

Abstract
INTRODUCTION
The thrombospondins (TSPs) are matricellular proteins that exert multifunctional effects by binding cytokines, cell-surface receptors and other proteins. TSPs play important roles in vascular pathobiology and are all expressed in arterial lesions. The differential effects of TSP-1, -2, and -5 represent a gap in knowledge in vascular smooth muscle cell (VSMC) physiology. Our objective is to determine if structural differences of the TSPs imparted different effects on VSMC functions critical to the formation of neointimal hyperplasia. We hypothesize that TSP-1 and -2 induce similar patterns of migration, proliferation and gene expression, while the effects of TSP-5 are different.
METHODS
Human aortic VSMC chemotaxis was tested for TSP-2 and TSP-5 (1 – 40μg/mL), and compared to TSP-1 and serum-free media (SFM) using a modified Boyden chamber. Next, VSMCs were exposed to TSP-1, TSP-2 or TSP-5 (0.2 – 40μg/mL). Proliferation was assessed by MTS assay. Finally, VSMCs were exposed to TSP-1, TSP-2, TSP-5 or SFM for 3, 6 or 24 hours. Quantitative real-time PCR was performed on 96 genes using a microfluidic card. Statistical analysis was performed by ANOVA or t-test, with p<0.05 being significant.
RESULTS
TSP-1, TSP-2 and TSP-5 at 20μg/mL all induce chemotaxis 3.1 fold compared to serum-free media. TSP-1 and TSP-2 induced proliferation 53% and 54% respectively, whereas TSP-5 did not. In the gene analysis, overall, cardiovascular system development and function is the canonical pathway most influenced by TSP treatment, and includes multiple growth factors, cytokines and proteases implicated in cellular migration, proliferation, vasculogenesis, apoptosis and inflammation pathways.
CONCLUSIONS AND RELEVANCE
The results of this study indicate TSP-1, -2, and -5 play active roles in VSMC physiology and gene expression. Similarly to TSP-1, VSMC chemotaxis to TSP-2 and -5 is dose-dependent. TSP-1 and -2 induces VSMC proliferation, but TSP-5 does not, likely due conservation of N-terminal domains in TSP-1 and -2. In addition, TSP-1, -2 and -5 significantly affect VSMC gene expression; however, little overlap exists in the specific genes altered. This study further delineates TSP-1, -2 and -5’s contributions to processes related to VSMC physiology.
KEYWORDS (MeSH): Thrombospondins, Gene Expression Profiling, Vascular Smooth Muscles, Cartilage Oligomeric Matrix Protein, Cell Migration, Cellular Proliferation
INTRODUCTION
Despite advances in angioplasty and stenting technology, restenosis secondary to neointimal hyperplasia remains a critical component of failure of both open and percutaneous revascularization procedures. Neointimal hyperplasia is characterized by vascular smooth muscle cell (VSMC) migration and proliferation following endothelial cell (EC) injury and induction of inflammatory pathways. Signaling between ECs, leukocytes, VSMCs and platelets in response to vessel injury have been shown to induce phenotypic changes in VSMCs, ultimately resulting in proliferation and deposition of extracellular matrix (ECM) [1]. One example of the proteins expressed in this injury pattern includes thrombospondins (TSPs), a family of multifunctional matricellular glycoproteins. While these proteins do not contribute to the normal ECM structural architecture, they do function in cellular signaling by interacting with other ECM proteins, such as cell surface receptors, proteases and cytokines [2].
The TSP family consists of 5 member proteins, which are divided into Groups A (TSP-1, TSP-2) and B (TSP-3, TSP-4, TSP-5), determined by the inclusion (Group A) or exclusion (Group B) of Type 1 repeats, domains found at the N-terminal side of the molecules [3]. TSP-1 is the most studied TSP, and is established as a potent anti-angiogenic protein [4] and an inducer of VSMC chemotaxis and proliferation [5]. Functions such as inducing EC apoptosis are experimentally attributed to CD36 binding of Type 1 repeats, while others, such as CD47 and integrin binding are attributed to the relatively conserved C-terminal domain [6]. Previous experiments indicate hyperglycemia, dyslipidemia, leptin, inflammatory mediators, such as TGFβ1, and statins drugs all regulate TSP-1 expression and function [7–10], implying TSP-1’s importance to vascular pathologies such as atherosclerosis and intimal hyperplasia. While TSP-1 is well characterized in the literature, less is known about the effects of other TSPs on VSMCs and their contributions to vascular disease progression.
TSP-2 is a Group A TSP, closely resembling TSP-1 in structure. Some of TSP-2’s functions are reported to mimic those of TSP-1, i.e. inhibiting angiogenesis, inducing EC apoptosis and participating in integrin-mediated signaling pathways [2]. However, some key differences distinguish TSP-1 from TSP-2. While TSP-1 and TSP-2 are both secreted by VSMCs, macrophages and fibroblasts, TSP-1 is stored in platelet α granules, and released in relatively large quantities following arterial injury, whereas TSP-2 is not. In one study, a wound healing assay, the release of TSP-2 from VSMCs was not detected until 3 days following injury, and peaked as far out as 10 days post-injury [11], whereas TSP-1 was detected early after injury, peaking at 3 days and subsiding by day 7. This finding suggests TSP-1 may function more as an acute phase reactant, while TSP-2 may be more important during the subsequent inflammation and remodeling phases.
TSP-5, also known as cartilage oligomeric matrix protein (COMP), is a Group B TSP notably different in structure compared to Group A TSPs. While the C-terminus of TSPs is relatively conserved, the N-terminus of TSP-5 lacks Type 1 repeats, and is notably truncated compared to other TSPs [12]. Previous studies have identified TSP-5 in normal rat aorta [13], human VSMCs [12] and the atherosclerotic plaques of ApoE−/− mice [14], implicating an importance to vascular pathobiology and a potential target for therapy; however, few experiments exploring TSP-5 and its relevance to arterial disease have been performed to date.
In the present study, we sought to identify the differences Group A and B TSPs impart on VSMC migration, proliferation, and gene expression. Specifically, we hypothesized that: (1) similarities in TSP-1 and TSP-2 structure would induce similar VSMC chemotaxis and proliferation, as well as similar alterations in pro-migratory and pro-inflammatory genes; and (2) structural differences of TSP-5 would induce VSMC chemotaxis, but not migration, and provide a distinct pattern of gene expression.
METHODS
Materials
TSP-1 was obtained from Athens Research (Athens, GA). Recombinant TSP-2 and TSP-5 were obtained from R&D Systems (Minneapolis, MN). Smooth muscle cell growth medium was purchased from Cell Applications, Inc. (San Diego, CA, USA). Dulbecco’s Modified Eagle Medium (DMEM), used as serum-free media (SFM), trypsin and trypsin neutralizing solution were purchased from Lonza (Walkersville, MD, USA).
Cell Culture
Human aortic VSMCs were obtained from Cell Applications, Inc. (San Diego, CA, USA) and used in early passage (P3-5). Cells were made quiescent by incubation in SFM for 48 hours.
Migration and Proliferation assays
Chemotaxis to SFM or TSP-2 or TSP-5 (1μg/mL – 40μg/mL) was assessed using a modified Boyden chemotaxis chamber (4 hours at 37° C). Results were recorded as cells migrated per 5 high power fields (400x). TSP-1 at a concentration of 20μg/mL was used as a positive control, as maximal migration at this concentration was previously established [9].
For proliferation, quiescent cells were exposed to SFM or TSP-1, TSP-2 or TSP-5 (0.2μg/mL – 40μg/mL) for 72 hours. Proliferation was assessed by MTS tetrazolium absorbance assay (Cell Titer 96 Aqueous One Solution, Promega, Madison, WI).
RNA isolation and cDNA reverse transcription
Quiescent cells were exposed to SFM, TSP-1, TSP-2 or TSP-5 (20μg/mL) for 3, 6 or 24 hours. Media was decanted and cells frozen to −80°C. RNA was isolated with RNeasy mini kit (Qiagen, Germantown, MD). Quality of the RNA samples was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). cDNA was generated using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Grand Island, NY) using a C1000 Touch Thermocycler (Bio Rad, Hercules, CA).
Real-time quantitative reverse transcriptase-polymerase chain reaction
TaqMan® Gene Signature Human Angiogenesis Array microfluidic assay cards run on a QuantStudio 7Flex Real-time PCR system (Applied Biosystems, Grand Island, NY) were used to determine gene expression in the SFM, TSP-1, TSP-2 and TSP-5 samples at 3, 6 and 24 hours.
Statistical analysis
The migration and proliferation assays were performed three times in triplicate, and analyzed by ANOVA in StatView (SAS Institute, Cary, NC). The gene expression assays were performed in duplicate as per the manufacturer’s recommendation and interpreted by t-test using Expression Suite 7 software (Applied Biosystems, Grand Island, NY); p values <0.05 were considered significant.
RESULTS
Migration
TSP-2 induced VSMC migration at all concentrations tested (p <0.05, figure 1). The 20μg/mL concentration caused the greatest increase in migration by 3.11 (± 0.20) fold. The 5μg/mL and 10μg/mL concentrations increased migration 2.26 (± 0.19) −2.60 (± 0.11) fold, but were not significantly different from each other. The 1μg/mL and 40μg/mL concentrations induced migration 1.72 (± 0.17) and 2.12 (± 0.88) fold.
Figure 1.
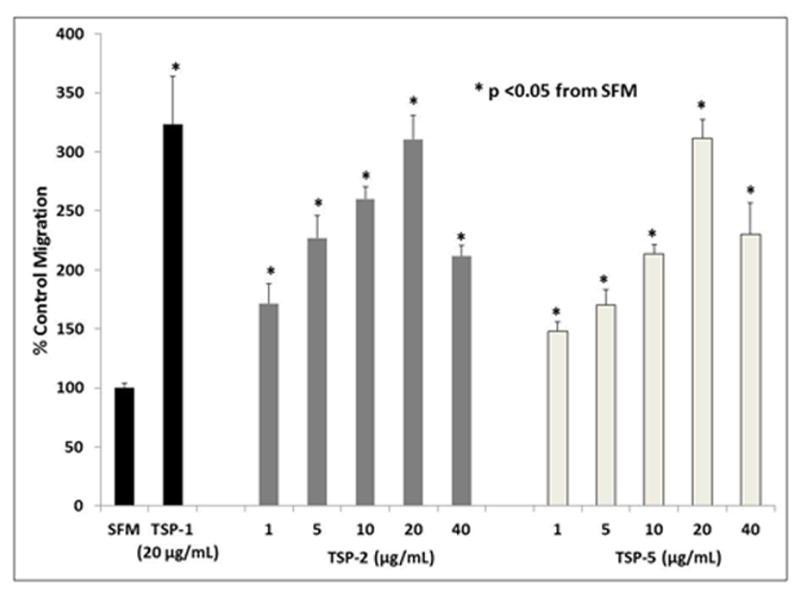
VSMC migration curve for TSP-2 and TSP-5, compared to SFM and TSP-1. Quiescent VSMCs were plated in a micro-Boyden chemotaxis chamber for 4 hours, using 1μg/mL to 40 μg/ml of TSP-2 or TSP-5 as chemoattractants. SFM and 20μg/mL TSP-1 acted as negative and positive controls respectively. Migration was assessed as cells counted per five high-powered fields, normalized to SFM, analyzed by ANOVA (p <0.05).
TSP-5 induced VSMC migration at all concentrations tested (p<0.05, Figure 1). The 20μg/mL concentration induced the greatest increase in migration at 3.11 (± 0.16) fold. The 10μg/mL and 40μg/mL concentrations increased migration 2.13 (± 0.83) and 2.30 (± 0.27) fold respectively, but were not significantly different from each other. Lastly, the 1μg/mL and 5μg/mL concentrations each induced less than two-fold change (1.48 ± 0.08 and 1.70 ± 0.13, respectively).
Proliferation
TSP-1 at a concentration of 0.2μg/mL increased absorbance at 490nm from 0.416 (± 0.018) to 0.637 (± 0.067). At 40μg/mL, absorbance was decreased to 0.191 (± 0.003). This corresponds to an increase in proliferation by 53% at a concentration of 0.2μg/mL and decreased proliferation by 54% at 40μg/mL. TSP-2 at 2μg/mL increased absorbance to 0.475 (± 0.35), corresponding to an increase in proliferation by 48%. No significant change in proliferation was noted at other concentrations for either TSP-1 or TSP-2. TSP-5 did not significantly induce or inhibit VSMC proliferation at any concentration tested (p<0.05, Figure 2).
Figure 2.
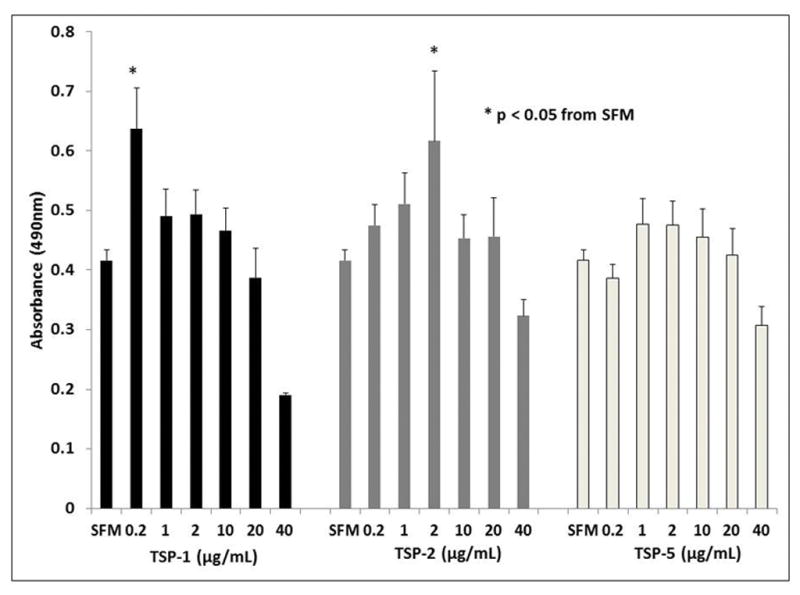
VSMC proliferation curves for TSP-1, TSP-2, and TSP-5 compared to SFM. Quiescent VSMCs were cultured in the presence of TSP-1, TSP-2 or TSP-5 over a range of concentrations from 0.2μg/mL to 40μg/mL. After 72 hours, the cells were incubated in MTS for 2 hours and absorbance measured. The data was analyzed by ANOVA (p <0.05).
Gene expression
The complete list of altered genes and the associated relative quantity (RQ) and p-values appears in the Supplemental Data. The list of significantly altered genes and the associated RQ appears in Table 1. In summary, TSP-1 altered the expression of 4 genes at three hours; 7 genes at six hours and 14 genes at 24 hours (p <0.05). TSP-2 altered the expression of 9 genes at three hours; 15 genes at six hours; and 5 genes at 24 hours (p< 0.05). TSP-5 altered the expression of 31 genes at three hours; 3 genes at six hours; and 19 genes at 24 hours (p <0.05). There was limited overlap across the different TSPs tested and time points within the same TSP treatment.
Table 1.
List of significantly up and down-regulated genes for each protein and time point.
| Up-regulated | TSP-1 | TSP-2 | TSP-5 |
|---|---|---|---|
| 3 hours |
3 genes ANGPTL4 (2.404), PTN (1.476), VEGF (1.672) |
8 genes ADAMTS1 (1.743), CXCL2 (3.805), FOXC2 (2.427), ITGA4 (1.190), HEY1 (4.943), IL8 (5.050), TIMP3 (1.916), VEGF (2.868) |
24 genes ADAMTS1 (4.336), ANGPTL1 (6.027), ANGPTL2 (1.335), CD44 (1.939), COL15A1 (7.005), COL4A1 (10.247), EDIL3 (1.879), FGF2 (16.467), FN1 (3.361), FOXC2 (6.477), FST (5.560), HEY1 (10.890), HSPG2 (4.550), IL12A (14.154), ITGA4 (3.568), ITGAV (2.359), KIT (6.751), NRP1 (2.061), PF4 (2.536), PGK1 (4.187), THBS2 (3.142), TIMP2 (2.481), TIMP3 (7.629), VEGF (7.394) |
| 6 hours |
2 genes IL12A (10.068) THBS1 (1.720) |
15 genes ADAMTS1 (2.579), BAI1 (7.877), FBLN5 (1.341), FGF1 (2.196), FGF2 (1.923), FOXC2 (1.897), FST (1.438), HGF (1.580), ITGA4 (1.640), KIT (2.757), NRP1 (1.348), PF4 (3.173), THBS1 (2.196), TIMP2 (1.352), VEGF (1.959) |
3 genes ANGPTL4 (1.792), IL12A (2.079), THBS2 (1.622) |
| 24 hours |
12 genes ANGPT2 (2.138), ANGPTL4 (1.221), CD44 (2.328), FGF2 (1.739), FST (1.835), HEY1 (4.281), NRP1 (1.952), NRP2 (1.816), PROX1 (11.076), TEK (1.957), TGFB1 (1.648), TIMP3 (3.483) |
3 genes ANGPTL4 (3.845) EDIL3 (1.554) ENPP2 (1.465) |
12 genes ANGPTL4 (2.218), COL15A1 (9.337), COL4A1 (10.505), EDIL3 (2.105), GRN (2.809), HSPG2 (4.835), KIT (7.016), NRP1 (2.850), PDGFRB (2.980), THBS1 (10.518), THBS2 (2.816), VEGFB (2.371) |
| Down-regulated | |||
| 3 hours |
1 gene VEGFC (0.702) |
1 gene COL15A1 (0.696) |
7 genes COL18A1 (0.352), ENPP2 (0.609) HGF (0.367), ITGB3 (0.331) MMP2 (0.334), PTN (0.348) SERPINF1 (0.225) |
| 6 hours |
5 genes HGF (0.588), IL8 (0.512) ITGB3 (0.578), MDK (0.793), PECAM1 (0.369) |
- | - |
| 24 hours |
2 genes CTGF (0.188), IL8 (0.347) |
2 genes CTGF (0.615), IL8 (0.259) |
7 genes EDG1 (0.356), IL8 (0.039) ITGB3 (0.412), MDK (0.298) MMP2 (0.371), PTN (0.241) SERPINF1 (0.205) |
Quiescent VSMCs were exposed to TSP-1, TSP-2, TSP-5 (20μg/mL) or SFM for 6 hours and RNA was extracted. qrt-PCR was performed using a microfluidic card. Significantly up and down-regulated genes (t-test, p <0.05) are shown above with the relative quantities (as compared to SFM) in parentheses.
A number of genes became detectable following TSP exposures were not initially detectable in SFM samples and are listed in Table 2 under “detectable”. This occurred for 16 genes in total for TSP-1; 12 genes in total for TSP-2; and 20 genes in total for TSP-5. In addition, a number of genes became undetectable following TSP exposure whereas they were initially detected in SFM and are listed in Table 2 under “undetectable”. This occurred in 10 genes in total for TSP-1; 14 genes in total for TSP-2; and 18 genes for TSP-5. Statistical comparison could not be performed for these data, as one of the comparative RQ values was zero (either the experimental group or SFM).
Table 2.
List of detectable and undetectable genes compared to SFM.
| Detectable | TSP-1 | TSP-2 | TSP-5 |
|---|---|---|---|
| 3 hours |
6 genes CEACAM1 F2 LEP PDGFB PROK1 TNNI1 |
4 genes F2 FTL4 IFNG PDGFB |
7 genes ANGPTL3 F2 FTL4 LEP PDGFB PROK1 SERPINB5 |
| 6 hours |
5 genes F2 LEP PDGFB PRL PROK1 |
3 genes CEACAM1 FTL4 PDGFB |
5 genes CEACAM1 CHGA FTL4 LEP PDGFB |
| 24 hours |
5 genes CEACAM1 CHGA F2 IFNG SERPINCB5 |
5 genes CEACAM1 FTL1 LEP PDGFB PROK1 |
8 genes ANGPTL3 F2 FTL4 FOXC2 LECT1 LEP PROK1 TNNI1 |
| Undetectable | |||
| 3 hours |
3 genes AMOT FTL1 TNMD |
3 genes COL4A3 FTL3 TNMD |
4 genes CSF3 FTL3 TGFA TNF |
| 6hours |
3 genes AMOT FTL1 FTL4 |
5 genes FTL1 FTL3 LEP TNMD TNF |
9 genes AMOT COL4A3 CSF3 CXCL10 FTL3 PRL PROX1 TNF TNMD |
| 24 hours |
4 genes BAI1 CSF3 FTL1 TNMD |
6 genes AMOT CXCL10 FTL3 SERPINC1 TNMD TNF |
5 genes CDH5 CSF3 FTL3 TGFA TNF |
Genes that were detectable in the experiment conditions, but not in SFM are listed above as “detectable”. Genes that were undetectable in the experimental conditions, but present in SFM samples are listed above as “undetectable”.
DISCUSSION
Neointimal hyperplasia remains a significant factor in the durability and failure of open or endovascular revascularization procedures. VSMCs are exposed to ECM proteins, cytokines, growth factors and many other molecules influencing the phenotypic change required for migration and proliferation. Thus, identifying genes and proteins as targets to prevent this phenotypic change are crucial to developing therapies to mitigate restenosis.
In the current work, we showed TSP-2 and TSP-5, in addition to TSP-1, are significant chemoattractants of VSMCs. We are the first to demonstrate that TSP-2-induced chemotaxis is dose-dependent, with peak chemotaxis occurring at 20μg/mL. Analysis of the TSP family molecules provides some insight into the explanation of these results. The C-terminus of the five TSP proteins, containing EGF-like and Ca2+ binding domains, is highly conserved. The CD47 receptor, to which the C-terminal domains bind, has been shown to mediate TSP-1 induced chemotaxis [6]. Chemotaxis due to TSP-2 has been previously demonstrated in ECs, but not VSMCs, and can be attributed to CD47 receptor binding. Our migration experiments also agree with a previous study that TSP-5 supports the chemotaxis of VSMCs at concentrations lower than 50μg/mL [12]. In the current study, we demonstrated that TSP-5 induced chemotaxis starting as low as 1μg/mL, with an optimal effect occurring at 20μg/mL. The concentrations required for chemotaxis are similar for other ECM proteins as well, such as vitronectin and fibronectin [15].
In addition, we demonstrated TSP-2 induced VSMC proliferation, and TSP-5 did not. Previous studies on TSP-1 demonstrate a modest proliferative effect mediated by CD36 receptor binding to the N-terminal type 1 repeats. TSP-2, although inducing proliferation at a higher concentration, had similar effects as TSP-1. By contrast, TSP-5 lacks these domains and as such, lacks the capacity to bind CD36, reinforcing our hypothesis TSP-5 should have chemotactic, but not proliferative effects on VSMCs.
With the ability to target specific genes for study and manipulation growing increasingly prevalent, characterizing effects on gene expression identifies potential targets for further study and intervention. Immediately noticeable is disparity in number of genes up and down-regulated when comparing across different TSPs or at different time points for the same TSP. Our study demonstrates TSP-1 and TSP-2, despite structural and functional similarities, do not induce similar patterns of gene expression in VSMCs. This finding indicates that although TSP-1 and TSP-2 may have overlapping functions, they are not interchangeable. Our study also demonstrates that while the functions of TSP-5 in VSMCs are not fully understood, TSP-5 has a clear influence on gene expression relevant to vascular disease. The pattern of genes up and down-regulated is distinct from TSP-1 and TSP-2, and the limited number of genes overlapping also suggests the function of TSP-5 is independent and different from either TSP.
When analyzing the TSP gene expression data with the IPA software, the most involved pathways and cellular functions highlight the importance of TSPs to vascular physiology. “Cardiovascular Disease” was one of the top involved disease processes for all three proteins, based on the significantly up and down-regulated genes, involving 17 of the genes on the TSP-1 list, 13 genes for TSP-2 and 28 genes for TSP-5 (p values ranging from 10−5 to 10−13). In addition, “Cardiovascular System Development and Function” is the top Physiologic System Development and Function pathway noted, involving 22–36 genes for each protein. Further investigation into each category reveals a list of annotated functions. “Vascular disease”, “disorders of artery”, “aortic disorder”, “vascular lesion”, and “atherosclerosis” were top listed functions for TSP-1, TSP-2 and TSP-5, despite limited overlap in the genes up and down-regulated. The IPA software also identifies “thrombosis”, “thrombosis of vein”, “atherosclerotic lesion” and “myocardial infarction” as significantly involved functions for TSP-1 and TSP-5. The individual processes listed in the “Cardiovascular System Development and Function” annotation heavily represent the migration, proliferation and differentiation and activation of ECs. While VSMC-induced signaling of ECs is a well-documented phenomenon [16], these findings may also represent a bias due to a much greater body of research on apoptosis-related genes and their functions in ECs.
In addition to the above canonical pathways, which represent overarching physiologic processes, specific genes involved in pathways relevant to the progression of vascular disease can also be identified in the gene expression data. Growth factors, such as PDGFβ, and cytokines such as interleukins, are key components in the signaling pathways between arterial wall cells that influence behavior following injury. Our study provides evidence these pathways may, in part, be regulated by TSPs. For example, inflammation has been implicated as one of the initiating factors of neointimal hyperplasia, whether in the setting of atherosclerosis or neointimal hyperplasia. IL12A, encoding interleukin-12, is one of the most highly up-regulated genes for TSP-1 (10-fold) and TSP-5 (14-fold), and has been recently implicated in the phenotypic transformation of VSMCs [17]. As for growth factors, PDGFB, encoding PDGFβ, was expressed at the 3 and 6 hour time points for all three TSPs and at 24 hours for TSP-2, however undetectable in the SFM-only samples. Other up-regulated genes related to growth factors include hepatocyte growth factor (HGF), transforming growth factor–beta (TGFB1), and fibroblast growth factors 1 and 2 (FGF1, FGF2). Up-regulation of growth factor genes potentially indicates the TSPs may play an integral role not only in direct stimulation of VSMCs, but also indirect influence of VSMC and neighboring cells via inflammatory mediators.
The genes down-regulated or undetectable following TSP treatment are also areas for further analysis. Of interest, MMP2 is down-regulated by TSP-5 at the 3 and 24 hour time points. MMP2 has been previously established to be inversely regulated by TSP-2, as a wound healing studying showed enhanced angiogenesis and reduced fibroblast contraction in association with increased MMP2 levels in TSP-2-null mice [11]. Down-regulation of MMP2, in conjunction with the up-regulation of various collagen and integrin-related genes, may indicate TSP-5 plays an important role in tissue remodeling.
Interestingly, the genes encoding the Group A TSPs, TSP-1 (THBS1) and TSP-2 (THBS2), are significantly up-regulated at some of the time points for each of the proteins. THBS1 is up-regulated by TSP-1 and TSP-2 about 2 fold at 6 hours. THBS2 is up-regulated nearly 3 fold by TSP-5 at 3 and 24 hours and 1.6 fold at 6 hours. The suggested positive feedback between the TSPs may imply repeated stimuli for VSMC migration and proliferation, making the process of intimal hyperplasia difficult to stop once started. Of note, the gene encoding TSP-5 (COMP) was not tested in this assay, but will be an area for further study.
The observational nature of our study poses several limitations. First, the VSMCs used in the study were cultured in isolation of ECs, platelets, monocytes and a variety of other factors that would be present in an in vivo specimen. Also, although the approach uses rt-PCR, mitigating a need for further confirmatory testing such is necessary with microarrays, the up or down-regulation of any particular gene does not indicate the level of protein expression, which could be contrary to the gene expression results and is an area for further study.
In summary, TSP-1, TSP-2 and TSP-5 independently induce VSMC chemotaxis, while TSP-1 and TSP-2 alone are able to induce proliferation, indicating a likely structural component to this disparity. Additionally, TSP-1, TSP-2 and TSP-5 all produce markedly different patterns of gene expression, both comparing across proteins as well as along different time points for the same protein. As technology improves, the complexities of vascular pathobiology are continually revealed. The above study provides the foundation for future investigations into potential therapeutic targets for mitigating the detrimental effects of intimal hyperplasia.
Supplementary Material
Acknowledgments
This study was supported by a VA Merit Award #BX001243. The funding source had no involvement in any portions of study design, data collection or analysis or decision to submit. We thank Karen Gentile for her assistance extracting RNA and assessing quality and Dr. Lorena Gonzalez for manuscript edits.
ABBREVIATIONS
- EC
endothelial cell
- SFM
serum-free media
- TSP
thrombospondin
- VSMC
vascular smooth muscle cell
Footnotes
Disclosures: Alex Helkin, Kristopher Maier and Vivian Gahtan have no financial conflicts of interest to disclose.
None of the authors have any conflicts of interest to report.
References
- 1.Collins MJ, Li X, Lv W, et al. Therapeutic strategies to combat neointimal hyperplasia in vascular grafts. Expert Rev Cardiovasc Ther. 2012;5:635–47. doi: 10.1586/erc.12.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adams JC, Lawler J. The thrombospondins. Int J Biochem Cell Biol. 2004;6:961–8. doi: 10.1016/j.biocel.2004.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stenina-Adognravi O. Invoking the power of thrombospondins: regulation of thrombospondins expression. Matrix Biol. 2014;37:69–82. doi: 10.1016/j.matbio.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rodriguez-Manzaneque JC, Lane TF, Ortega MA, et al. Thrombospondin-1 suppresses spontaneous tumor growth and inhibits activation of matrix metalloproteinase-9 and mobilization of vascular endothelial growth factor. Proc Natl Acad Sci U S A. 2001;98:12485–90. doi: 10.1073/pnas.171460498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patel MK, Lymn JS, Clunn GF, et al. Thrombospondin-1 is a potent mitogen and chemoattractant for human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1997;17:2107–14. doi: 10.1161/01.atv.17.10.2107. [DOI] [PubMed] [Google Scholar]
- 6.Krishna SM, Golledge J. The role of thrombospondin-1 in cardiovascular health and pathology. Int J Cardiol. 2013;168:692–706. doi: 10.1016/j.ijcard.2013.04.139. [DOI] [PubMed] [Google Scholar]
- 7.Maier KG, Han X, Sadowitz B, Gentile KL, Middleton FA, Gahtan V. Thrombospondin-1: a proatherosclerotic protein augmented by hyperglycemia. J Vasc Surg. 2010;51:1238–47. doi: 10.1016/j.jvs.2009.11.073. [DOI] [PubMed] [Google Scholar]
- 8.Chavez RJ, Haney RM, Cuadra RH, et al. Upregulation of thrombospondin-1 expression by leptin in vascular smooth muscle cells via JAK2- and MAPK-dependent pathways. Am J Physiol Cell Physiol. 2012;303:C179–91. doi: 10.1152/ajpcell.00008.2012. [DOI] [PubMed] [Google Scholar]
- 9.Seymour K, Stein J, Han X, Maier KG, Gahtan V. Statins and nitric oxide donors affect thrombospondin 1-induced chemotaxis. Vasc Endovascular Surg. 2014;48:470–5. doi: 10.1177/1538574414554718. [DOI] [PubMed] [Google Scholar]
- 10.Yabkowitz R, Mansfield PJ, Ryan US, et al. Thrombospondin mediates migration and potentiates platelet-derived growth factor-dependent migration of calf pulmonary artery smooth muscle cells. J Cell Physiol. 1993;157:24–32. doi: 10.1002/jcp.1041570104. [DOI] [PubMed] [Google Scholar]
- 11.Agah A, Kyriakides TR, Lawler J, Bornstein P. The lack of thrombospondin-1 (TSP1) dictates the course of wound healing in double-TSP1/TSP2-null mice. Am J Pathol. 2002;161:831–9. doi: 10.1016/S0002-9440(10)64243-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Riessen R, Fenchel M, Chen H, et al. Cartilage oligomeric matrix protein (thrombospondin-5) is expressed by human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2001;21:47–54. doi: 10.1161/01.atv.21.1.47. [DOI] [PubMed] [Google Scholar]
- 13.Oldberg A, Antonsson P, Lindblom K, Heinegård D. COMP (cartilage oligomeric matrix protein) is structurally related to the thrombospondins. J Biol Chem. 1992;267:22346–50. [PubMed] [Google Scholar]
- 14.Frolova EG, Pluskota E, Krukovets I, et al. Thrombospondin-4 regulates vascular inflammation and atherogenesis. Circ Res. 2010;107:1313–25. doi: 10.1161/CIRCRESAHA.110.232371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Willis AI, Sadowitz B, Fuse S, et al. Thrombospondin 1, fibronectin, and vitronectin are differentially dependent upon RAS, ERK1/2, and p38 for induction of vascular smooth muscle cell chemotaxis. Vasc Endovascular Surg. 2011;45:55–62. doi: 10.1177/1538574410387677. [DOI] [PubMed] [Google Scholar]
- 16.Lacolley P, Regnault V, Nicoletti A, et al. The vascular smooth muscle cell in arterial pathology: a cell that can take on multiple roles. Cardiovasc Res. 2012;95:194–204. doi: 10.1093/cvr/cvs135. [DOI] [PubMed] [Google Scholar]
- 17.Karagiannis GS, Weile J, Bader GD, Minta J. Integrative pathway dissection of molecular mechanisms of moxLDL-induced vascular smooth muscle phenotype transformation. BMC Cardiovasc Disord. 2013;16:13–24. doi: 10.1186/1471-2261-13-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.