.1111/j.1742-4658.2011.08131.x. [DOI] [PubMed] [Google Scholar]
- 55.Landete JM, Garcia-Haro L, Blasco A, Manzanares P, Berbegal C, Monedero V, Zuniga M. 2010. Requirement of the Lactobacillus casei MaeKR two-component system for l-malic acid utilization via a malic enzyme pathway. Appl Environ Microbiol 76:84–95. doi: 10.1128/AEM.02145-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martino PD, Fursy R, Bret L, Sundararaju B, Phillips RS. 2003. Indole can act as an extracellular signal to regulate biofilm formation of Escherichia coli and other indole-producing bacteria. Can J Microbiol 49:443–449. doi: 10.1139/w03-056. [DOI] [PubMed] [Google Scholar]
- 57.Griffith DP, Musher DM, Itin C. 1976. Urease. The primary cause of infection-induced urinary stones. Invest Urol 13:346–350. [PubMed] [Google Scholar]
- 58.Minuth JN, Musher DM, Thorsteinsson SB. 1976. Inhibition of the antibacterial activity of gentamicin by urine. J Infect Dis 133:14–21. doi: 10.1093/infdis/133.1.14. [DOI] [PubMed] [Google Scholar]
- 59.Cho GS, Krauss S, Huch M, Du Toit M, Franz CM. 2011. Development of a quantitative PCR for detection of Lactobacillus plantarum starters during wine malolactic fermentation. J Microbiol Biotechnol 21:1280–1286. doi: 10.4014/jmb.1107.07003. [DOI] [PubMed] [Google Scholar]
- 60.Lemme A, Sztajer H, Wagner-Dobler I. 2010. Characterization of mleR, a positive regulator of malolactic fermentation and part of the acid tolerance response in Streptococcus mutans. BMC Microbiol 10:58. doi: 10.1186/1471-2180-10-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sheng J, Marquis RE. 2007. Malolactic fermentation by Streptococcus mutans. FEMS Microbiol Lett 272:196–201. doi: 10.1111/j.1574-6968.2007.00744.x. [DOI] [PubMed] [Google Scholar]
- 62.Sheng J, Baldeck JD, Nguyen PT, Quivey RG Jr, Marquis RE. 2010. Alkali production associated with malolactic fermentation by oral streptococci and protection against acid, oxidative, or starvation damage. Can J Microbiol 56:539–547. doi: 10.1139/W10-039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ragnarsdottir B, Fischer H, Godaly G, Gronberg-Hernandez J, Gustafsson M, Karpman D, Lundstedt AC, Lutay N, Ramisch S, Svensson ML, Wullt B, Yadav M, Svanborg C. 2008. TLR- and CXCR1-dependent innate immunity: insights into the genetics of urinary tract infections. Eur J Clin Invest 38(Suppl 2):12–20. doi: 10.1111/j.1365-2362.2008.02004.x. [DOI] [PubMed] [Google Scholar]
- 64.Ragnarsdottir B, Samuelsson M, Gustafsson MC, Leijonhufvud I, Karpman D, Svanborg C. 2007. Reduced toll-like receptor 4 expression in children with asymptomatic bacteriuria. J Infect Dis 196:475–484. doi: 10.1086/518893. [DOI] [PubMed] [Google Scholar]
- 65.Ragnarsdottir B, Svanborg C. 2012. Susceptibility to acute pyelonephritis or asymptomatic bacteriuria: host-pathogen interaction in urinary tract infections. Pediatr Nephrol 27:2017–2029. doi: 10.1007/s00467-011-2089-1. [DOI] [PubMed] [Google Scholar]
- 66.Duell BL, Carey AJ, Tan CK, Cui X, Webb RI, Totsika M, Schembri MA, Derrington P, Irving-Rodgers H, Brooks AJ, Cripps AW, Crowley M, Ulett GC. 2012. Innate transcriptional networks activated in bladder in response to uropathogenic Escherichia coli drive diverse biological pathways and rapid synthesis of IL-10 for defense against bacterial urinary tract infection. J Immunol 188:781–792. doi: 10.4049/jimmunol.1101231. [DOI] [PubMed] [Google Scholar]
- 67.Ulett GC, Totsika M, Schaale K, Carey AJ, Sweet MJ, Schembri MA. 2013. Uropathogenic Escherichia coli virulence and innate immune responses during urinary tract infection. Curr Opin Microbiol 16:100–107. doi: 10.1016/j.mib.2013.01.005. [DOI] [PubMed] [Google Scholar]
- 68.Hannan TJ, Totsika M, Mansfield KJ, Moore KH, Schembri MA, Hultgren SJ. 2012. Host-pathogen checkpoints and population bottlenecks in persistent and intracellular uropathogenic Escherichia coli bladder infection. FEMS Microbiol Rev 36:616–648. doi: 10.1111/j.1574-6976.2012.00339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sivick KE, Mobley HL. 2010. Waging war against uropathogenic Escherichia coli: winning back the urinary tract. Infect Immun 78:568–585. doi: 10.1128/IAI.01000-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bouatra S, Aziat F, Mandal R, Guo AC, Wilson MR, Knox C, Bjorndahl TC, Krishnamurthy R, Saleem F, Liu P, Dame ZT, Poelzer J, Huynh J, Yallou FS, Psychogios N, Dong E, Bogumil R, Roehring C, Wishart DS. 2013. The human urine metabolome. PLoS One 8:e73076. doi: 10.1371/journal.pone.0073076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Regueiro J, Vallverdu-Queralt A, Simal-Gandara J, Estruch R, Lamuela-Raventos R. 2013. Development of a LC-ESI-MS/MS approach for the rapid quantification of main wine organic acids in human urine. J Agric Food Chem 61:6763–6768. doi: 10.1021/jf401839g. [DOI] [PubMed] [Google Scholar]
- 72.Golby P, Davies S, Kelly DJ, Guest JR, Andrews SC. 1999. Identification and characterization of a two-component sensor-kinase and response-regulator system (DcuS-DcuR) controlling gene expression in response to C4-dicarboxylates in Escherichia coli. J Bacteriol 181:1238–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tanaka K, Kobayashi K, Ogasawara N. 2003. The Bacillus subtilis YufLM two-component system regulates the expression of the malate transporters MaeN (YufR) and YflS, and is essential for utilization of malate in minimal medium. Microbiology 149:2317–2329. doi: 10.1099/mic.0.26257-0. [DOI] [PubMed] [Google Scholar]
- 74.London J, Meyer EY. 1969. Malate utilization by a group D streptococcus: physiological properties and purification of an inducible malic enzyme. J Bacteriol 98:705–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kawai S, Suzuki H, Yamamoto K, Kumagai H. 1997. Characterization of the l-malate permease gene (maeP) of Streptococcus bovis ATCC 15352. J Bacteriol 179:4056–4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Alteri CJ, Mobley HL. 2012. Escherichia coli physiology and metabolism dictates adaptation to diverse host microenvironments. Curr Opin Microbiol 15:3–9. doi: 10.1016/j.mib.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Alteri CJ, Mobley HL. 2007. Quantitative profile of the uropathogenic Escherichia coli outer membrane proteome during growth in human urine. Infect Immun 75:2679–2688. doi: 10.1128/IAI.00076-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ribéreau-Gayon P, Glories Y, Maujean A, Dubourdieu D. 2006. Handbook of enology, 2nd ed, vol 2, p 1–49. John Wiley & Sons, Ltd, Chichester, United Kingdom. [Google Scholar]
- 79.Nawirska-Olszanska A, Biesiada A, Sokol-Letowska A, Kucharska AZ. 2014. Characteristics of organic acids in the fruit of different pumpkin species. Food Chem 148:415–419. doi: 10.1016/j.foodchem.2013.10.080. [DOI] [PubMed] [Google Scholar]
- 80.Zaura DS, Metcoff J. 1969. Quantification of seven tricarboxylic acid cycle and related acids in human urine by gas-liquid chromatography. Anal Chem 41:1781–1787. doi: 10.1021/ac60282a034. [DOI] [PubMed] [Google Scholar]
- 81.Renault PP, Heslot H. 1987. Selection of Streptococcus lactis mutants defective in malolactic fermentation. Appl Environ Microbiol 53:320–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chang GG, Tong L. 2003. Structure and function of malic enzymes, a new class of oxidative decarboxylases. Biochemistry 42:12721–12733. doi: 10.1021/bi035251+. [DOI] [PubMed] [Google Scholar]
- 83.Kline KA, Schwartz DJ, Gilbert NM, Hultgren SJ, Lewis AL. 2012. Immune modulation by group B streptococcus influences host susceptibility to urinary tract infection by uropathogenic Escherichia coli. Infect Immun 80:4186–4194. doi: 10.1128/IAI.00684-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Delannoy CM, Zadoks RN, Crumlish M, Rodgers D, Lainson FA, Ferguson HW, Turnbull J, Fontaine MC. 15 November 2014. Genomic comparison of virulent and non-virulent Streptococcus agalactiae in fish. J Fish Dis doi: 10.1111/jfd.12319. [DOI] [PubMed] [Google Scholar]
- 85.Rosinski-Chupin I, Sauvage E, Mairey B, Mangenot S, Ma L, Da Cunha V, Rusniok C, Bouchier C, Barbe V, Glaser P. 2013. Reductive evolution in Streptococcus agalactiae and the emergence of a host adapted lineage. BMC Genomics 14:252. doi: 10.1186/1471-2164-14-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Richards VP, Lang P, Bitar PD, Lefebure T, Schukken YH, Zadoks RN, Stanhope MJ. 2011. Comparative genomics and the role of lateral gene transfer in the evolution of bovine adapted Streptococcus agalactiae. Infect Genet Evol 11:1263–1275. doi: 10.1016/j.meegid.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sorensen UB, Poulsen K, Ghezzo C, Margarit I, Kilian M. 2010. Emergence and global dissemination of host-specific Streptococcus agalactiae clones. mBio 1:e00178-10. doi: 10.1128/mBio.00178-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dando SJ, Mackay-Sim A, Norton R, Currie BJ, St John JA, Ekberg JA, Batzloff M, Ulett GC, Beacham IR. 2014. Pathogens penetrating the central nervous system: infection pathways and the cellular and molecular mechanisms of invasion. Clin Microbiol Rev 27:691–726. doi: 10.1128/CMR.00118-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Okumura CY, Nizet V. 2014. Subterfuge and sabotage: evasion of host innate defenses by invasive gram-positive bacterial pathogens. Annu Rev Microbiol 68:439–458. doi: 10.1146/annurev-micro-092412-155711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Landwehr-Kenzel S, Henneke P. 2014. Interaction of Streptococcus agalactiae and cellular innate immunity in colonization and disease. Front Immunol 5:519. doi: 10.3389/fimmu.2014.00519. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.