

Abstract
SOX9 haploinsufficiency underlies campomelic dysplasia (CD) with or without testicular dysgenesis. Current understanding of the phenotypic variability and mutation spectrum of SOX9 abnormalities remains fragmentary. Here, we report three patients with hitherto unreported SOX9 abnormalities. These patients were identified through molecular analysis of 33 patients with 46,XY disorders of sex development (DSD). Patients 1–3 manifested testicular dysgenesis or regression without CD. Patients 1 and 2 carried probable damaging mutations p.Arg394Gly and p.Arg437Cys, respectively, in the SOX9 C‐terminal domain but not in other known 46,XY DSD causative genes. These substitutions were absent from ~120,000 alleles in the exome database. These mutations retained normal transactivating activity for the Col2a1 enhancer, but showed impaired activity for the Amh promoter. Patient 3 harbored a maternally inherited ~491 kb SOX9 upstream deletion that encompassed the known 32.5 kb XY sex reversal region. Breakpoints of the deletion resided within nonrepeat sequences and were accompanied by a short‐nucleotide insertion. The results imply that testicular dysgenesis and regression without skeletal dysplasia may be rare manifestations of SOX9 abnormalities. Furthermore, our data broaden pathogenic SOX9 abnormalities to include C‐terminal missense substitutions which lead to target‐gene‐specific protein dysfunction, and enhancer‐containing upstream microdeletions mediated by nonhomologous end‐joining.
Keywords: Campomelic dysplasia, deletion, enhancer, mutation, testis
Introduction
SOX9 (OMIM *608160) controls embryonic development by transactivating several genes such as COL2A1 involved in skeletal formation and AMH involved in testicular development. Known SOX9 mutations include various missense substitutions in the high‐mobility group or dimerization domains, as well as several nonsense, frameshift, and splice‐site mutations widely distributed in the coding region (Meyer et al. 1997; Bernard et al. 2003; Harley et al. 2003; Michel‐Calemard et al. 2004; Staffler et al. 2010). Patients with SOX9 mutations manifest campomelia, hypoplastic scapulae, pelvic anomalies, micrognathia, and cleft palate, collectively referred to as campomelic dysplasia (CD), although a certain percentage of mutation‐positive patients show a mild variant of CD that lacks campomelia (acampomelic CD: ACD) (Bernard et al. 2003; Michel‐Calemard et al. 2004; Staffler et al. 2010). SOX9 mutations also result in complete or partial gonadal dysgenesis in individuals with 46,XY karyotype (Meyer et al. 1997; Michel‐Calemard et al. 2004). As CD/ACD‐compatible skeletal abnormalities were described in all patients with SOX9 mutations and disorders of sex development (DSD) were shared only by ~70% of 46,XY patients (Mansour et al. 1995), it seems that skeletal tissues are more vulnerable than testis to impaired SOX9 function. Kwok et al. (1996) suggested that SOX9 mutations are unlikely to underlie 46,XY DSD in the absence of skeletal abnormalities.
Recent studies have identified submicroscopic deletions in the SOX9 upstream region in six patients with isolated 46,XY DSD (Pop et al. 2004; Lecointre et al. 2009; Kim et al. 2015). These patients shared a 32.5 kb overlapping region of deletion at a position 607–640 kb upstream of the SOX9 start codon, which was designated as the XY sex reversal region (XYSR). Since SOX9 expression is regulated by multiple tissue‐specific enhancers (Bagheri‐Fam et al. 2006), XYSR likely contains a testis‐specific enhancer. Considering the limited number of reported patients, further studies are necessary to clarify the phenotypic variability and mutation spectrum of SOX9 abnormalities. Furthermore, the genomic basis of SOX9 upstream deletions remains to be investigated. Here, we report three unique cases with SOX9 abnormalities.
Materials and Methods
Subjects
This study was approved by the Institutional Review Board Committee at the National Center for Child Health and Development. The study group consisted of 33 Japanese patients with 46,XY DSD. All patients showed genital abnormalities at birth; of these, 29 had isolated DSD, whereas the remaining patients manifested DSD with additional clinical features. Eleven and 22 patients were raised as a female and male, respectively. Patients with apparent chromosomal abnormalities were excluded from this study.
Mutation analysis
After obtaining written informed consent from the patients or their parents, genomic DNA samples were collected from the patients. Mutation analysis was performed by next‐generation sequencing (NGS). Genomic DNA samples were isolated from peripheral leukocytes. Target regions in the human genome were amplified with the SureSelect Target Enrichment system (G7531C or all exome v5; Agilent Technologies, Palo Alto, CA) and sequenced on a HiSeq 2000 sequencer (Illumina, San Diego, CA). Nucleotide alterations were called by Avadis NGS 1.3.1 (DNA Chip Research, Yokohama, Japan) or SAMtools 0.1.17 software (http://samtools.soursefrge.net/). In this study, we focused on protein‐altering substitutions and splice‐site mutations of 27 known causative genes for 46,XY DSD, that is, AKR1C2, AKR1C4, AMH, AMHR2, AR, ATF3, ATRX, BNC2, CYP11A1, DHH, DMRT1, GATA4, HSD3B2, HSD17B3, INSL3, INSR, LHCGR, MAP3K1, NR5A1, POR, RXFP2, SOX9, SRD5A2, SRY, STAR, TSPYL1, and WT1. Nucleotide substitutions of allele frequency 1% or higher in the Japanese population (Human Genetic Variation Browser, http://www.genome.med.kyoto-u.ac.jp/SnpDB) were excluded as polymorphisms. SOX9 (NM_000346.3) mutations indicated by NGS were confirmed by Sanger sequencing using a primer pair: SOX9‐exon3FW2 (5′‐CAGGCGCACACGCTGACCAC‐3′) and SOX9‐exon3RV (5′‐CCTCTCTTTCTTCGGTTAT‐3′). Furthermore, PCR products carrying the nucleotide alterations were subcloned into the TOPO TA cloning vector (Life Technologies, Carlsbad, CA) and the mutant and wild‐type alleles were sequenced separately. Whenever possible, parental samples of mutation‐positive patients were also subjected to molecular analysis.
Functional analyses of SOX9 substitutions
Conservation and functional consequences of SOX9 substitutions were predicted using Polyphen‐2 (http://genetics.bwh.harvard.edu/pph2/) (Adzhubei et al. 2010). Population frequencies of the substitutions were analyzed using the Exome Aggregation Consortium Browser (http://exac.broadinstitute.org/).
The transactivating activity of the substitutions was assessed by a previously reported method with modifications (Kelberman et al. 2006). Briefly, an expression vector for wild‐type SOX9 was purchased from Origene Technologies (RC208944, Rockville, MD), and each SOX9 mutation was introduced into the expression vector by site‐directed mutagenesis (PrimeSTAR Mutagenesis Basal Kit; Takara Bio, Ohtsu, Japan). In this study, we compared the transactivating activity of newly identified mutants to that of the known ACD‐associated SOX9 mutant c.527C>T (p.Pro176Leu) (Michel‐Calemard et al. 2004). We used a PGL3 reporter vector (Promega, Madison, WI) containing the murine Amh promoter sequence (from −231 to 0 to the transcription start site of Amh, NC_000076.6) and a PGL4 reporter vector (Promega) containing the murine Col2a1 enhancer sequence (from +1958 to +2485 to the transcription start site of Col2a1, NC_000081.6). An expression vector for NR5A1 was kindly provided by Professor Toshihiko Yanase (Fukuoka University, Fukuoka, Japan). Luciferase assays for the Amh promoter and for the Col2a1 enhancer were carried out using COS‐1 (RIKEN, Ibaraki, Japan) and HEK293 cells (Health Science Research Resources Bank, Tokyo, Japan), respectively. The cells were seeded in 6‐well dishes and treated with Lipofectamine 2000 Reagent (Life Technologies). For Amh promoter assays, we transfected cells with 40 ng SOX9 expression vector, 100 ng NR5A1 expression vector, 1 μg reporter vector, and 3 ng pCMV‐PRL control vector (Promega). For Col2a1 enhancer assays, we used 200 ng SOX9 expression vector, 500 ng reporter vector, and 3 ng pCMV‐PRL vector. At 48 h after transfection, luciferase activity was measured using the Dual‐Luciferase Reporter Assay System (Promega) with Lumat LB9507 (Berthold, Oak Ridge, TN). Every assay was performed in triplicate and all experiments were repeated at least three times. The results are expressed as the mean ± one standard deviation, and statistical significance was calculated by the Student t test. P values of <0.005 were considered significant.
Copy‐number analysis
Copy‐number alterations were analyzed by comparative genomic hybridization using a catalog human array (4 × 180 k format) or a custom‐made array (design ID, 031687) (Agilent Technologies). The deletion breakpoints were determined by direct sequencing of PCR products harboring the fusion junction. The products were generated using a primer pair: 5′‐TTTTTTCCTTGAAGTTAATG‐3′ and 5′‐AATGTAGTGCTATATATTGC‐3′. Sizes and genomic positions of the deletions were analyzed using the UCSC genome browser (http://genome.ucsc.edu/; GRCh37/hg19) and the presence or absence of repeat sequences was examined with RepeatMasker (http://www.repeatmasker.org). We referred to the Database of Genomic Variants (http://projects.tcag.ca/variation/) to exclude known benign variants.
Results
Mutation analysis
We identified two heterozygous missense substitutions c.1180C>G (p.Arg394Gly) and c.1309C>T (p.Arg437Cys) in patients 1 and 2, respectively, in SOX9 (Fig. 1). These substitutions have not been reported previously. Patients 1 and 2 carried no mutations in the other genes examined or in other nucleotides of SOX9. The substitution of patient 2 was shared by the phenotypically normal mother, whereas parental samples of patient 1 were not available for genetic analysis. The p.Arg394Gly and p.Arg437Cys substitutions resided within the proline/glutamine/serine (PQS)‐rich domain (also known as SPQ‐rich domain) at the C‐terminus (McDowall et al. 1999) (Fig. 1A).
Figure 1.
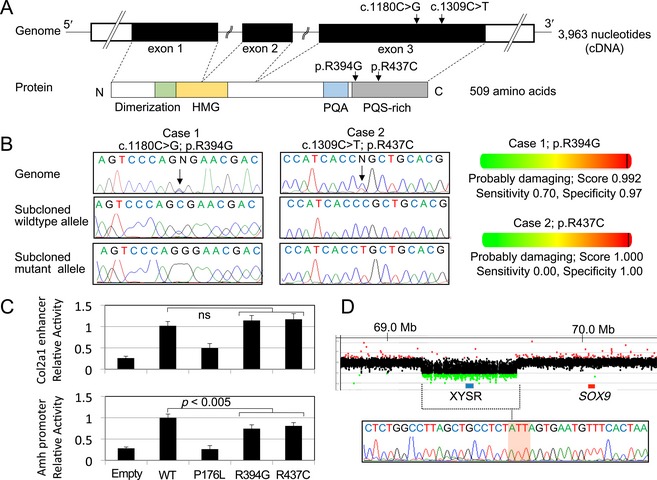
SOX9 abnormalities in patients 1–3. (A) Genomic and protein structures of SOX9/SOX9. The positions of the c.1180C>G (p.Arg394Gly) and c.1309C>T (p.Arg437Cys) mutations are indicated by arrows. White and black boxes in the upper panel indicate the untranslated and coding regions, respectively. Colored boxes in the lower panel indicate dimerization (codon 60–101) (Bernard et al. 2003), high‐mobility group (HMG: codon 101–184), proline/glutamine/alanine (PQA: codon 339–379), and proline/glutamine/serine‐rich (PQS‐rich: codon 386–509) domains (McDowall et al. 1999). (B) Nucleotide substitutions detected in patients 1 and 2. Left panel: electro chromatograms of the mutations. The mutated nucleotides are indicated by arrows. Right panel: in silico functional prediction of mutant proteins. (C) In vitro assays using reporters containing the Col2a1 enhancer or Amh promoter. The transactivating activity of p.Arg394Gly and p.Arg437Cys mutants was compared to that of the known ACD‐associated SOX9 mutant p.Pro176Leu (Michel‐Calemard et al. 2004). The results are expressed as the mean ± one standard deviation. Relative transactivating activities of the SOX9 mutants against the wild‐type are shown. Empty: empty expression vector; ns: not significant. (D) SOX9 upstream deletion in patient 3. Upper panel: array‐based comparative genomic hybridization analysis. The black, red, and green dots denote signals indicative of the normal, increased (> +0.5) and decreased (< −1.0) copy‐numbers, respectively. The blue and red boxes represent previously reported XY sex reversal region (XYSR) (Kim et al. 2015) and SOX9 exons, respectively. Genomic positions refer to the UCSC database (http://genome.ucsc.edu/; GRCh37/hg19). Lower panel: sequence of the fusion junction. The junction is accompanied by a short‐nucleotide insertion of unknown origin (the red‐shaded area).
Functional analysis of SOX9 substitutions
The c.1180C>G (p.Arg394Gly) and c.1309C>T (p.Arg437Cys) substitutions involved highly conserved amino acids, and were predicted as “probably damaging” by in silico analyses (Fig. 1B). These substitutions were absent from ~120,000 alleles of the exome database. The p.Arg394Gly and p.Arg437Cys mutants retained normal in vitro transactivating activity for the Col2a1 enhancer (relative fold activation: 1.14 and 1.17, respectively), but exerted impaired activity for the Amh promoter (relative fold activation: 0.74 and 0.81, respectively) (Fig. 1C). In contrast, the previously reported ACD‐associated p.Pro176Leu mutant showed markedly reduced activity for both reporters (relative fold activation: 0.50 for the Col2al enhancer and 0.26 for the Amh promoter).
Copy‐number analysis
We identified a heterozygous deletion in the upstream region of SOX9 in patient 3 (Fig. 1D). The deletion was 490,990 bp in physical length and started at a position 380,011 bp upstream to the SOX9 start codon (chr17: 69,246,534–69,737,523; GRCh37/hg19). The deletion encompassed the known XYSR (Figs. 1D, 2). The deletion breakpoints resided in nonrepeat sequences and shared no homology (Fig. 1D). The fusion junction was accompanied by a short‐nucleotide insertion of unknown origin that was indicative of an “information scar” of nonhomologous end‐joining (NHEJ) (Lieber 2008). Patient 3 carried no sequence alteration in all genes examined. The SOX9 upstream deletion was identified in the phenotypically normal mother of patient 3.
Figure 2.
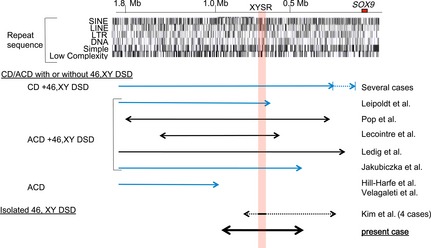
Schematic representation of the SOX9 upstream region. Upper panel represents positions of SOX9 and repeat sequences (UCSC database, http://genome.ucsc.edu/; GRCh37/hg19). The numbers indicate the distance from SOX9. Lower panel represents genomic rearrangements in the present and previous cases. Blue and black arrows indicate chromosomal translocations and deletions, respectively. Broken arrows indicate breakpoint regions of multiple patients. The red‐shaded area represents the XYSR reported by Kim et al. (2015). XYSR, XY sex reversal region; CD, campomelic dysplasia; ACD, acampomelic CD; DSD, disorders of sex development.
Clinical features of the mutation‐positive patients
Physical and hormonal findings of patients 1–3 are summarized in Table 1. Blood inhibin B levels were not determined in these cases. Patient 1 was a 19‐year‐old individual raised as a male. He manifested hypospadias and bilateral cryptorchidism at birth and underwent surgical intervention at 5 years of age. At 14 years of age, he was subjected to endocrine evaluation because of a lack of pubertal sexual development. Blood examinations revealed increased levels of gonadotropins and mildly decreased levels of testosterone, indicating testicular dysfunction. He began to receive testosterone supplementation therapy at age 14, and human chorionic gonadotropin and human menopausal gonadotropin therapy at age 16. He underwent surgical intervention for gynecomastia at 14 and 16 years of age. Abdominal ultrasound at 18 years of age showed small testes with focal microlithiasis. Müllerian duct derivatives were absent. He had no skeletal abnormalities except for spina bifida occulta, a relatively common neural tube anomaly of that has not been associated with SOX9 mutations (Greene and Copp 2014).
Table 1.
Molecular and clinical findings of patients 1–3
| Patient 1 | Patient 2 | Patient 3 | ||||
|---|---|---|---|---|---|---|
| Karyotype | 46,XY | 46,XY | 46,XY | |||
| Molecular defects in SOX9 (NM_000346.3) | c.1180C>G (p.Arg394Gly) | c.1309C>T (p.Arg437Cys) | Upstream deletion | |||
| Physical findings at birth | ||||||
| External genitalia | Male‐type genitalia with hypospadias and unpalpable testes | Male‐type genitalia with micropenis and unpalpable testes | Complete female‐type genitalia | |||
| Physical findings at later ages | ||||||
| Age at exam. (year) | 19 | 2.8 | 22 | |||
| Penile size (cm)a | 5.7 b (8.6–10.0) | 2.5 (3.0–3.6) | Not examined | |||
| Testicular size (mL) | 5 (right), 3–4 (left)c | Not palpable | Not palpable | |||
| Gonadal histology | Not analyzed | Fibrous tissues | Streak gonad with seminoma | |||
| Uterus | Absent | Absent | Present | |||
| Additional findings | Spina bifida occulta | None | None | |||
| Hormonal findingsa | ||||||
| Age at exam. (year) | 19 | 2.8 | 13 | |||
| B | S | B | S | B | S | |
| LH (mIU/mL)d | 37.5 (0.5–5.0) | 126.6 (6.0–21.0) | 2.8 (<0.3–1.3) | Not analyzed | 13.8 (<0.2–2.1) | 119.4 (1.3–7.0) |
| FSH (mIU/mL)d | 17.7 (0.8–4.4) | 24.3 (1.5–8.0) | 109.8 (0.4–1.5) | Not analyzed | 82.5 (<0.3–3.0) | 135.8 (1.3–3.9) |
| Testosterone (ng/mL)e | 5.3 (2.5–11.0) | 4.9 (6.3–9.8) | <0.03 (0.06–0.16) | <0.03 (>0.2) | 0.09 (0.68–1.22) | 0.16 (2.96–5.58) |
| AMH (ng/mL) | Not analyzed | Not analyzed | <0.1 (74.1–148.1) | <0.1 (no data) | Not analyzed | Not analyzed |
The conversion factor to the SI unit: LH 1.0 (IU/L), FSH 1.0 (IU/L), testosterone 3.47 (nmol/L), and AMH 7.14 (pmol/L). Penile size and hormone values below the reference range are boldfaced, and hormone values above the reference range are italicized. B, basal; S, stimulated; LH, luteinizing hormone; FSH, follicle‐stimulating hormone; AMH, anti‐Müllerian hormone.
Reference ranges are shown in parentheses.
After hormone replacement therapy.
After surgical interventions.
Gonadotropin releasing hormone stimulation test (100 μg/m2, max. 100 μg bolus i.v.; blood sampling at 0, 30, 60, 90, and 120 min).
Human chorionic gonadotropin stimulation test (3000 IU/m2, max. 5000 IU i.m. for three consecutive days; blood sampling on days 1 and 4).
Patient 2 was a 5‐year‐old male individual. At birth, he showed male‐type external genitalia; however, bilateral testes were not palpable in the scrotum or in the inguinal region. At 2.6 years of age, laparoscopic examination detected possible gonadal remnants with spermatic cord in the bilateral inguinal canals. Histological examination of biopsied samples showed that the possible remnants were fibrous tissues without germ cells. At 2.8 years of age, he was referred to our clinic for further evaluation. He showed normal skeletal features and borderline micropenis. Endocrine analyses revealed increased gonadotropin levels and undetectable levels of testosterone and anti‐Müllerian hormone. Abdominal imaging did not detect a uterus or gonads. Thus, this patient was diagnosed with testicular regression. At 5 years of age, he was capable of voiding in a standing position.
Patient 3 was a 22‐year‐old individual with a female phenotype. Her growth and development were uneventful until pubertal age. At 13 years of age, she visited our clinic because of a lack of pubertal sexual development. She showed normal skeletal features and female‐type external genitalia. Endocrine evaluation indicated severe gonadal dysfunction. Abdominal magnetic resonance imaging detected a uterus of a prepubertal size. Estrogen supplementation therapy from 14 years of age successfully induced breast budding and vaginal bleeding. Gonadectomy was performed at 17 years of age. Histological examination revealed bilateral dysgenic gonads with seminoma.
Discussion
This study provides several notable findings. This is the first report documenting the association between SOX9 intragenic mutations and isolated 46,XY DSD. In vitro assays confirmed the target‐specific functional impairment of the c.1180C>G (p.Arg394Gly) and c.1309C>T (p.Arg437Cys) mutants, which were not observed in the known ACD‐associated mutant c.527C>T (p.Pro176Leu). Notably, unlike other known pathogenic SOX9 missense mutations, p.Arg394Gly and p.Arg437Cys resided within the C‐terminal PQS‐rich domain. As the PQS‐rich domain is required for SOX9 interaction with other proteins (Tsuda et al. 2003), the two mutations may affect SOX9‐mediated protein–protein interactions in the developing testis. Physical and hormonal findings of patients 1 and 2 with these mutations were indicative of impaired testicular development, although blood inhibin B levels, a sensitive marker for the function of the testis (Grinspon et al. 2012), were not determined in these patients.
It is worth mentioning that patient 2 manifested bilateral testicular regression, a rare form of 46,XY DSD that probably occurs as a result of the disturbance of developmental processes during testicular tubule formation (Mizuno et al. 2012). The genetic basis of testicular regression remains unknown, with the exception of NR5A1 mutations that account for a minor fraction of cases (Philibert et al. 2007). It has been suggested that testicular regression and gonadal dysgenesis are a continuum of a disorder (Marcantonio et al. 1994). Indeed, NR5A1 mutations are known to underlie both conditions (Ferraz‐de‐Souza et al. 2011). Animal studies suggested that SOX9 plays a role in testicular tubule differentiation through the interaction with SOX8 (Barrionuevo et al. 2009). Moreover, SOX9 plays a critical role not only in testicular development but also in the maintenance of differentiated status of the testes (Sekido and Lovell‐Badge 2009; Veitia 2010). Thus, testicular regression may be a rare manifestation in patients with SOX9 mutations. However, this notion is based on the findings of a single individual, and therefore awaits further investigation.
One may argue against p.Arg394Gly and p.Arg437Cys being responsible for the severe DSD in patients 1 and 2 because these mutations resulted in only modest decrease in the transactivating activity for the AMH promoter. The discrepancy between the phenotypic severities and the results of in vitro assays can be explained by assuming that some SOX9 target genes other than AMH are more sensitive to defective function of SOX9. Actually, a number of testicular genes are known to be regulated by SOX9 (Sekido and Lovell‐Badge 2009; Veitia 2010). Alternatively, in the developing testis, the p.Arg394Gly and p.Arg437Cys mutations may disrupt the synergic interaction between SOX9 and certain cofactors. It is known that SOX9 synergizes with other proteins to transactivate target genes (Sekido and Lovell‐Badge 2009; Veitia 2010). On the other hand, we cannot exclude the possibility that genetic variations in other genes or some environmental factors affected sexual development in patients 1 and 2, although mutations in known 46,XY DSD causative genes were excluded in these patients. Further studies, including in vitro assays using reporter vectors containing various SOX9 target promoters and expression vectors for several SOX9 cofactors, and whole exome sequencing of patients 1 and 2, will clarify the precise functional consequences of the mutants.
The findings in patient 3 support the notion that the 32.5 kb XYSR contains a DNA element(s) essential for testicular development. Moreover, exclusion mapping indicates that SOX9 enhancers for skeletal tissues and craniofacial regions are located outside of the ~491 kb region deleted in patient 3 (Fig. 2) (Pop et al. 2004; Hill‐Harfe et al. 2005; Velagaleti et al. 2005; Lecointre et al. 2009; Jakubiczka et al. 2010; Ledig et al. 2010; Kim et al. 2015). In addition, the results of this study, together with those of previous studies (Pop et al. 2004; Lecointre et al. 2009; Kim et al. 2015) provide evidence of the genomic heterogeneity of SOX9 upstream deletions. The breakpoint sequences in patient 3 suggest that the deletion resulted from NHEJ. Actually, genomic regions around SOX9 are not enriched with repeat sequences that serve as substances of nonallelic homologous recombination (Fig. 2). Thus, NHEJ may be the major cause of SOX9 upstream microdeletions, although other mechanisms such as microhomology‐mediated replication errors may also be involved in the development of such deletions. Since all known pathogenic deletions in the SOX9 upstream region, including that in our patient, were transmitted from phenotypically normal mothers of the patients (Kim et al. 2015), de novo occurrence of such deletions seems to be a rare event.
In summary, the results indicate that the phenotypic consequences of SOX9 mutations are broader than previously reported and include testicular dysgenesis and regression without skeletal dysplasia. Furthermore, our data suggest that DSD‐associated SOX9 abnormalities include C‐terminal missense substitutions that lead to target‐specific protein dysfunction, and NHEJ‐mediated upstream microdeletions encompassing XYSR.
Conflict of Interest
None declared.
Acknowledgments
This study was funded by the Grant‐in‐Aid from the Ministry of Education, Culture, Sports, Science and Technology; by the Grant‐in‐Aid from the Japan Society for the Promotion of Science; by the Grants from the Ministry of Health, Labour and Welfare, from the National Center for Child Health and Development and from the Takeda Foundation.
References
- Adzhubei, I. A. , Schmidt S., Peshkin L., Ramensky V. E., Gerasimova A., Bork P., et al. 2010. A method and server for predicting damaging missense mutations. Nat. Methods 7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagheri‐Fam, S. , Barrionuevo F., Dohrmann U., Gunther T., Schule R., Kemler R., et al. 2006. Long‐range upstream and downstream enhancers control distinct subsets of the complex spatiotemporal Sox9 expression pattern. Dev. Biol. 291:382–397. [DOI] [PubMed] [Google Scholar]
- Barrionuevo, F. , Georg I., Scherthan H., Lecureuil C., Guillou F., Wegner M., et al. 2009. Testis cord differentiation after the sex determination stage is independent of Sox9 but fails in the combined absence of Sox9 and Sox8. Dev. Biol. 327:301–312. [DOI] [PubMed] [Google Scholar]
- Bernard, P. , Tang P., Liu S., Dewing P., Harley V. R., and E. Vilain . 2003. Dimerization of SOX9 is required for chondrogenesis, but not for sex determination. Hum. Mol. Genet. 12:1755–1765. [DOI] [PubMed] [Google Scholar]
- Ferraz‐de‐Souza, B. , Lin L., and Achermann J. C.. 2011. Steroidogenic factor‐1 (SF‐1, NR5A1) and human disease. Mol. Cell. Endocrinol. 336:198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene, N. D. , and Copp A. J.. 2014. Neural tube defects. Annu. Rev. Neurosci. 37:221–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinspon, R. P. , Loreti N., Braslavsky D., Bedecarrás P., Ambao V., Gottlieb S., et al. 2012. Sertoli cell markers in the diagnosis of paediatric male hypogonadism. J. Pediatr. Endocrinol. Metab. 25:3–11. [DOI] [PubMed] [Google Scholar]
- Harley, V. R. , Clarkson M. J., and Argentaro A.. 2003. The molecular action and regulation of the testis‐determining factors, SRY (sex‐determining region on the Y chromosome) and SOX9 [SRY‐related high‐mobility group (HMG) box 9]. Endocr. Rev. 24:466–487. [DOI] [PubMed] [Google Scholar]
- Hill‐Harfe, K. L. , Kaplan L., Stalker H. J., Zori R. T., Pop R., Scherer G., et al. 2005. Fine mapping of chromosome 17 translocation breakpoints > or = 900 Kb upstream of SOX9 in acampomelic campomelic dysplasia and a mild, familial skeletal dysplasia. Am. J. Hum. Genet. 76:663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubiczka, S. , Schroder C., Ullmann R., Volleth M., Ledig S., Gilberg E., et al. 2010. Translocation and deletion around SOX9 in a patient with acampomelic campomelic dysplasia and sex reversal. Sex Dev. 4:143–149. [DOI] [PubMed] [Google Scholar]
- Kelberman, D. , Rizzoti K., Avilion A., Bitner‐Glindzicz M., Cianfarani S., Collins J., et al. 2006. Mutations within Sox2/SOX2 are associated with abnormalities in the hypothalamo‐pituitary‐gonadal axis in mice and humans. J. Clin. Invest. 116:2442–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, G. J. , Sock E., Buchberger A., Just W., Denzer F., Hoepffner W., et al. 2015. Copy number variation of two separate regulatory regions upstream of SOX9 causes isolated 46, XY or 46, XX disorder of sex development. J. Med. Genet. 52:240–247. [DOI] [PubMed] [Google Scholar]
- Kwok, C. , Goodfellow P. N., and Hawkins J. R.. 1996. Evidence to exclude SOX9 as a candidate gene for XY sex reversal without skeletal malformation. J. Med. Genet. 33:800–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecointre, C. , Pichon O., Hamel A., Heloury Y., Michel‐Calemard L., Morel Y., et al. 2009. Familial acampomelic form of campomelic dysplasia caused by a 960 kb deletion upstream of SOX9. Am. J. Med. Genet. A 149A:1183–1189. [DOI] [PubMed] [Google Scholar]
- Ledig, S. , Hiort O., Scherer G., Hoffmann M., Wolff G., Morlot S., et al. 2010. Array‐CGH analysis in patients with syndromic and non‐syndromic XY gonadal dysgenesis: evaluation of array CGH as diagnostic tool and search for new candidate loci. Hum. Reprod. 25:2637–2646. [DOI] [PubMed] [Google Scholar]
- Lieber, M. R. 2008. The mechanism of human nonhomologous DNA end joining. J. Biol. Chem. 283:1–5. [DOI] [PubMed] [Google Scholar]
- Mansour, S. , Hall C. M., Pembrey M. E., and Young I. D.. 1995. A clinical and genetic study of campomelic dysplasia. J. Med. Genet. 32:415–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcantonio, S. M. , Fechner P. Y., Migeon C. J., Perlman E. J., and Berkovitz G. D.. 1994. Embryonic testicular regression sequence: a part of the clinical spectrum of 46, XY gonadal dysgenesis. Am. J. Med. Genet. 49:1–5. [DOI] [PubMed] [Google Scholar]
- McDowall, S. , Argentaro A., Ranganathan S., Weller P., Mertin S., Mansour S., et al. 1999. Functional and structural studies of wild type SOX9 and mutations causing campomelic dysplasia. J. Biol. Chem. 274:24023–24030. [DOI] [PubMed] [Google Scholar]
- Meyer, J. , Sudbeck P., Held M., Wagner T., Schmitz M. L., Bricarelli F. D., et al. 1997. Mutational analysis of the SOX9 gene in campomelic dysplasia and autosomal sex reversal: lack of genotype/phenotype correlations. Hum. Mol. Genet. 6:91–98. [DOI] [PubMed] [Google Scholar]
- Michel‐Calemard, L. , Lesca G., Morel Y., Boggio D., Plauchu H., and Attia‐Sobol J.. 2004. Campomelic acampomelic dysplasia presenting with increased nuchal translucency in the first trimester. Prenat. Diagn. 24:519–523. [DOI] [PubMed] [Google Scholar]
- Mizuno, K. , Kojima Y., Kamisawa H., Kurokawa S., Moritoki Y., Nishio H., et al. 2012. Feasible etiology of vanishing testis regarding disturbance of testicular development: histopathological and immunohistochemical evaluation of testicular nubbins. Int. J. Urol. 19:450–456. [DOI] [PubMed] [Google Scholar]
- Philibert, P. , Zenaty D., Lin L., Soskin S., Audran F., Leger J., et al. 2007. Mutational analysis of steroidogenic factor 1 (NR5a1) in 24 boys with bilateral anorchia: a French collaborative study. Hum. Reprod. 22:3255–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop, R. , Conz C., Lindenberg K. S., Blesson S., Schmalenberger B., Briault S., et al. 2004. Screening of the 1 Mb SOX9 5′ control region by array CGH identifies a large deletion in a case of campomelic dysplasia with XY sex reversal. J. Med. Genet. 41:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekido, R. , and Lovell‐Badge R.. 2009. Sex determination and SRY: down to a wink and a nudge? Trends Genet. 25:19–29. [DOI] [PubMed] [Google Scholar]
- Staffler, A. , Hammel M., Wahlbuhl M., Bidlingmaier C., Flemmer A. W., Pagel P., et al. 2010. Heterozygous SOX9 mutations allowing for residual DNA‐binding and transcriptional activation lead to the acampomelic variant of campomelic dysplasia. Hum. Mutat. 31:E1436–E1444. [DOI] [PubMed] [Google Scholar]
- Tsuda, M. , Takahashi S., Takahashi Y., and Asahara H.. 2003. Transcriptional co‐activators CREB‐binding protein and p300 regulate chondrocyte‐specific gene expression via association with Sox9. J. Biol. Chem. 278:27224–27229. [DOI] [PubMed] [Google Scholar]
- Veitia, R. A. 2010. FOXL2 versus SOX9: a lifelong “battle of the sexes”. Bioessays 32:375–380. [DOI] [PubMed] [Google Scholar]
- Velagaleti, G. V. , Bien‐Willner G. A., Northup J. K., Lockhart L. H., Hawkins J. C., Jalal S. M., et al. 2005. Position effects due to chromosome breakpoints that map approximately 900 Kb upstream and approximately 1.3 Mb downstream of SOX9 in two patients with campomelic dysplasia. Am. J. Hum. Genet. 76:652–662. [DOI] [PMC free article] [PubMed] [Google Scholar]