

Abstract
Bacterial pathogens can exploit metabolic pathways to facilitate their successful infection cycles, but little is known about roles of d‐galactosamine (GalN)/N‐acetyl‐d‐galactosamine (GalNAc) catabolism pathway in bacterial pathogenesis. Here, we report the genomic reconstruction of GalN/GalNAc utilization pathway in Streptococci and the diversified aga regulons. We delineated two new paralogous AgaR regulators for the GalN/GalNAc catabolism pathway. The electrophoretic mobility shift assays experiment demonstrated that AgaR2 (AgaR1) binds the predicted palindromes, and the combined in vivo data from reverse transcription quantitative polymerase chain reaction and RNA‐seq suggested that AgaR2 (not AgaR1) can effectively repress the transcription of the target genes. Removal of agaR2 (not agaR1) from Streptococcus suis 05ZYH33 augments significantly the abilities of both adherence to Hep‐2 cells and anti‐phagocytosis against RAW264.7 macrophage. As anticipated, the dysfunction in AgaR2‐mediated regulation of S. suis impairs its pathogenicity in experimental models of both mice and piglets. Our finding discovered two novel regulators specific for GalN/GalNAc catabolism and assigned them distinct roles into bacterial infections. To the best of our knowledge, it might represent a first paradigm that links the GalN/GalNAc catabolism pathway to bacterial pathogenesis.
Keywords: AgaR, amino sugars, d‐galactosamine, N‐acetyl‐d‐galactosamine, Streptococcus suis, virulence.
Introduction
Amino sugars are referred to a variety of diversified/complex monosaccharides in which a hydroxyl group is chemically replaced with the amine group. Most of current knowledge on the metabolism of amino sugars comes from studies with Escherichia coli (Reizer et al. 1996), Bacillus subtilis (Freymond et al. 2006; Gaugue et al. 2014; Plumbridge 2015) and Streptomycetes coelicolor (Rigali et al. 2008). Relative to the best‐known examples of amino sugars, glucosamine (GlcN) and N‐acetylglucosamine (GlcNAc), the investigations on the other two amino sugar derivatives of galactose, N‐acetyl‐d‐galactosamine (GalNAc) and d‐galactosamine (GalN), are relatively limited, but increasingly accumulated (Abu‐Qarn et al. 2008; Leyn et al. 2012). It seemed very likely that GalN/GalNAc amino sugars as common components participate in the formation of various cell structures/constitutes in three domains of life (Abu‐Qarn et al. 2008; Plumbridge 2015). In general, not only does GalNAc act as an element of lipopolysaccharide displayed on bacterial cell wall (Bernatchez et al. 2005; Leyn et al. 2012), and but also connects carbohydrate chains in mammalian mucins (Carraway and Hull 1991). Additionally, it functions as the substrate in N‐acetyl β‐galactosidation, a new type of post‐translational modification of protein in organisms, including bacterial pathogens (Sadler et al. 1979; Barr and Nordin 1980; Davis et al. 1986; Abu‐Qarn et al. 2008). Given the multiple roles played by GalN/GalNAc, we therefore anticipated a hypothesis that GalN/GalNAc metabolism might be linked to bacterial infectivity.
The paradigm pathway for GalN/GalNAc utilization/catabolism, which was initially proposed for E. coli in 1996 (Reizer et al. 1996), contained the following five steps (Fig. 1): (1) the transport and phosphorylation of GalN/GalNAc substrates (catalyzed by phosphortransferase system [PTS] systems AgaBCD and AgaVWEF, respectively) (Brinkkotter et al. 2000), (2) the AgaA‐mediated deacetylation of GalNAc‐6‐P (Reizer et al. 1996), (3) the deamination/isomerization of GalN‐6‐P by the bi‐functional enzyme AgaS into Tag‐6‐P (Reizer et al. 1996), (4) the AgaZ kinase‐aided phosphorylation of Tag‐1,6‐P from Tag‐6‐P (Reizer et al. 1996), and (5) cleavage of Tag‐1,6‐PP by the class II aldolase, AgaY (Brinkkotter et al. 2002), to produce glyceraldehyde 3‐phospahte and glycerone phosphate (PEP). Interestingly, a recent comparative genomics‐based study suggested an extensive diversity in GalN/GalNAc utilization pathways of Proteobacteria such as Shewanella (Leyn et al. 2012). In particular note the first two steps of this pathway exhibit of high variability, while the latter three steps are largely conserved (Leyn et al. 2012). In E. coli, not only does AgaR regulator that belongs to the DeoR family of transcriptional factors, act as an autoregulator, but also negatively controls the expression of two aga genes (agaZ and agaS) of GalN/GalNAc catabolism pathway via direct binding of the specific palindromes in front of these target genes (Ray and Larson 2004). Although the fact that both GalN and GalNAc can induce activities of these aga genes‐encoding protein products is aware, the physiological ligands for AgaR repressor remain unclear (Leyn et al. 2012).
Figure 1.
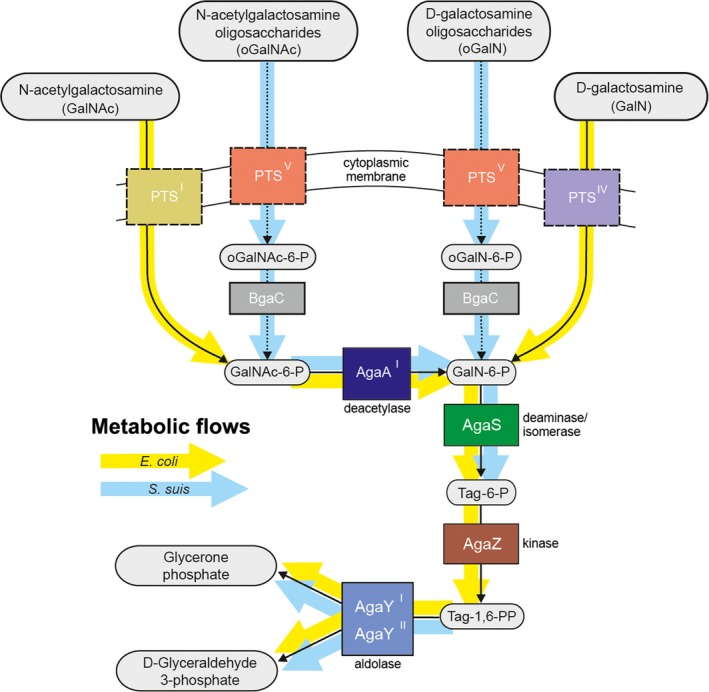
Reconstruction for GalNAc/GalN utilization pathways in Streptococcus suis. The variants of GalNAc/GalN pathway in Escherichia coli and S. suis are highlighted with yellow and light‐blue background arrows, respectively. GalNAc, N‐acetyl‐d‐galactosamine; GalN, galactosamine; PTS, phosphotransferase system; BgaC, β‐galactosidase; AgaA, GalNAc‐6‐P deacetylase; AgaS, GalN‐6‐P deaminase/isomerase; Tag, Tagtose; AgaZ, Tag‐6‐P kinase; AgaY, Tag‐1,6‐PP aldolase.
Streptococcus suis, a Gram‐positive bacterium, is a zoonotic agent with the ability to infect both its natural host swine and human individuals with close contact with swine/pork‐related products (Feng et al. 2010). According to the differentiation in their bacterial capsule structure, this Streptococcus species is categorized into 35 serotypes (Feng et al. 2010, 2014b). Among them, serotype 2 (SS2) is generally believed to be most virulent, in that it is frequently isolated from clinical diseased swine (Ma et al. 2009; Feng et al. 2014b) and human sporadic cases (Feng et al. 2009b) and/or big‐scale outbreaks (Tang et al. 2006; Chen et al. 2007). We are aware that SS2 has spread to more than 30 countries/regions and claims nearly 1600 cases of human infections worldwide (Feng et al. 2014b). The molecular mechanism underlying bacterial pathogenicity has been partially delineated thus far (Feng et al. 2014b), and these identified virulence determinants include the previously‐known virulence factors exemplified with capsule (Benga et al. 2008; Seitz et al. 2014) and suilysin (Jacobs et al. 1996; Lun et al. 2003; Takeuchi et al. 2014). In addition to the regulatory networks amongst the newly identified factors (e.g., the two‐component systems SalK/SalR [Li et al. 2008], NisK/NisR [Xu et al. 2014], CiaRH [Li et al. 2011], etc.), it would be of particular interest to note the contributions of enzymes from central metabolisms (enolase [Eno] [Esgleas et al. 2008; Feng et al. 2009a; Zhang et al. 2009a; Lu et al. 2012], glutamine synthase [GlnA] [Si et al. 2009], Inosine 5‐monophosphate dehydrogenase [Impdh] [Zhang et al. 2009b; Zhou et al. 2014], etc.) to bacterial pathogenesis (Feng et al. 2014b). However, nothing is aware regarding the potential relevance of bacterial GalN/GalNAc metabolism and/or its regulation to S. suis infection.
In this work, we employed the integrative approaches combining the bioinformatics and comparative genomics to conduct the genomic reconstruction of the GalN/GalNAc utilization pathway in Lactobacillaceae, including the zoonotic pathogen S. suis. Also, we are the first to report the functional definition of two new GntR‐type transcription factors (referred to AgaR2 and/or AgaR1) with involvement of GalN/GalNAc catabolism. More intriguingly, we observed that AgaR2‐dependent regulation of GalN/GalNAc utilization pathway is required for bacterial virulence of S. suis serotype 2. To the best of our knowledge, it represents the first example that the genetic control of GalN/GalNAc catabolism is linked to bacterial infectivity of Streptococcus.
Materials and Methods
Bacterial strains, cell lines and growth conditions
Bacterial strains used here included derivatives of either E. coli or S. suis 2 (Table S1). Escherichia coli Topo10 and BL21 (DE3) are applied for gene cloning, and protein expression, respectively. The growth medium for E. coli and S. suis 2 is separately Luria‐Bertani (LB) broth and Todd‐Hewitt broth (THB; Difco Laboratories, Detroit, MI). These bacteria are grown at 37°C overnight. Given the selective pressure to ensure the replication of recombinant plasmids or the maintenance of engineered strains, appropriate antibiotics (Sigma, St. Louis, MO) were supplemented as follows: 100 μg/mL of Spectinomycin for S. suis; either 100 μg/mL of Ampicillin or 50 μg/mL of Kanamycin for E. coli. The two kinds of cell lines used here corresponded to the human laryngeal epithelial cell Hep‐2 (CCTCC GDC004) and the mouse macrophagocyte Raw 264.7 (ATCC TIB‐71, Rockville, MD), respectively (Table S1) (Hu et al. 2014), and cultivated at 37°C in the presence of 5% CO2 in Dulbecco's modified Eagle's medium with 10% fetal bovine serum (Roche, Indianapolis, IN, USA),100 μg/mL gentamycin, and 5 μg/mL penicillin G (Feng et al. 2012).
Plasmids and DNA manipulations
The S. suis agaR2 (SSU05_0447) gene was amplified using polymerase chain reaction (PCR) with primers SSU05_0447‐F plus SSU05_0447‐R (Table S2) and ligated into the BamHI and XhoI sites of pET28a(+) expression vector (Feng et al. 2008), resulting in the recombinant plasmid pET28‐447 (Table S1). Similarly, the other expression plasmid pET28‐448 was given through direct cloning of S. suis agaR1 (SSU05_0448) gene carrying BamHI and SalI sites introduced by primers (SSU05_0448‐F plus SSU05_0448‐R) into the expression vector pET28a(+) with the same cuts (Tables S1, S2). The above two plasmids (pET28‐447 and pET28‐448) are designed to prepare in vitro proteins of AgaR2 and AgaR1, respectively. For functional complementation, the two genes (SSU05_0447 and SSU05_0448) were separately inserted into the low‐copy shuttle vector pVA838 (Romero et al. 1987), giving the plasmids pVA838‐447 and pVA838‐448, respectively (Table S2). All the recombinant plasmids involved in this study were confirmed with both PCR assays and direct DNA sequencing.
Expression and purification of two AgaR proteins
The two recombinant plasmids (pET28a‐448 and pET28a‐447) were separately transformed into BL21 (DE3), giving the engineered strains FYJ356, and FYJ536, respectively (Table S1). The two versions of hexahistidine‐tagged AgaR protein (referred to AgaR1 [SSU05_0448] and AgaR2 [SSU05_0447]) were produced using the above two engineered strains FYJ356 and FYJ536, respectively. In brief, when bacterial optical density at wave‐length of 600 nm (OD600) reached 0.6–1.0, the bacterial cultures with an appropriate plasmid (Table S1) were induced with 0.5 mmol/L isopropyl‐β‐d‐thiogalactopyranoside (IPTG) at 30°C for 3–5 h. As we described before (Feng and Cronan 2009), two rounds of French pressure‐based lysis were conducted for release of the recombinant protein AgaR1 (and/or AgaR2), and subsequent procedures of protein purification included the nickel column‐based affinity purification followed by fast phase liquid chromatography (FPLC) (Feng et al. 2008; Feng and Cronan 2012). Consequently, the protein of interest was concentrated via ultrafiltration (Feng et al. 2008), and the purity was judged by 12% sodiumdodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE).
Western blotting
Western blot was performed routinely to further verify the hexahistidine‐tagged AgaR1 (and/or AgaR2) protein. The protein samples were separated with 12% SDS‐PAGE and thereafter transferred to a nitrocellulose membrane (Amersham, GE Healthcare, Piscataway, NJ, USA). The primary antibody was an anti‐hexahistidine mouse monoclonal antibody, and the secondary antibody was a peroxidase‐conjugated goat anti‐mouse immunoglobulin G. The signal of target protein band was captured by an exposure to the high‐performance chemiluminescence ECL film (Amersham, GE Healthcare, Piscataway, NJ, USA).
Liquid chromatography quadrupole time‐of‐flight mass spectrometry
To verify the identity of the recombinant AgaR1 (and/or AgaR2), the resultant peptides by digestion with Sequencing Grade Trypsin (G‐Biosciences, St. Louis, MO, 12.5 ng μL−1 in 25 mmol/L ammonium bicarbonate) were subjected for analyses of A Waters Q‐Tof API‐US Quad‐ToF mass spectrometer linked to a Waters nano Acquity UPLC (Feng et al. 2013b). The mass data acquired were assayed through Waters Protein Lynx Global Server 2.2.5, Mascot (Matrix Science, Boston, MA, USA) combined with BLAST against NCBI nr database (Feng et al. 2014a).
Size exclusion chromatography and chemical cross‐linking assays
The nickel column‐based purified AgaR1 (and/or AgaR2) protein was further assayed by gel filtration chromatography using a Superdex 75 column (Amersham, GE Healthcare, Piscataway, NJ, USA) run on an Äkta fast protein liquid chromatography system (GE Healthcare) as described previously (Feng and Cronan 2010, 2011). The column effluent was evaluated at a flow rate of 0.5 mL/min in PBS buffer (10 mmol/L Na2HPO4, 2 mmol/L KH2PO4, 20 mmol/L Tris‐HCl, 137 mmol/L NaCl, 2.7 mmol/L KCl, pH 7.4). The protein peak at the position of expected elution volume was sampled and verified by 12% SDS‐PAGE.
To further elucidate the solution structure of AgaR1 (and/or AgaR2) protein from S. suis, we conducted chemical cross‐linking experiments in which ethylene glycol bis‐succinimidylsuccinate (EGS) (Pierce, Rockford, IL, USA) was added (Feng et al. 2008). In each reaction system (15 μL in total), the interested protein (~5 mg/mL) was separately incubated with the EGS cross‐linker (at different levels [0.1, 0.2, 0.5, 1.0, 2.5, 5, 10, 20 μmol/L for AgaR1 protein; 0, 2.5, 5, 10, 20 μmol/L, for AgaR2 protein]) for 1 h at room temperature. Finally, the reaction products were visualized via 12% SDS‐PAGE followed by commassiee brilliant blue staining.
Electrophoretic mobility shift assays
The function of the predicted AgaR2 and AgaR1 binding sites were proved using electrophoretic mobility shift assays (EMSA) as we established earlier (Feng and Cronan 2011; Feng et al. 2013a,b) with minor modifications. All the double‐strand DNA probes were produced in vitro by annealing two complementary oligonucleotides in TEN buffer (10 mmol/L Tris‐HCl, 1 mmol/L Ethylene Diamine Tetraacetic Acid (EDTA), 100 mmol/L NaCl; pH 8.0), and labeled with DIG‐ddUTP (Roche, Indianapolis, IN, USA) by the terminal transferase (Feng and Cronan 2011, 2012). Three AgaR1‐specific DNA probes used here included SSU05_0448/9 site1 probe (38 bp), EF814 site1 probe (38 bp), and EF1809 site1 probe (38 bp). In addition to SSU05_0447 probe (36 bp) as the negative control, the other four tested AgaR2‐recognizable sites corresponded to SSU05_0195 probe (36 bp), SSU05_1259 probe (36 bp), 5 SSU05_0448/9 site2 (36 bp), and EF1809 site2 probe (36 bp) (Table S2). After 20 min of incubation of the DIG‐labeled DNA probes (0.2 pmol) with or without AgaR2 (and/or AgaR1) protein in the binding buffer (Roche) at room temperature, the DNA–protein complexes were separated by the native 7% PAGE gel and transferred onto an equilibrated, positively charged nylon membrane (Roche) by contact blotting followed by UV cross‐linking (120 mJ for 180 sec) (Feng and Cronan 2011; Feng et al. 2013b). Finally, the signals were captured by exposure to the high‐performance chemiluminescence film (Amersham Hyperfilm ECL) (Feng and Cronan 2009, 2010).
RNA isolation and real‐time qPCR
Mid‐log phase cultures of S. suis 2 strains (wild type [WT], ΔagaR1, ΔagaR2, CΔagaR1 and CΔagaR2) grown in THB media (with/without GalNAc‐6P) were collected to prepare the total bacterial RNA using an RNeasy bacterial RNA isolation kit (Qiagen, Hilden, Germany) (Feng et al. 2008; Li et al. 2008). As we performed before (Feng and Cronan 2009, 2010), RNA integrity/quality was validated by separation of 1.0% agarose gel electrophoresis. The possible contamination of trace genomic DNA in the RNA samples was ruled out by PCR‐based detections, using the total RNA as the template (Feng and Cronan 2009, 2010, 2011).
Using the qualified RNA preparations, complementary DNAs (cDNAs) were synthesized by reverse transcription (RT). Then, the real‐time quantitative PCR (qPCR) combined with the SYBR green method (Feng et al. 2008; Feng and Cronan 2009) was carried out to probe possible relevance of agaR2 (and/or agaR1) to the altered expression profile of genes encoding GalNAc utilization pathway. The method of 2−ΔΔCT (34) was applied to determine the relative level of the target genes associated with GalNAc utilization in which the 16S rRNA‐encoding gene 16S rDNA as the internal reference (Table S2). Target genes tested here are agaS (SSU05_0195), agaAY (SSU05_1258/9), bgaC (SSU05_0449), and gadVWEF (SSU05_0450, SSU05_0451, SSU05_0452 and SSU05_0453), respectively.
Construction of ΔagaR2 (and ΔagaR1) mutants and functional complementation
The agaR2 (or agaR1) gene from the S. suis 2 strain 05ZYH33 was replaced with the spectinomycin resistance (SpcR) cassette by homologous recombination (Feng et al. 2008, 2012; Hu et al. 2014). Briefly, the SpcR cassette from pSET2 (Takamatsu et al. 2001) was cloned into the pUC19 vector (Invitrogen) to give the intermediate plasmid pUC19‐Spc (Table S1), and then the two DNA fragments adjacent to the agaR2 (or agaR1) gene were separately inserted into the pUC19‐Spc vector, giving the knockout plasmid pUC::447 and pUC::448, respectively (Table S1). As we did before (Feng et al. 2008; Li et al. 2008), the plasmid of pUC::447 (or pUC::448) was electroporated into the competent cells of S. suis 05ZYH33 to acquire positive transformants with SpcR. The multiplex‐PCR techniques were adopted to screen the ΔagaR2 (and/or ΔagaR1) mutant (Table S2). Consequently, the mutants we acquired for functional experiments were further proved by direct DNA sequencing. The two plasmids of pVA838‐447 and pVA838‐448 were separately transformed into ΔagaR2 and ΔagaR1 mutants to give the complemented strains CΔagaR2 and CΔagaR1, respectively (Table S2).
Assays for ability of bacterial adherence and phagocytosis
As Hytönen et al. (Hytonen et al. 2006) reported, S. suis bacteria (WT, ΔagaR2 and CΔagaR2) grown in the mid‐log phase were subjected to cell lines‐based analyses. The two cell lines are Hep‐2 (human laryngeal epithelial cell line) and murine macrophage Raw 264.7 cells (Feng et al. 2012; Hu et al. 2014). Bacterial adherence was tested with Hep‐2 cell line, and the evaluation for ability of anti‐phagocytosis was based on the Raw264.7 cells.
Infection assays of experimental animals
To reveal the role of agaR2 (and/or agaR1) in bacterial pathogenesis/virulence, two different kinds of experimental animals were employed, including BALB/c (4‐week old, female) mice and SPF‐piglets. In the infection experiment of mice, totally 60 animals were challenged that are classified into six groups (10 mice/group). Except that THB acted as a negative control, the other five groups infected with S. suis 2 (at a dose of 1 × 108 CFU per mouse) corresponded to WT, ΔagaR2, CΔagaR2, ΔagaR1, and CΔagaR1, respectively.
The result obtained from the experiment of mice infection was further checked using the infection test of piglets, its natural host of S. suis. Given the fact that only agaR2 (not agaR1) plays a role in bacterial infectivity, three groups of piglets (six piglets/group) were rechallenged by WT, ΔagaR2, and CΔagaR2, respectively. Clinical syndromes of the infected mice/piglets were monitored for 72 h. Of particular note, deaths were recorded and moribund animals were humanely killed. All experiments on live vertebrates in this study were approved by the Ethics Committee of Research Institute for Medicine of Nanjing Command and performed in accordance with the relevant guidelines and regulations (Cao et al. 2011; Feng et al. 2012; Hu et al. 2014).
Bioinformatics analyses
Genome sequences were downloaded from the MicrobesOnline genomic data base (Dehal et al. 2010). Identification of orthologs was performed using the BLASTP search in the non‐redundant database (Altschul et al. 1997) and MicrobesOnline tree browser. For functional protein annotations by distant homology to characterize proteins, BLAST search in the SwissProt/UniProt database was used. Analysis of chromosomal gene clustering was performed by MicrobesOnline and SEED web resources (Dehal et al. 2010; Disz et al. 2010). The GalNAc utilization subsystem curation and analysis were conducted, using the SEED platform (Disz et al. 2010). Protein domains were determined by protein similarity search tools in the Pfam database (Sonnhammer et al. 1998). Multiple sequence alignments were constructed by either ClustalW (http://www.ebi.ac.uk/Tools/clustalw2/index.html) (Thompson et al. 2002) or MUSCLE (Edgar 2004). Phylogenetic trees were constructed using the maximum likelihood algorithm implemented in the PHYLIP package (Felsenstein 1996) and visualized via the dendroscope tool (Huson et al. 2007). Sequences Logos were constructed using WebLogo package (Crooks et al. 2004).
For genomic reconstruction of the regulons, we used the well‐established comparative genomics approach (Rodionov 2007). The approach includes inference of transcriptional factor‐binding sites (TFBSs), construction of nucleotide positional weight matrices (PWMs) for TFBSs motifs, and reconstruction of regulons in complete genomes on the basis of prediction of putative TFBSs in the promoter gene regions. First, in the studied Lactobacillales genomes, we revealed orthologs of previously known genes for GalNAc utilization (Table S3). Second, we predicted possible transcriptional regulators for the uncovered GalNAc utilization. Candidate regulators were attributed to the regulons by using a genomic colocalization and co‐occurence of a putative regulator with the identified GalNAc utilization genes. Such analysis defined two groups of transcriptional regulators belonging to the HutC subfamily of the GntR family (Fig. 7). For each group of AgaR proteins, we identified putative binding motifs analyzing upstream regions of presumably regulated gene by the Discover Profile tool implemented in the RegPredict Web server (Novichkov et al. 2010). In this approach, putative TFBSs were determined as overrepresented words in upstream regions of putatively co‐regulated genes. Based on the genomic co‐occurrence and co‐presence of genes, it has been proposed that AgaR1‐binding sites should be located upstream hydrolase and PTS genes while AgaR2 sites ought to be upstream of deacetylase, isomerase, and aldolase genes. For the prediction of putative TFBSs, we analyzed upstream regions of probably related operons, expanding −400 to +100 bp relative to start codon of the first gene of the operon. Both AgaR1 and AgaR2 belong to HutC subfamily. TFBSs for HutC subfamily proteins have structure of an even palindrome (Rigali et al. 2002; Suvorova et al. 2015). Thus, for prediction of the AgaR1 and AgaR2 TFBSs motifs, we searched for even palindromic DNA motifs of 14–24 bp. Among all motifs found for each set of upstream regions, we selected the longest motif with the highest information content. The selected motif was quality controlled by two approaches, (1) consistency check and (2) phylogenetic footprinting. Consistency check (Mironov et al. 1999; Rodionov 2007) was done, that is, motif was checked for the presence in multiple number of genomes. High quality motif should find predicted TFBSs in upstream regions of predicted regulated operons in genomes having the analyzed regulator, but not in genomes lacking the regulator. Phylogenetic footprinting technique is based on analysis of multiple alignments for upstream regions of orthologous genes (Shelton et al. 1997). High quality motif should find predicted TFBSs that are located inside conserved islands of multiple alignments. When the high quality of the motif was confirmed by both consistency check and phylogenetic footprinting, the identified regulatory motifs were used for the construction of the PWMs (profiles) and the obtained matrices were used to determine additional candidate regulatory sites in the analyzed genomes, using the Run Profile tool in the RegPredict Web server. The scores of sites were calculates as a sum of nucleotide weights for each position.
Figure 7.
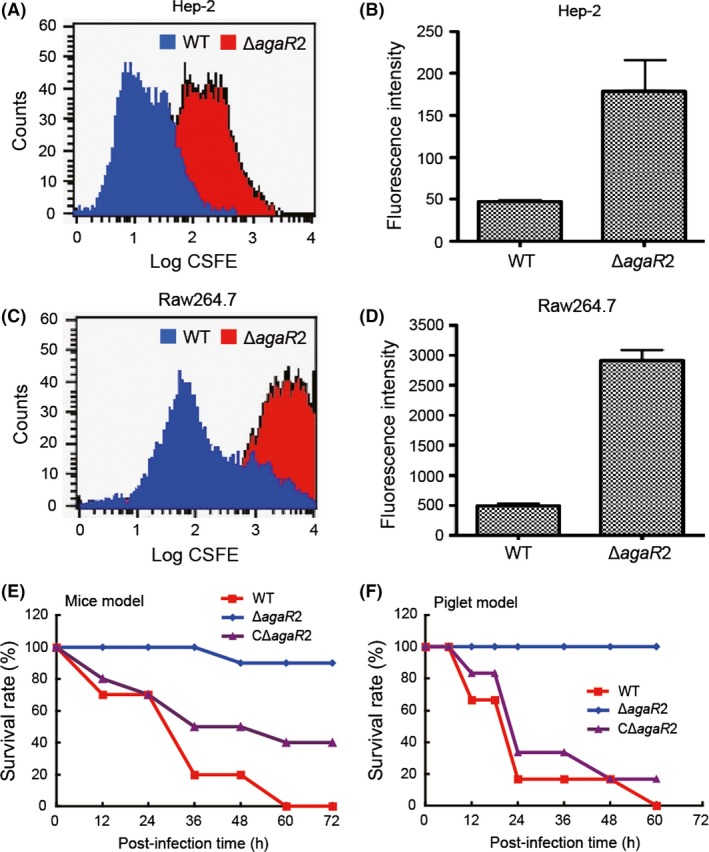
Role of AgaR2‐mediated regulation of GalNAc/GalN catabolism in bacterial virulence. (A) FACS‐based visualization for a role of agaR2 (ZYH05_0447) gene in adherence of Streptococcus suis 2 to Hep‐2 cells. (B) Quantitative analyses for effects of agaR2 deletion on adherence of S. suis 2 to Hep‐2 cells. (C) FACS‐based visualization for the relevance of agaR2 gene to the ability of bacterial anti‐phagocytosis against macrophage Raw264.7 cells. (D) Quantitative analyses for effects of agaR2 gene exerted on the ability of bacterial anti‐phagocytosis against macrophage Raw264.7 cells. (E) Evaluation for a role of agaR2 gene in bacterial virulence using infection model of mice. (F) Use of experimental infection of piglets to assay the relevance of agaR2 gene to bacterial pathogenesis. Note: P < 0.01. GalNAc, N‐acetyl‐d‐galactosamine; GalN, galactosamine; FACS, fluorescence‐activated cell sorting.
Statistics
The data used here were expressed as mean ± SD. Unless specified, data were analyzed by two‐tailed, unpaired t test, and all assays were repeated no less than three times. The threshold for significance refers to the P < 0.05.
Results
Reconstruction of the GalNAc utilization pathway in Lactobacillaceae
The GalNAc utilization pathway has been previously reconstructed by the comparative genomics approach in a large number of Proteobacteria species (Leyn et al. 2012) and experimentally analyzed in E. coli (Brinkkotter et al. 2000) and Shewanella sp. ANA‐3 (Leyn et al. 2012). Here we used the same approach for reconstruction of the GalNAc utilization pathway in the zoonotic agent, S. suis (Fig. 1 and Table S1). For integrated genomic reconstruction of GalNAc utilization pathways and concordant transcriptional regulation, we searched orthologs of known GalNAc utilization genes (Leyn et al. 2012) in complete Lactobacillales genomes. As a result, the genes encoding, this metabolic pathway was identified in 16 genomes representing four families, Streptococcaceae, Lactobacillaceae, Enterobacteraceae, and Carnobacteriaceae (Table S1). The minimal number of genes in the reconstructed regulons was detected in Lactobacillus helveticus and Streptococcus pyogenes. In these organisms, only one or two genes from the GalNAc utilization pathway were found because these genes were insufficient for GalNAc utilization. Most probably, these organisms cannot utilize GalNAc. The minimal gene set allowing utilization of GalNAc is present in a group of closely related genomes including Streptococcus gordonii, Streptococcus mitis, and Streptococcus pneumonia. This gene set contains genes for regulator (agaR), galactosamine‐6‐phosphate deaminase/isomerase (agaS), glycoside hydrolase (bgaC), and PTS (gadVWEF). The other well‐studied Streptococcus genomes also contain genes for the tagatose‐1,6‐diphosphate aldolase (agaY). In contrast, all the analyzed Lactobacillus lack agaY gene but contain the gene for N‐acetylgalactosamine‐phosphate deacetylase (agaA). Amongst Streptococcaceae, agaA gene was identified only in the S. suis genome. The gene for tagatose‐6‐phosphate kinase (agaZ) was found only in Enterococcus faecalis and Carnobacterium sp. 17‐4.
Previously agaY gene was shown to be not obligatory for the GalNAc utilization and function of agaZ was proposed to be actualized by other genes, such as lacC (EC 2.7.1.144) or pfk (EC 2.7.1.11). Orthologs of these genes were found in the studied genomes, for example in S. suis LacC is encoded by gene with locus tag SSU05_1041 and Pfk is encoded by SSU05_0543.
The presence or absence of agaA gene is the crucial point for the ability or disability to utilize GalNAc. Thus, the absence of this gene in the Aga regulons points to the ability of the organism to utilize only GalN, but not GalNAc utilization. Thus, organisms lacking agaA gene should have GalN‐specific transporters whereas organism having agaA gene should be able to transport GalNAc (Leyn et al. 2012). To predict the specificity of the Lactobacillaceae transport systems, we compared all the identified PTSs to the previously described ones. The phylogenetic analysis revealed that all the Lactobacillaceae PTSs are orthologous to the transport system from Haemophilus parasuis (Fig. S4). This PTS was previously proposed to be specific to GalN‐containing oligosaccharides. Co‐occurrence and co‐localization of this PTS with the hydrolase genes in the most studied genomes confirms its specificity to the oligosaccharides. On the other hand, PTS genes are co‐localized also with the agaA gene in the large number of studied genomes (Table S3) that designate to the possibility of this system to transport GalNAc or both GalNAc and GalN containing oligomers. However, we cannot have success in predicting the precise specificity of the gadVWEF encoded PTS, using only the comparative genomics approach.
Comparative genomics‐based insights into the regulation of GalNAc utilization in Lactobacillales
For the integrated genomic reconstruction of GalNAc utilization pathways and the concordant transcriptional regulation, we searched orthologs of known GalNAc utilization genes (Leyn et al. 2012) in complete Lactobacillales genomes. As a result, the genes encoding this metabolic pathway were identified in 16 genomes representing four families, Streptococcaceae, Lactobacillaceae, Enterobacteraceae, and Carnobacteriaceae (Table S3). Analysis of conserved chromosomal loci with the GalNAc utilization genes detected the presence of one or two suggested transcriptional regulators per genome (Fig. 2 and Table S3). All predicted regulators belong to the GntR family of transcription factors (Hoskisson and Rigali 2009). Phylogenetic analysis revealed that the predicted regulators form two separate orthologous groups that we named AgaR1 and AgaR2 (Fig. 3). A strong tendency of agaR1 and agaR2 genes to cluster onto the chromosome with the GalNAc utilization genes suggests conservation of their function (Fig. 2). Two Lactobacillaceae and four Streptococcaceae genomes have both agaR1 and agaR2 genes, whereas in other genomes only one of the regulator genes was present (Fig. 2 and Table S3).
Figure 2.
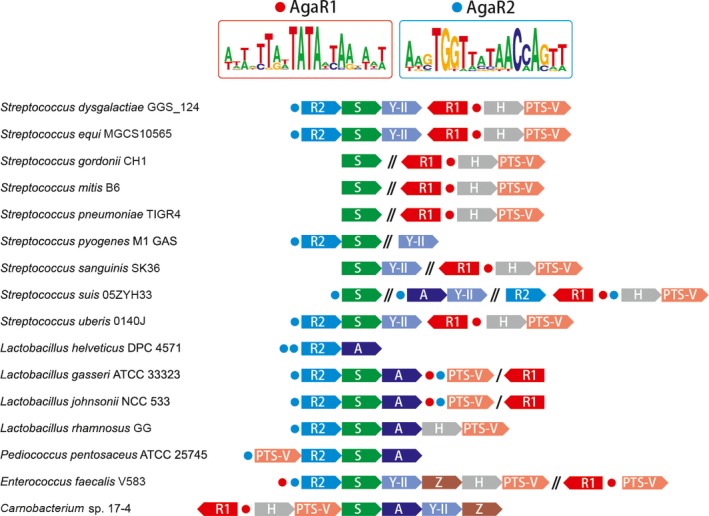
Discovery of the genome‐wide regulons encoding the GalNAc/GalN utilization pathways in Lactobacillales Arrows represent the GalNAc/GalN catabolism‐related genes, and circles denote the predicted AgaR‐recognizable sites. The genes are variably colored according to differential functions assigned in Figure 1 and labeled with the last letter of the corresponding protein. Genes from the same genetic loci that are not adjacent/neighbored each other are separated by a slash. Similarly, genes from different genetic loci are separated by a double slash. Given the two types of putative AgaR‐binding palindromes shown on the top (details in Table S4), AgaR1 and AgaR2 sites are accordingly colored with red and blue. Sequence logos were given using the WebLogo package (http://weblogo.berkeley.edu/logo.cgi). The detailed information on the displayed loci is listed in Table S3 and Figure S4. GalNAc, N‐acetyl‐d‐galactosamine; GalN, galactosamine.
Figure 3.
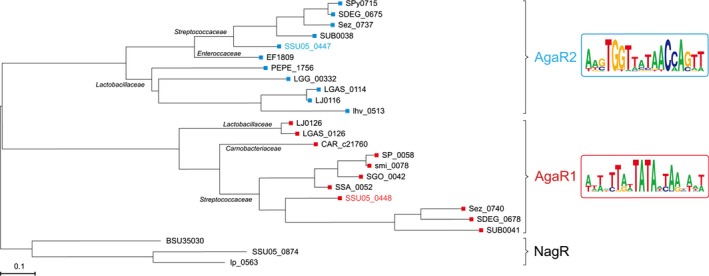
Maximum‐likelihood phylogenetic tree of AgaR proteins. AgaR1 (SSU05_0448) is given in red, and AgaR2 (SSU05_0447) is highlighted in blue. The NagR proteins from Bacillus subtilis, Streptococcus suis, and Lactobacillus plantarum are used as outtree. The logos for the predicted palindromes recognized by AgaR1 (AgaR2) is presented on the right hand. All the homologs of AgaR1 (AgaR2) is accessed to Table S3. NagR proteins from B. subtilis 168 (Bertram et al. 2011), L. plantarum WCFS1, and S. suis 05ZYH33 were used as an outgroup. NagR was selected because, as like AgaR1 and AgaR2, this protein is a member of the HutC subfamily of the GntR family and a regulator of aminosugar metabolism.
To infer the AgaR1/AgaR2 regulons in Lactobacillales, we used the comparative genomics approach applied in the RegPredict Web server. This approach combines prediction of candidate regulator‐binding sites with cross‐genomics comparison of regulons. Since AgaR1 and AgaR2 protein orthologous groups are distantly related to each other (30% identity), we propose that their binding motifs should also be different. In most analyzed genomes, agaR1 is co‐localized with genes for glycoside hydrolase and PTS, whereas agaR2 tends to be clustered onto the chromosome with the genes for deacetylase, isomerase, and aldolase (Fig. 2 and Table S3). Thus, we proposed that AgaR1‐binding sites should be located upstream hydrolase and PTS genes while AgaR2 sites ought to be upstream of deacetylase, isomerase, and aldolase genes. Upstream regions of genes presumably regulated by each transcriptional factor were analyzed using Discover Profile tool of the RegPredict Web resource. After the identification of putative‐binding motif, we searched for additional regulatory sites in the analyzed genomes and finally reconstructed AgaR1 and AgaR2 regulons.
The candidate motifs for both the studied regulators have an even palindrome structure (Figs. 2, 5A and Table S4) that is in good agreement with previous observations on binding motifs for proteins of the HutC subfamily of GntR family (Hoskisson and Rigali 2009). Predicted AgaR1‐binding sites have a length 20 bp, whereas the predicted‐binding sites for AgaR2 are 18 bp sequences (Fig. 5A and Table S4). Both AgaR1 and AgaR2‐binding motifs have a palindrome AT‐rich central part. The AgaR1‐binding motif is AT‐rich at all times and quite degenerated, that is, has moderate information content. On the other hand, the AgaR2‐binding motif is more CG‐rich and conserved and has a higher information content than AgaR1‐binding motif. Additionally, AgaR2‐binding motif demonstrate similarities with the motifs of some regulators, previously characterized experimentally or in silico, such as GnbR of Lactobacillus casei BL23 (Bidart et al. 2014) and NagR proteins of B. subtilis (Bertram et al. 2011; Leyn et al. 2013) and various Lactoacillales (Ravcheev et al. 2013). Composition of the AgaR1 and AgaR2 regulons varies between species (Fig. 2). In Lactobacillus rhamnosus (L. rhamnosus), Enterococcus faecalis (E. faecalis), and Carnobacterium sp. 17‐4, all genes for GalNAc utilization are organized in a single operon that is regulated by a single regulator, AgaR2 in the first two genomes and AgaR1 in the last one (Fig. 2 and Table S3). In the most part of the Streptococcaceae and Lactobaciilaceae genomes, we observed a strong “division of labor” between the two regulators (Fig. 2). Thus, AgaR1‐binding sites were found upstream of hydrolase/PTS genes, whereas AgaR2 looks to regulate genes for deacetylase/isomerase/aldolase (Fig. 2). In the genomes of S. suis, Lactobacillus gasseri (L. gasseri) and Lactobacillus johnsonii (L. johnsonii), overlapping of regulons was detected (Fig. 2). In all these genomes, genes for the transport system seemed likely to be under double regulation by AgaR1 and AgaR2. Also, in the vast majority of the analyzed genomes autoregulation was identified for both agaR1 and agaR2 genes (Table S4).
Figure 5.
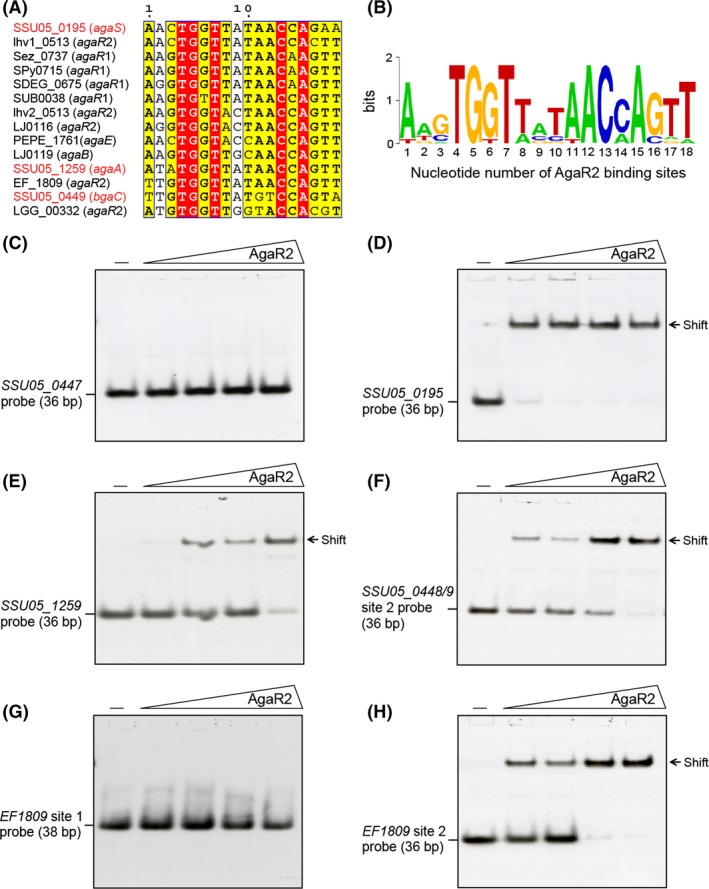
Binding of AgaR2 to the predicted cognate sites. Alignment of the AgaR2‐binding sites from species of Lactobacillales (A) and the resulting sequence logo (B). In (A) the identical residues are white letters in red background, similar residues are black letters in yellow background, and varied residues are in black letters. Names and locus tags for genes from Streptococcus suis are shown by red font. In (B) the sequence logo is generated using WebLogo (http://weblogo.berkeley.edu/logo.cgi). The detailed information of the cognate sites is seen in Table S4. (C) AgaR2 does not bind its own promoter. (D) Binding of agaS (SSU05_0195) promoter to AgaR2 proteinInteraction of AgaR2 protein with the promoter regions of both agaA (SSU05_1259) (in E) and bgaC (SSU05_0449) (in F) AgaR2 protein from Streptococcus suis cannot bind to the AgaR1 site of EF1809 of Enterococcus faecalis V583 (in G), whereas it binds to its AgaR2 site (in H). The minus sign denotes no protein of AgaA2 added. The protein levels of AgaR2 (in the right hand four lanes of each panel [left to right]) were 0.5, 1, 2 and 5 pmol. The protein samples were incubated with 0.2 pmol of DIG‐labeled probe in a total volume of 20 μL. A representative result from three independent gel shift assays (7% native PAGE) is given.
Characterization of AgaR1 and AgaR2, two novel GntR‐type regulators
To probe the putative function of the two new members (S. suis AgaR2 and AgaR1) of the GntR family of transcription factors, we employed BL21(DE3)/pET28(a) a prokaryotic expression system to prepare the above two proteins in vitro. Consequently, the N‐terminal hexahistidine tagged S. suis AgaR2 protein was purified to homogeneity and gave a single‐protein band of appropriate molecular mass (~31 kDa for monomer) (Fig. 4A). The 6xHis tagged version of this recombinant S. suis AgaR2 protein was also determined by Western blot using the anti‐6xHis tag primary antibody (Fig. 4B). Chemical cross‐linking assays with the EGS cross‐linker visualized clearly the EGS dose‐dependent dimerization of AgaR2, indicating its predominant solution structure of this protein is a dimer (Fig. 4C). Liquid chromatography mass spectrometry‐based determination of tryptic peptides of the recombinant AgaR2 protein band excised from an SDS‐PAGE gel (Fig. 4A) verified its identity, in that the peptides matched S. suis SSU05_0447 protein with 77% coverage of the expected peptides (Fig. 4D).
Figure 4.
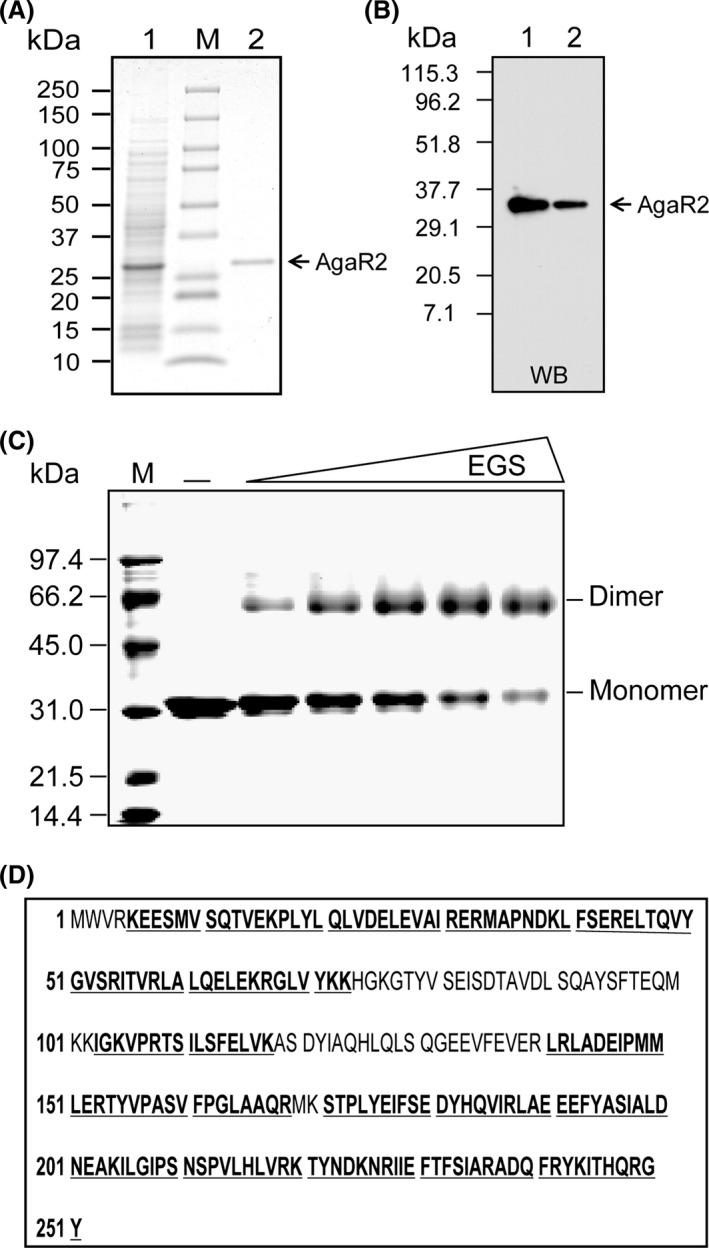
Preparation, identification, and characterization of the AgaR2 (SSU05_0447) protein. (A) 12% SDS‐PAGE profile of the purified AgaR2 (SSU05_0447) protein from Streptococcus suis. The protein with expected size (~31 kDa) is indicated with an arrow. “M” is the abbreviation for protein molecular weight. The numbers on left hand represent protein size (kDa). 1, crude extract of Escherichia coli lysate expressing the recombinant AgaR2 protein; 2, the purified form of the recombinant AgaR2 protein. (B) Western blotting analyses for the N‐terminal 6x his tagged AgaR2 protein using the anti‐6xHis tag primary antibody. WB, western blot; 1, the crude extract of E. coli lysate expressing the recombinant AgaR2 protein; 2, the purified form of AgaR2 protein. (C) Chemical cross‐linking assays for the solution structure of the AgaR2 protein. The chemical cross‐linker used here is EGS. The triangle on the top represents the addition of EGS cross‐linker in varied concentrations (0, 2.5, 5, 10, 20 μmol/L in the right‐hand five lanes [left to right]). (M) Molecular weight. The molecular weight of the monomeric AgaR2 protein is estimated to be ~31 kDa, and the dimeric form is ~62 kDa. The protein sample was judged with 12% SDS‐PAGE. (D) MS‐based identification of the purified AgaR2 protein. The tryptic peptides with hits to the AgaR2 sequence are given in bold and underlined type. SDS‐PAGE, sodiumdodecyl sulfate polyacrylamide gel electrophoresis; EGS, ethylene glycol bis‐succinimidylsuccinate.
Somehow different from the AgaR2 protein, two forms of AgaR1 protein (monomer, the predominant form, and trace amount of dimer) still can be detected by the separation with 12% SDS‐PAGE (Fig. S2A). Given that two possibilities are present (either the contaminated protein with the molecular mass at the dimeric position, or the dimeric form of AgaR1), Western blotting with the anti‐6xHis tag primary antibody was conducted. As anticipated, it clearly showed that two protein bands with an appropriate molecular mass (~31 kDa for monomer and ~60 kDa for dimer), ruling out the possibility of protein contamination (Fig. S2B). This observation is unexpected, but not without precedent. In fact we recently encountered a similar scenario in the case of the Brucella BioR regulator that is also a member of the GntR family transcription factor (Feng et al. 2013a). The essence of BioR1 forming a dimer was further proved by chemical cross‐linking assays (Fig. S2C). MS‐based analyses of two protein bands of AgaR1 (one is cut from a dimer, the other is collected from a monomer) validated exactly the identity wherein the two forms of peptides covered S. suis SSU05_0448 protein at the level of no less than 80% (Fig. S2D and E).
Streptococcus suis AgaR2 (AgaR1) binds the predicted cognate palindromes
It seemed very likely that the genome of S. suis 05ZYH33 (Accession no.: CP000407.1) encodes a fully functional GalNAc utilization machinery and most of the genes encoding this pathway are regulated by AgaR2 (and/or AgaR1) (Fig. 1). Thereby, we employed EMSA to probe the binding of the AgaR2 (and/or AgaR1) protein to the cognate palindromes (Table S4, Fig. 5A and B). On the S. suis chromosome, totally three putative AgaR2‐binding sites (SSU05_0195 probe, SSU05_1259 probe and SSU05_0448/9 site 2 probe) were localized, whereas only one possible AgaR1‐recognizable site (SSU05_0448/9 site 1 probe) was detected. Because of the absence of the predicted AgaR2‐binding site upstream of the agaR2 gene, we expected that this gene is not autoregulated. Indeed, EMSA experiments suggested that AgaR2 protein cannot bind to its own promoter region (SSU05_0447 probe), ruling out the possibility of autoregulation by AgaR2 regulator (Fig. 5C). By contrast, EMSA tests revealed clearly that AgaR2 protein bound the other three AgaR2‐binding palindromes in a dose‐dependent manner (Fig. 5D–F). Additionally, we also used S. suis AgaR2 protein to evaluate the function of the two AgaR‐binding sites in front of the EF1809 locus of E. faecalis V583, a close relative of S. suis. As a result, AgaR2 physically interacted with the EF1809 site 2, TTGTGGTTATAACCAGTT (Fig. 5H), but not the EF1809 site 1, TTTATTGACAAAATAAAAAA (Fig. 5G) further validated the specificity of AgaR2 binding.
Different from the scenario with AgaR2, S. suis AgaR1 was only found to have the ability to interact with the predicted SSU05_0448/9 site 1, TCTATTAATATACTAACACT (Fig. S3A). As anticipated from the comparative genomic analysis of the regulons, no interplay was observed between the AgaR2‐specific SSU05_0448/9 site 2, TTGTGGTTATGTCCAGTA and S. suis AgaR1 protein (Fig. S3B), ruling out the possibility of cross‐regulation by AgaR2 and AgaR1. More importantly, the two putative AgaR1‐recognizable sites from E. faecalis V583 (referred to EF814 site 1, AAATTCAATATATTAAGATA and EF1809 site 1) were demonstrated to be functional, using gel shift assays with the S. suis AgaR1 protein (Fig. S3C and D).
Regulatory roles of AgaR2 (AgaR1) in Streptococcus GalNAc utilization
Given the observation that AgaR2 efficiently binds to the promoter regions of the three genes/operons (agaS (SSU05_0195), agaY (SSU05_1259), and bgaC (SSU05_0448)) encoding GalNAc utilization pathway in S. suis (Figs. 5, 6A), we therefore attempted to elucidate its possible function in modulating Streptococcus GalNAc utilization machinery. The ΔagaR2 (Δ447) isogenic mutant was constructed, using an approach of homologous recombination, and its complementary strain CΔagaR2 (CΔ447) was generated through the low‐copy plasmid pVA838‐borne expression of agaR2 gene (Table S1). We noted that the deletion of agaR2 does not affect its growth (Fig. S1A). Additionally, no obvious alteration of capsule was observed in the Δ agaR2 mutant in comparison with the WT strain 05ZYH33 (Fig. S1B and C). The qPCR‐based transcriptional analyses showed that removal of agaR2 gene gave at least five‐fold increment of agaS (agaAY and bgaC‐gadVWEF operon) expression (Fig. 6B). Whereas the transcription of the above target genes was restored to the level seen in its parental strain S. suis 05ZYH33 (Fig. 6B). Similar results were also observed in altered expression profile of the ΔagaR2 mutant revealed by the RNA‐Seq (e.g., expression of the three genes SSU05_0195 (agaS), SSU05_1259 (agaA), and SSU05_1258 (agaY) are elevated upon removal of agaR2, Table 1). Therefore, it seemed true that AgaR2 is a functional repressor for the Streptococcus GalNAc utilization pathway. In contrast, we failed to visualize any significant alteration of the bgaC (SSU05_0448) expression level in the ΔagaR1 mutant in comparison with the WT strain and its complementary strain CagaR1 (not shown). The suggested possibility that agaR1 might play an uncovered role or be an evolutional relic with loss of physiological demand and/or advantage, in that (1) there is no in vivo regulatory role attributed to its protein product AgaR1 although it retains DNA‐binding activity to its own promoter, (2) the fact that all the other GalNAc metabolism‐related genes are without control by AgaR1 is due to the lack of the predicted AgaR1‐recognizable palindromes, (3) no physiological function can be present in GalNAc metabolism, even AgaR1 can autoregulate itself. Thus, our subsequent interests focused on AgaR2 regulator.
Figure 6.
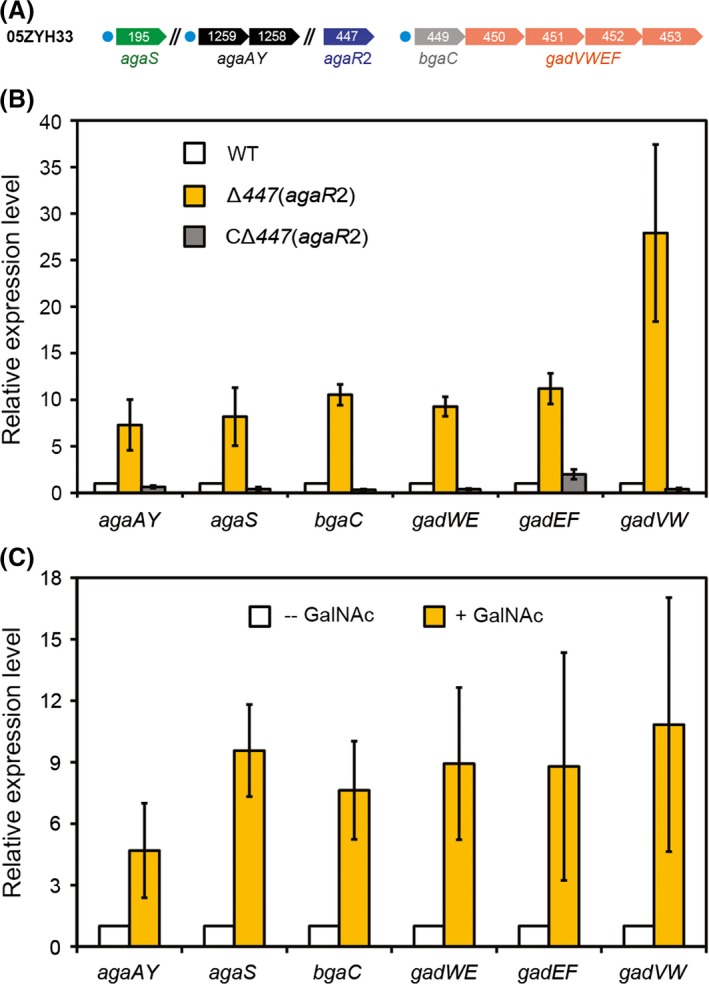
GalNAc/GalN utilization pathway is repressed by AgaR2 and induced by addition of GalNAc. (A) Schematic diagram for agaR2 and its cognate target genes.Circles denote the AgaR2‐recognizable sites, and the arrows represent the genes of the GalNAc utilization pathway. Note: SSU05_1259 and SSU05_1258 constitutes an operon agaAY, whereas the four ordered genes (SSU05_0449, SSU05_0450, SSU05_0451, and SSU05_0452) might form an operon of bgaC‐gadVWEF. (B) Real‐time qPCR assays for effects of agaR2 on expression profile of genes of GalNAc/GalN utilization pathway in Streptococcus suis. (C) Addition of GalNAc increases expression level of GalNAc/GalN utilization pathway‐encoding genes in S. suis.The data are expressed as averages ± SD, and error bars mean SD. No less than five independent experiments were carried out here. The minus sign denotes no addition of GalNAc into growth media, whereas the plus sign denotes addition of GalNAc in vitro. Note: P < 0.005. GalNAc, N‐acetyl‐d‐galactosamine; GalN, galactosamine; qPCR, quantitative PCR; SD, standard deviations.
Table 1.
RNA‐Seq assays for expression profile of genes encoding N‐acetyl‐d‐galactosamine utilization pathway
| Altered expression profile | ||||
|---|---|---|---|---|
| Gene ID | Function | Ratio (ΔagaR2/WT) | Up/down regulation | P‐value |
| 05SSU0195 (agaS) | Phosphosugar isomerase | 6.20 | Up | 3.12836E‐261 |
| 05SSU1259 (agaA) | N‐acetylglucosamine‐6‐phosphate deacetylase | 3.12 | Up | 6.71E‐98 |
| 05SSU1258 (agaY) | Tagatose 1,6‐diphosphate aldolase | 2.98 | Up | 1.27E‐151 |
Considering the direct relevance of AgaR2 to GalNAc utilization in S. suis, it is reasonable to probe the possibility whether this regulator acts to be responsive to the presence of GalNAc. We thereby grew WT strain S. suis 05ZYH33 in the medium with/without the supplementation of GalNAc, and compared the expression profile of relevant target genes like agaS. As anticipated, the expression level of all the tested GalNAc catabolism pathway‐encoding genes (including agaS and bgaC) was given a 5‐ to 10‐fold increment upon the addition of 10 mmol/L GalNAc into growth conditions (Fig. 6C). Together, we concluded that the GalNAc/GalN utilization pathway is repressed mainly by the AgaR2 regulator, whereas induced by an addition of GalNAc.
Contribution of AgaR2 (AgaR1) to bacterial pathogenesis
As we knew that amino sugars (GalN and/or GalNAc) constitute common residues for surface structure of various bacterial cell walls (Bernatchez et al. 2005; Freymond et al. 2006), we thus ambitiously reasoned that (1) such a bacterial surface structure might be involved in mutual communication/crosstalk between bacterial pathogens and the inhabited/infected hosts, (2) the maintenance and regulation of GalN/GalNAc catabolism is probably implicated into successful infections of some bacterial pathogens. To probe possible interference between GalN/GalNAc catabolism and the formation of virulence‐associated surface structures, we systemically employed two lines of approaches including cell lines‐based tests and infections of experimental animals.
First, Hep‐2 cell line was used to evaluate the ability of bacterial adherence (Fig. 7A and B), and RAW264.7 macrophage was subjected to dissecting its capability of anti‐phagocytosis (Fig. 7C and D). Fluorescence‐activated cell sorting (FACS)‐based experiments elucidated that around threefold increment of bacterial adherence potential to Hep‐2 cells was given in the ΔagaR2 mutant relative to the WT 05ZYH33 strain (Fig. 7A and B). Furthermore, the deletion of agaR2 gene was found to give nearly fivefold increment of anti‐phagocytosis ability against RAW264.7 macrophage (Fig. 7C and D). This finding was in much similarity to scenarios seen with both ΔneuB and Δcps2B mutants of S. suis 2 (note: the two genes are involved in bacterial surface architecture) (Feng et al. 2012). Thereby, we anticipated that the enhancement in abilities of bacterial attachment and anti‐phagocytosis might be partially due to the altered carbohydrates with GalN/GalNAc residues on bacterial cell wall surface and this kind of alteration could be attributed to the dysfunction in AgaR2‐mediated regulation of GalN/GalNAc utilization pathway. In contrast, no obvious difference between the ΔagaR1 mutant and its parental strain 05ZYH33 was observed on either bacterial adherence to Hep‐2 cell line or bacterial anti‐phagocytosis against RAW264.7 macrophage (not shown). In fact, such a different effect exerted by AgaR2 (and AgaR1) is not very surprising, in that only Aga2 (not AgaR1) evolved to possess a regulatory role in controlling GalNAc catabolism in S. suis.
Given the fact that not only the two mutants (ΔneuB and Δcps2B) of S. suis 2 feature the potential of both increased adherence/anti‐phagocytosis, but also exhibit dramatically reduced virulence in infection models of both mice and piglets (Feng et al. 2012), it is of much interest to further probe a possible role of agaR2 in bacterial virulence. In the mice infection assays, all the 10 BALB/c (4‐week old, female) mice infected with the WT strain 05ZYH33 were sick shortly, and most of the rats died within 60 h (Fig. 7E). By contrast, mice of the negative control group inoculated with THB survived (not shown). In particular note, nearly all the mice infected with the ΔagaR2 mutant survived, while most of animals injected with the complementary strain CΔagaR2 died within 3 days. To verify the results obtained from the above model of mice, we repeated the infection tests using the model of SPF‐piglets, natural hosts for S. suis. As a result, we observed that the six SPF‐piglets inoculated with WT virulent strain developed most of the typical disease symptoms (high fever, limping, swollen joints, etc.), and most of them died on day 1 (Fig. 7F). As expected, all the piglets infected with the ΔagaR2 mutant survived during the period of entire experiment. However, the re‐introduction of agaR2 gene into the ΔagaR2 mutant restored fully its strong virulence (Fig. 7F). Collectively, we believed that the prevalent regulator AgaR2 for GalN/GalNAc utilization pathway contributes to bacterial infectivity of S. suis, although we failed to note an apparent role of the other secondary regulator AgaR1 in bacterial pathogenicity (not shown).
Discussion
The data shown here represents a first report that illustrated the genomic reconstruction of GalN/GalNAc catabolism pathway in Streptococci and Firmicutes. Different from the scenarios described in E. coli (Reizer et al. 1996) and Shewanella (Leyn et al. 2012), a significant variation of this pathway was proposed for Streptococci (Fig. 1 and Table S4). As you can see from the working model, AgaZ is absent in S. suis. It seemed likely that our continued genomic analyses pointed out that S. suis actually lacks any ortholog of this gene. Thereby, we speculated that the loss of AgaZ function in S. suis might be compensated by two other genes that are not in aga regulon: (1) SSU05_1041 (lacC2; tagatose 6‐phosphate kinase, EC 2.7.1.144), (2) SSU05_0543 (pfk, 6‐phosphofructokinase, EC 2.7.1.11). This hypothesis required further experimental verification.
Through extensive analyses for genomic contexts from Firmicutes, we dissected two GalN/GalNAc pathway‐specific regulators namely AgaR2 and AgaR1. The agaR1 gene is divergently transcribed into genes forming an operon and encoding a β‐galactosidase (BgaC) (Hu et al. 2014) and a PTS sugar uptake system (named agaBCDE that could be a GalN uptake system). The agaR2 gene in many Streptococcus species is located in front of the genes encoding the predicted galactosamine‐6‐phosphate isomerase and Tagatose 1,6‐diphosphate aldolase, two key enzymes in the AGA catablic pathway. In S. suis, the locus SSU05_0448 is annotated to be agaR1, whereas SSU05_0447 is referred to agaR2. Somewhat evolutionally distinct from the paradigm regulator (the E. coli agaR protein product) that belongs to the DeoR family of transcription factor, the two newly identified regulators AgaR2 and AgaR1 are classified into the GntR family of transcription repressors (Figs. 3, S1). Additionally, it seemed likely that the two GntR‐like regulators (AgaR2 and AgaR1) themselves fit into two different subgroups and might possess the varied preference in their DNA‐binding sites (Figs. 3, S4). The fact that AgaR2‐recognizable palindrome is predicted to be present in front of multiple target genes whereas AgaR1 has only one site located in its own promoter region (Fig. 2) raised the possibility that AgaR2 could have been evolved into a prevalent/major regulator for GalN/GalNAc utilization pathway (AgaR1 might be a minor/and even a cryptic regulator). Our in vitro gel shift assays validated the direct interaction between the AgaR2 (and/or AgaR1) protein and its binding sites (Figs. 5, S3). Given the facts that a significant regulated expression of GalN/GalNAc utilization‐related genes (such as agaS, agaAY , and bgaC) by AgaR2 was observed, however, no in vivo role could be attributed to AgaR1, we came to be more confident that AgaR2 relative to AgaR1 acts as a prevalent/leading player in modulating expression of genes encoding bacterial GalN/GalNAc catabolism in most Firmicutes. However, we did not obtain evidence for physiological ligand/molecular effector of AgaR2 repressor, although we observed that GalNAc does induce the expression of GalN/GalNAc catabolism‐related genes such as agaS (Fig. 6C).
The essence that amino sugars (GalN and/or GalNAc) are common components on the surface of bacterial cell walls (Bernatchez et al. 2005; Freymond et al. 2006) has driven us to test possible roles for the regulation of GalN/GalNAc catabolism in the infectivity of bacterial pathogens. As expected, an interference of GalN/GalNAc catabolism/utilization by disrupting agaR2 encoding a prevalent regulator, significantly altered abilities of both bacterial adherence to Hep‐2 cells and anti‐phagocytosis against RAW264.7 macrophage (Fig. 7). Given the fact that in this alteration we observed a similar scenario seen with the two virulence‐associated determinants (neuB and cps2B) with biological roles in constitution/development of bacterial surface architecture (Feng et al. 2012), we reasoned that regulation/maintenance of GalN/GalNAc catabolism pathway is essential for full virulence of S. suis. In fact, this proposal was subsequently proved in that the dysfunction in AgaR2‐mediated regulation impairs bacterial infectivity of S. suis in both mice and piglets (Fig. 7). Also, we are not surprised to establish the relevance of GalN/GalNAc catabolism to bacterial pathogenicity, in that the alteration of carbohydrates with GalN/GalNAc residues on bacterial cell surface interferes with the bacterial pathogen‐binding host cells.
Taken together, we are first to report the genomic reconstruction of the GalN/GalNAc utilization pathway in S. suis and its novel regulatory network with variations. More intriguingly, we revealed that the interference of GalN/GalNAc utilization pathway by the inactivation of the AgaR2 regulator attenuates greatly the bacterial infectivity of the zoonotic pathogen S. suis. Our finding might provide a metabolic basis for design of small molecule drugs (inhibitors)‐based therapeutics against S. suis infection through targeting the regulatory network of GalN/GalNAc utilization pathway.
Conflict of Interest
None declared.
Supporting information
Table S1. Strains and plasmids used in this study.
Table S2. DNA primers used in this study.
Table S3. AgaR regulons in Firmicutes.
Table S4. AgaR1 (AgaR2) binding sites.
Figure S1. Multiple sequence alignments of SSU05_0447 (AgaR2) with two other bacterial homologs. The three homologous proteins used here included Bacillus subtilus NagR (NC_018520.1), SSU05_0447 (AgaR2) of Streptococcus suis 05ZYH33 (NC_009442.1), and Escherichia coli AgaR (NC_007779.1). The program of ClustalW2 (http://www.ebi.ac.uk/Tools/clustalw2/index.html) was applied to conduct the multiple alignment of protein sequences, and the final output is generated by the ESPript 2.2 program (http://espript.ibcp.fr/ESPript/cgi-bin/ESPript.cgi). Identical residues are in white letters with a red background, similar residues are in red letters with a white background, varied residues are in black letters, and dots represent gaps. The predicted protein secondary structure is given on the top. Designations: NagR, N‐acetylglucosamine repressor; AgaR, acetyl‐galactosamine repressor; bs, Bacillus subtilus; ec, E. coli, α, α‐helix; β, β‐sheet; T, β‐turns/coils.
Figure S2. Purification, verification, and characterization of the AgaR1 (SSU05_0448) protein. (A) 12% SDS‐PAGE profile of the purified AgaR1 (SSU05_0448) protein from Streptococcus suis. (B) Western blot analyses for the N‐terminal 6x his tagged AgaR1 protein, using the anti‐6xHis tag primary antibody. The monomeric protein with expected size of ~30 kDa is indicated with an arrow, whereas the dimer form (~60 kDa) is highlighted with an asterisk. Designations: M, protein standard marker; WB, western blot. (C) Determination for the solution structure of the AgaR1 protein, using chemical cross‐linking assays. The chemical cross‐linker used here is ethylene glycol bis‐succinimidylsuccinate (EGS). The triangle on the top represents the addition of the EGS cross‐linker in varied concentrations (0.1, 0.2, 0.5, 1.0, 2.5, 5, 10, 20 µmol/L in the right‐hand eight lanes [left to right]). The minus sign denotes no addition of EGS. The protein sample was separated with 12% SDS‐PAGE. The MS‐based identification of the purified AgaR1 protein with the solution structure of both monomer (in D) and dimer (in E). The tryptic peptides that match the AgaR1 protein are given in bold and under‐lined type.
Figure S3. Binding of AgaR1 to the predicted palindromes. The predicted AgaR1‐binding site (in A) of SSU05_0448/9 gene bound AgaR1 (SSU05_0448) protein, whereas the AgaR2‐binding site (in B) of this locus does not interact with AgaR1 protein. (C) Streptococcus suis AgaR1 protein bound to the predicted AgaR1 site of EF814 gene from Enterococcus faecalis V583. (D) S. suis AgaR1 protein bound to the putative AgaR1 site of EF1809 gene from E. faecalis V583. The minus sign denotes no addition of AgaA1 protein. The protein levels of AgaR2 (on the right‐hand four lanes of each panel [left to right]) were 0.5, 1, 2, and 5 pmol. The protein samples were incubated with 0.2 pmol of the DIG‐labeled probe in a total volume of 15 µL. A representative result from three independent gel shift assays (7% native PAGE) is given.
Figure S4. Phylogenetic analyses of phosphotransferase system (PTS). In total, the PTS system is classified into five sub‐groups, one of which is PTS‐V (highlighted in blue). Locus tag of PTS system is showed here for AgaC, SSU05_0451 is indicated in red.
Acknowledgments
This work was supported by the start‐up package of Zhejiang University (Youjun Feng), the Zhejiang Provincial Natural Science Foundation for Distinguished Young Scholars (grant no. LR15H190001), the National Natural Science Foundation of China (grant no. 31570027, 81501725, 31170124, 81371768, 81172794, and 81471920), the Russian Academy of Sciences (program “Molecular and Cellular Biology”.), the National Research Fund (#6847110), Luxembourg, and was co‐funded under the Marie Curie Actions of the European Commission (FP7‐COFUND). Dr. Feng is a recipient of the “Young 1000 Talents” Award.
MicrobiologyOpen 2015; 4(6): 983–1000
References
- Abu‐Qarn, M. , Eichler J., and Sharon N.. 2008. Not just for Eukarya anymore: protein glycosylation in Bacteria and Archaea. Curr. Opin. Struct. Biol. 18:544–550. [DOI] [PubMed] [Google Scholar]
- Altschul, S. F. , Madden T. L., Schaffer A. A., Zhang J., Z. Zhang , Miller W., et al. 1997. Gapped BLAST and PSI‐BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr, J. , and Nordin P.. 1980. Biosynthesis of glycoproteins by membranes of Acer pseudoplatanus. Incorporation of mannose and N‐acetylglucosamine. Biochem. J. 192:569–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benga, L. , Fulde M., Neis C., Goethe R., and Valentin‐Weigand P.. 2008. Polysaccharide capsule and suilysin contribute to extracellular survival of Streptococcus suis co‐cultivated with primary porcine phagocytes. Vet. Microbiol. 132:211–219. [DOI] [PubMed] [Google Scholar]
- Bernatchez, S. , Szymanski C. M., Ishiyama N., Li J., Jarrell H. C., Lau P. C., et al. 2005. A single bifunctional UDP‐GlcNAc/Glc 4‐epimerase supports the synthesis of three cell surface glycoconjugates in Campylobacter jejuni . J. Biol. Chem. 280:4792–4802. [DOI] [PubMed] [Google Scholar]
- Bertram, R. , Rigali S., Wood N., Lulko A. T., Kuipers O. P., and Titgemeyer F.. 2011. Regulon of the N‐acetylglucosamine utilization regulator NagR in Bacillus subtilis . J. Bacteriol. 193:3525–3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidart, G. N. , Rodriguez‐Diaz J., Monedero V., and Yebra M. J.. 2014. A unique gene cluster for the utilization of the mucosal and human milk‐associated glycans galacto‐N‐biose and lacto‐N‐biose in Lactobacillus casei . Mol. Microbiol. 93:521–538. [DOI] [PubMed] [Google Scholar]
- Brinkkotter, A. , Kloss H., Alpert C., and Lengeler J. W.. 2000. Pathways for the utilization of N‐acetyl‐galactosamine and galactosamine in Escherichia coli . Mol. Microbiol. 37:125–135. [DOI] [PubMed] [Google Scholar]
- Brinkkotter, A. , Shakeri‐Garakani A., and Lengeler J. W.. 2002. Two class II D‐tagatose‐bisphosphate aldolases from enteric bacteria. Arch. Microbiol. 177:410–419. [DOI] [PubMed] [Google Scholar]
- Cao, M. , Feng Y., Wang C., Zheng F., Li M., Liao H., et al. 2011. Functional definition of LuxS, an autoinducer‐2 (AI‐2) synthase and its role in full virulence of Streptococcus suis serotype 2. J. Microbiol. 49:1000–1011. [DOI] [PubMed] [Google Scholar]
- Carraway, K. L. , and Hull S. R.. 1991. Cell surface mucin‐type glycoproteins and mucin‐like domains. Glycobiology 1:131–138. [DOI] [PubMed] [Google Scholar]
- Chen, C. , Tang J., Dong W., Wang C., Feng Y., Wang J., et al. 2007. A glimpse of streptococcal toxic shock syndrome from comparative genomics of S. suis 2 Chinese isolates. PLoS One 2:e315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks, G. E. , Hon G., Chandonia J. M., and Brenner S. E.. 2004. WebLogo: a sequence logo generator. Genome Res. 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, C. G. , Elhammer A., Russell D. W., Schneider W. J., Kornfeld S., Brown M. S., et al. 1986. Deletion of clustered O‐linked carbohydrates does not impair function of low density lipoprotein receptor in transfected fibroblasts. J. Biol. Chem. 261:2828–2838. [PubMed] [Google Scholar]
- Dehal, P. S. , Joachimiak M. P., Price M. N., Bates J. T., Baumohl J. K., Chivian D., et al. 2010. MicrobesOnline: an integrated portal for comparative and functional genomics. Nucleic Acids Res. 38:D396–D400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disz, T. , Akhter S., Cuevas D., Olson R., Overbeek R., Vonstein V., et al. 2010. Accessing the SEED genome databases via Web services API: tools for programmers. BMC Bioinformatics 11:319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R. C. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esgleas, M. , Li Y., Hancock M. A., Harel J., Dubreuil J. D., and Gottschalk M.. 2008. Isolation and characterization of alpha‐enolase, a novel fibronectin‐binding protein from Streptococcus suis . Microbiology 154:2668–2679. [DOI] [PubMed] [Google Scholar]
- Felsenstein, J. 1996. Inferring phylogenies from protein sequences by parsimony, distance, and likelihood methods. Methods Enzymol. 266:418–427. [DOI] [PubMed] [Google Scholar]
- Feng, Y. , and Cronan J. E.. 2009. A new member of the Escherichia coli fad regulon: transcriptional regulation of fadM (ybaW). J. Bacteriol. 191:6320–6328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, Y. , and Cronan J. E.. 2010. Overlapping repressor binding sites result in additive regulation of Escherichia coli FadH by FadR and ArcA. J. Bacteriol. 192:4289–4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, Y. , and Cronan J. E.. 2011. The Vibrio cholerae fatty acid regulatory protein, FadR, represses transcription of plsB, the gene encoding the first enzyme of membrane phospholipid biosynthesis. Mol. Microbiol. 81:1020–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, Y. , and Cronan J. E.. 2012. Crosstalk of Escherichia coli FadR with global regulators in expression of fatty acid transport genes. PLoS One 7:e46275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, Y. , Li M., Zhang H., Zheng B., Han H., Wang C., et al. 2008. Functional definition and global regulation of Zur, a zinc uptake regulator in a Streptococcus suis serotype 2 strain causing streptococcal toxic shock syndrome. J. Bacteriol. 190:7567–7578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, Y. , Pan X., Sun W., Wang C., Zhang H., Li X., et al. 2009a. Streptococcus suis enolase functions as a protective antigen displayed on the bacterial cell surface. J. Infect. Dis. 200:1583–1592. [DOI] [PubMed] [Google Scholar]
- Feng, Y. , Shi X., Zhang H., Zhang S., Ma Y., Zheng B., et al. 2009b. Recurrence of human Streptococcus suis infections in 2007: three cases of meningitis and implications that heterogeneous S. suis 2 circulates in China. Zoonoses Public Health 56:506–514. [DOI] [PubMed] [Google Scholar]
- Feng, Y. , Zhang H., Ma Y., and Gao G. F.. 2010. Uncovering newly emerging variants of Streptococcus suis, an important zoonotic agent. Trends Microbiol. 18:124–131. [DOI] [PubMed] [Google Scholar]
- Feng, Y. , Cao M., Shi J., Zhang H., Hu D., Zhu J., et al. 2012. Attenuation of Streptococcus suis virulence by the alteration of bacterial surface architecture. Sci. Rep. 2:710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, Y. , Xu J., Zhang H., Chen Z., and Srinivas S.. 2013a. Brucella BioR regulator defines a complex regulatory mechanism for bacterial biotin metabolism. J. Bacteriol. 195:3451–3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, Y. , Zhang H., and Cronan J. E.. 2013b. Profligate biotin synthesis in alpha‐proteobacteria – a developing or degenerating regulatory system? Mol. Microbiol. 88:77–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, Y. , Napier B. A., Manandhar M., Henke S. K., Weiss D. S., and Cronan J. E.. 2014a. A Francisella virulence factor catalyses an essential reaction of biotin synthesis. Mol. Microbiol. 91:300–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, Y. , Zhang H., Wu Z., Wang S., Cao M., Hu D., et al. 2014b. Streptococcus suis infection: An emerging/reemerging challenge of bacterial infectious diseases? Virulence 5:477–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freymond, P. P. , Lazarevic V., Soldo B., and Karamata D.. 2006. Poly(glucosyl‐N‐acetylgalactosamine 1‐phosphate), a wall teichoic acid of Bacillus subtilis 168: its biosynthetic pathway and mode of attachment to peptidoglycan. Microbiology 152:1709–1718. [DOI] [PubMed] [Google Scholar]
- Gaugue, I. , Oberto J., and Plumbridge J.. 2014. Regulation of amino sugar utilization in Bacillus subtilis by the GntR family regulators, NagR and GamR. Mol. Microbiol. 92:100–115. [DOI] [PubMed] [Google Scholar]
- Hoskisson, P. A. , and Rigali S.. 2009. Chapter 1: variation in form and function the helix‐turn‐helix regulators of the GntR superfamily. Adv. Appl. Microbiol. 69:1–22. [DOI] [PubMed] [Google Scholar]
- Hu, D. , Zhang F., Zhang H., Hao L., Gong X., Geng M., et al. 2014. The β‐galactosidase (BgaC) of the zoonotic pathogen Streptococcus suis is a surface protein without the involvement of bacterial virulence. Sci. Rep. 4:4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson, D. H. , Richter D. C., Rausch C., Dezulian T., Franz M., and Rupp R.. 2007. Dendroscope: an interactive viewer for large phylogenetic trees. BMC Bioinformatics 8:460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hytonen, J. , Haataja S., and Finne J.. 2006. Use of flow cytometry for the adhesion analysis of Streptococcus pyogenes mutant strains to epithelial cells: investigation of the possible role of surface pullulanase and cysteine protease, and the transcriptional regulator Rgg. BMC Microbiol. 6:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs, A. A. , van den Berg A. J., and Loeffen P. L.. 1996. Protection of experimentally infected pigs by suilysin, the thiol‐activated haemolysin of Streptococcus suis . Vet. Rec. 139:225–228. [DOI] [PubMed] [Google Scholar]
- Leyn, S. A. , Gao F., Yang C., and Rodionov D. A.. 2012. N‐acetylgalactosamine utilization pathway and regulon in proteobacteria: genomic reconstruction and experimental characterization in Shewanella . J. Biol. Chem. 287:28047–28056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyn, S. A. , Kazanov M. D., Sernova N. V., Ermakova E. O., Novichkov P. S., and Rodionov D. A.. 2013. Genomic reconstruction of the transcriptional regulatory network in Bacillus subtilis . J. Bacteriol. 195:2463–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, M. , Wang C., Feng Y., Pan X., Cheng G., Wang J., et al. 2008. SalK/SalR, a two‐component signal transduction system, is essential for full virulence of highly invasive Streptococcus suis serotype 2. PLoS One 3:e2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , Tan C., Zhou Y., Fu S., Hu L., Hu J., et al. 2011. The two‐component regulatory system CiaRH contributes to the virulence of Streptococcus suis 2. Vet. Microbiol. 148:99–104. [DOI] [PubMed] [Google Scholar]
- Lu, Q. , Lu H., Qi J., Lu G., and Gao G. F.. 2012. An octamer of enolase from Streptococcus suis . Protein Cell 3:769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun, S. , Perez‐Casal J., Connor W., and Willson P. J.. 2003. Role of suilysin in pathogenesis of Streptococcus suis capsular serotype 2. Microb. Pathog. 34:27–37. [DOI] [PubMed] [Google Scholar]
- Ma, Y. , Feng Y., Liu D., and Gao G. F.. 2009. Avian influenza virus, Streptococcus suis serotype 2, severe acute respiratory syndrome‐coronavirus and beyond: molecular epidemiology, ecology and the situation in China. Philos. Trans. R. Soc. Lond. B Biol. Sci. 364:2725–2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironov, A. A. , Koonin E. V., Roytberg M. A., and Gelfand M. S.. 1999. Computer analysis of transcription regulatory patterns in completely sequenced bacterial genomes. Nucleic Acids Res. 27:2981–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novichkov, P. S. , Rodionov D. A., Stavrovskaya E. D., Novichkova E. S., Kazakov A. E., Gelfand M. S., et al. 2010. RegPredict: an integrated system for regulon inference in prokaryotes by comparative genomics approach. Nucleic Acids Res. 38:W299–W307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plumbridge, J. 2015. Regulation of the utilization of amino sugars by Escherichia coli and Bacillus subtilis: same genes, different control. J. Mol. Microbiol. Biotechnol. 25:154–167. [DOI] [PubMed] [Google Scholar]
- Ravcheev, D. A. , Best A. A., Sernova N. V., Kazanov M. D., Novichkov P. S., and Rodionov D. A.. 2013. Genomic reconstruction of transcriptional regulatory networks in lactic acid bacteria. BMC Genom. 14:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray, W. K. , and Larson T. J.. 2004. Application of AgaR repressor and dominant repressor variants for verification of a gene cluster involved in N‐acetylgalactosamine metabolism in Escherichia coli K‐12. Mol. Microbiol. 51:813–826. [DOI] [PubMed] [Google Scholar]
- Reizer, J. , Ramseier T. M., Reizer A., Charbit A., and Saier M. H. Jr.. 1996. Novel phosphotransferase genes revealed by bacterial genome sequencing: a gene cluster encoding a putative N‐acetylgalactosamine metabolic pathway in Escherichia coli . Microbiology 142(Pt 2):231–250. [DOI] [PubMed] [Google Scholar]
- Rigali, S. , Derouaux A., Giannotta F., and Dusart J.. 2002. Subdivision of the helix‐turn‐helix GntR family of bacterial regulators in the FadR, HutC, MocR, and YtrA subfamilies. J. Biol. Chem. 277:12507–12515. [DOI] [PubMed] [Google Scholar]
- Rigali, S. , Titgemeyer F., Barends S., Mulder S., Thomae A. W., Hopwood D. A., et al. 2008. Feast or famine: the global regulator DasR links nutrient stress to antibiotic production by Streptomyces . EMBO Rep. 9:670–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodionov, D. A. 2007. Comparative genomic reconstruction of transcriptional regulatory networks in bacteria. Chem. Rev. 107:3467–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero, D. A. , Slos P., Robert C., Castellino I., and A. Mercenier . 1987. Conjugative mobilization as an alternative vector delivery system for lactic streptococci . Appl. Environ. Microbiol. 53:2405–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadler, J. E. , Paulson J. C., and Hill R. L.. 1979. The role of sialic acid in the expression of human MN blood group antigens. J. Biol. Chem. 254:2112–2119. [PubMed] [Google Scholar]
- Seitz, M. , Beineke A., Singpiel A., Willenborg J., Dutow P., Goethe R., et al. 2014. Role of capsule and suilysin in mucosal infection of complement‐deficient mice with Streptococcus suis . Infect. Immun. 82:2460–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton, D. A. , Stegman L., Hardison R., Miller W., Bock J. H., Slightom J. L., et al. 1997. Phylogenetic footprinting of hypersensitive site 3 of the beta‐globin locus control region. Blood 89:3457–3469. [PubMed] [Google Scholar]
- Si, Y. , Yuan F., Chang H., Liu X., Li H., Cai K., et al. 2009. Contribution of glutamine synthetase to the virulence of Streptococcus suis serotype 2. Vet. Microbiol. 139:80–88. [DOI] [PubMed] [Google Scholar]
- Sonnhammer, E. L. , Eddy S. R., Birney E., Bateman A., and Durbin R.. 1998. Pfam: multiple sequence alignments and HMM‐profiles of protein domains. Nucleic Acids Res. 26:320–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suvorova, I. A. , Korostelev Y. D., and Gelfand M. S.. 2015. GntR family of bacterial transcription factors and their DNA binding motifs: structure, positioning and co‐evolution. PLoS One 10:e0132618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamatsu, D. , Osaki M., and Sekizaki T.. 2001. Construction and characterization of Streptococcus suis‐Escherichia coli shuttle cloning vectors. Plasmid 45:101–113. [DOI] [PubMed] [Google Scholar]
- Takeuchi, D. , Akeda Y., Nakayama T., Kerdsin A., Sano Y., Kanda T., et al. 2014. The contribution of suilysin to the pathogenesis of Streptococcus suis meningitis. J. Infect. Dis. 209:1509–1519. [DOI] [PubMed] [Google Scholar]
- Tang, J. , Wang C., Feng Y., Yang W., Song H., Chen Z., et al. 2006. Streptococcal toxic shock syndrome caused by Streptococcus suis serotype 2. PLoS Med. 3:e151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J. D. , Gibson T. J., and Higgins D. G.. 2002. Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinformatics Chapter 2:Unit 2.3. [DOI] [PubMed] [Google Scholar]
- Xu, J. , Fu S., Liu M., Xu Q., Bei W., Chen H., et al. 2014. The two‐component system NisK/NisR contributes to the virulence of Streptococcus suis serotype 2. Microbiol. Res. 169:541–546. [DOI] [PubMed] [Google Scholar]
- Zhang, A. , Chen B., Mu X., Li R., Zheng P., Zhao Y., et al. 2009a. Identification and characterization of a novel protective antigen, enolase of Streptococcus suis serotype 2. Vaccine 27:1348–1353. [DOI] [PubMed] [Google Scholar]
- Zhang, X. H. , He K. W., Duan Z. T., Zhou J. M., Yu Z. Y., Ni Y. X., et al. 2009b. Identification and characterization of inosine 5‐monophosphate dehydrogenase in Streptococcus suis type 2. Microb. Pathog. 47:267–273. [DOI] [PubMed] [Google Scholar]
- Zhou, J. , Zhang X., He K., Wang W., Ni Y., Zhu H., et al. 2014. Characterization and proteome analysis of inosine 5‐monophosphate dehydrogenase in epidemic Streptococcus suis serotype 2. Curr. Microbiol. 68:663–669. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Strains and plasmids used in this study.
Table S2. DNA primers used in this study.
Table S3. AgaR regulons in Firmicutes.
Table S4. AgaR1 (AgaR2) binding sites.
Figure S1. Multiple sequence alignments of SSU05_0447 (AgaR2) with two other bacterial homologs. The three homologous proteins used here included Bacillus subtilus NagR (NC_018520.1), SSU05_0447 (AgaR2) of Streptococcus suis 05ZYH33 (NC_009442.1), and Escherichia coli AgaR (NC_007779.1). The program of ClustalW2 (http://www.ebi.ac.uk/Tools/clustalw2/index.html) was applied to conduct the multiple alignment of protein sequences, and the final output is generated by the ESPript 2.2 program (http://espript.ibcp.fr/ESPript/cgi-bin/ESPript.cgi). Identical residues are in white letters with a red background, similar residues are in red letters with a white background, varied residues are in black letters, and dots represent gaps. The predicted protein secondary structure is given on the top. Designations: NagR, N‐acetylglucosamine repressor; AgaR, acetyl‐galactosamine repressor; bs, Bacillus subtilus; ec, E. coli, α, α‐helix; β, β‐sheet; T, β‐turns/coils.
Figure S2. Purification, verification, and characterization of the AgaR1 (SSU05_0448) protein. (A) 12% SDS‐PAGE profile of the purified AgaR1 (SSU05_0448) protein from Streptococcus suis. (B) Western blot analyses for the N‐terminal 6x his tagged AgaR1 protein, using the anti‐6xHis tag primary antibody. The monomeric protein with expected size of ~30 kDa is indicated with an arrow, whereas the dimer form (~60 kDa) is highlighted with an asterisk. Designations: M, protein standard marker; WB, western blot. (C) Determination for the solution structure of the AgaR1 protein, using chemical cross‐linking assays. The chemical cross‐linker used here is ethylene glycol bis‐succinimidylsuccinate (EGS). The triangle on the top represents the addition of the EGS cross‐linker in varied concentrations (0.1, 0.2, 0.5, 1.0, 2.5, 5, 10, 20 µmol/L in the right‐hand eight lanes [left to right]). The minus sign denotes no addition of EGS. The protein sample was separated with 12% SDS‐PAGE. The MS‐based identification of the purified AgaR1 protein with the solution structure of both monomer (in D) and dimer (in E). The tryptic peptides that match the AgaR1 protein are given in bold and under‐lined type.
Figure S3. Binding of AgaR1 to the predicted palindromes. The predicted AgaR1‐binding site (in A) of SSU05_0448/9 gene bound AgaR1 (SSU05_0448) protein, whereas the AgaR2‐binding site (in B) of this locus does not interact with AgaR1 protein. (C) Streptococcus suis AgaR1 protein bound to the predicted AgaR1 site of EF814 gene from Enterococcus faecalis V583. (D) S. suis AgaR1 protein bound to the putative AgaR1 site of EF1809 gene from E. faecalis V583. The minus sign denotes no addition of AgaA1 protein. The protein levels of AgaR2 (on the right‐hand four lanes of each panel [left to right]) were 0.5, 1, 2, and 5 pmol. The protein samples were incubated with 0.2 pmol of the DIG‐labeled probe in a total volume of 15 µL. A representative result from three independent gel shift assays (7% native PAGE) is given.
Figure S4. Phylogenetic analyses of phosphotransferase system (PTS). In total, the PTS system is classified into five sub‐groups, one of which is PTS‐V (highlighted in blue). Locus tag of PTS system is showed here for AgaC, SSU05_0451 is indicated in red.