

Abstract
The opportunistic pathogen Pseudomonas aeruginosa is a leading cause of nosocomial infections. Its relatively impermeable outer membrane (OM) limits antibiotic entry, and a chromosomally encoded AmpC β‐lactamase inactivates β‐lactam antibiotics. AmpC expression is linked to peptidoglycan (PG) recycling, and soluble (sLT) or membrane‐bound (mLT) lytic transglycosylases are responsible for generating the anhydromuropeptides that induce AmpC expression. Thus, inhibition of LT activity could reduce AmpC‐mediated β‐lactam resistance in P. aeruginosa. Here, we characterized single and combination LT mutants. Strains lacking SltB1 or MltB had increased β‐lactam minimum inhibitory concentrations (MICs) compared to wild type, while only loss of Slt decreased MICs. An sltB1 mltB double mutant had elevated β‐lactam MICs compared to either the sltB1 or mltB single mutants (96 vs. 32 μg/mL cefotaxime), without changes to AmpC levels. Time–kill assays with β‐lactams suggested that increased MIC correlated with a slower rate of autolysis in the sltB1 mltB mutant – an antisuicide phenotype. Strains lacking multiple mLTs were more sensitive to β‐lactams and up to 16‐fold more sensitive to vancomycin, normally incapable of crossing the OM. Multi‐mLT mutants were also sensitive to bile salts and osmotic stress, and were hyperbiofilm formers, all phenotypes consistent with cell envelope compromise. Complementation with genes encoding inactive forms of the enzymes – or alternatively, overexpression of Braun's lipoprotein – reversed the mutants' cell envelope damage phenotypes, suggesting that mLTs help to stabilize the OM. We conclude that P. aeruginosa mLTs contribute physically to cell envelope stability, and that Slt is the preferred target for future development of LT inhibitors that could synergize with β‐lactams.
Keywords: Antibiotic resistance, Braun's lipoprotein, cell envelope permeability, peptidoglycan‐associated lipoprotein, β‐lactam
Introduction
Pseudomonas aeruginosa is a Gram‐negative opportunistic pathogen and frequent cause of hospital‐acquired infections. It belongs to the ESKAPE group of pathogens (with Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumanii, and Enterobacter spp.) for which treatment options are dwindling (Rice 2008). The discovery and development of novel antibiotics and antibiotic adjuvants is urgently required for treatment of these and other pathogens (Rice 2008; Boucher et al. 2009; Davies and Davies 2010). Among the mechanisms that contribute to antibiotic resistance in P. aeruginosa are the inducible expression of a chromosomally encoded AmpC β‐lactamase and reduced outer membrane (OM) permeability (Lister et al. 2009). The factors influencing OM characteristics (Cascales et al. 2002; Nikaido 2005; Ruiz et al. 2005) and AmpC induction (Jacobs et al. 1994; Kraft et al. 1999; Moya et al. 2009) remain incompletely understood.
In most Gram‐negative bacteria, the cell wall consists of a thin layer of peptidoglycan (PG) composed of alternating N‐acetylmuramic acid (NAM) and N‐ acetylglucosamine (NAG) glycan polymers, cross‐linked by stem peptides on the NAM residues (Johnson et al. 2013). The PG is located in the periplasm between the inner membrane (IM) and OM, and is connected to both via interactions with IM PG biosynthetic complexes and OM lipoproteins such as Braun's lipoprotein (Lpp) (Silhavy et al. 2010). PG maintenance and turnover are dynamic processes that rely on the coordinated efforts of multiple enzymes, including high‐molecular‐weight penicillin‐binding proteins (PBPs) that form the β(1,4)‐glycosidic bond between NAM and NAG, and cross‐link the stem peptides via their transglycosylase and transpeptidase activities, respectively. Low‐molecular‐weight PBPs have endopeptidase and/or carboxypeptidase activities that control the extent of crosslinking, while lytic transglycosylases (LTs) cleave the glycosidic bond between NAM and NAG with the concomitant formation of 1,6‐anhydromuropeptides (anhMPs), allowing for elongation and separation of daughter cells after cell division (Priyadarshini et al. 2006; Scheurwater et al. 2008). Under normal conditions, a proportion of anhMPs are shuttled back to the cytoplasm via the IM permease, AmpG, where they undergo processing by the β‐N‐acetylglucosaminidase, NagZ, and the amidase, AmpD, for recycling into nascent PG (Jacobs et al. 1994).
Changes in PG metabolism can impact antibiotic susceptibility in multiple ways. Inhibition of PBP transpeptidase function by β‐lactams leads to an imbalance between insertion of new material and cleavage of the existing cell wall by lytic enzymes, ultimately leading to loss of cellular integrity (Cho et al. 2014). In some Gram‐negative species, including P. aeruginosa, this autolytic activity leads to increased cytosolic accumulation of anhMPs that bind to the transcriptional activator, AmpR, inducing AmpC expression (Jacobs et al. 1994; Mark et al. 2011). Inactivation of AmpD – or its homologs AmpDh2 or AmpDh3 – increases cytosolic anhMP concentration and induces AmpC expression (Juan et al. 2006), while inactivation of AmpG prevents anhMPs from entering the cytoplasm, thereby preventing AmpC induction (Zhang et al. 2010; Zamorano et al. 2011). Inactivation of NagZ also prevents AmpC induction (Zamorano et al. 2010) by preventing processing of anhMPs to the form required for AmpR activation. Given the essential role of anhMPs in AmpC expression, inactivation of LTs has the potential to reduce the rate of anhMP production, thus preventing induction. However, LT inactivation could also delay autolysis, increasing the length of time that it takes for cells to accumulate sufficient damage to cause death and thus effectively increasing resistance. A clearer picture of the contributions of individual LTs to resistance would allow for more targeted drug development.
In P. aeruginosa, chemical or genetic inactivation of PBP4 – a D,D‐carboxypeptidase – also leads to increased β‐lactam resistance via induction of AmpC expression (Moya et al. 2009; Alvarez‐Ortega et al. 2010), presumably due to an increase in cytoplasmic levels of anhMPs with pentapeptide stems. Combining a PBP4 (dacB) mutation with loss of a specific LT – SltB1 – further increased resistance and AmpC expression via mechanisms that remain unclear, as sltB1 mutants had wild‐type AmpC expression (Cavallari et al. 2013). Whether the loss of other LTs in a PBP4‐deficient background might similarly amplify β‐lactam resistance is unknown.
LTs are potential targets for novel antibiotics or antibiotic adjuvants (Bernal et al. 2013), but lack of systematic analyses of LT function, difficulties associated with studying enzymes that act on an insoluble substrate, and the apparent redundancy of these enzymes in many bacteria has hampered inhibitor development (Lee et al. 2013). LTs were classified into four families based on sequence similarity and the presence of consensus motifs (Blackburn and Clarke 2001; Scheurwater et al. 2008). The physiological roles of individual LTs – or of LTs belonging to specific families – remain largely unknown. Loss of specific Family 1 LTs in P. aeruginosa increased β‐lactam sensitivity, while loss of specific Family 3 LTs decreased β‐lactam sensitivity (Cavallari et al. 2013). Whether membrane‐bound (mLTs) and soluble (sLTs) lytic transglycosylases in general have different roles in antibiotic resistance is not clear.
Here, we examined the contributions of individual LTs, LT families, and mLTs versus sLTs to β‐lactam sensitivity in P. aeruginosa by systematically deleting the genes encoding these enzymes. In most cases, the loss of one or few LTs caused modest changes in minimum inhibitory concentrations (MICs), while the loss of all mLTs caused extreme β‐lactam and vancomycin sensitivity. Slt was the only enzyme whose individual loss decreased β‐lactam MICs (Cavallari et al. 2013; Cho et al. 2014). Increased β‐lactam MICs due to loss of Family 3 LTs – all but one of which are sLTs – were associated with slower rates of death. This study suggests that modest changes in MICs caused by loss of single enzymes contribute to stepwise changes in overall resistance (Juan et al. 2006; Alvarez‐Ortega et al. 2010; Breidenstein et al. 2011; Fernandez et al. 2011) and reveals the potential of Slt as a target for combination therapies with β‐lactam antibiotics.
Materials and Methods
Bacterial strains, plasmids, and mutants
The nine P. aeruginosa LT genes previously identified by bioinformatic methods (Blackburn and Clarke 2001; Legaree and Clarke 2008) were analyzed in this study. A 10th LT, RlpA (rare lipoprotein A) was identified by Jorgenson et al. (2014) and despite having only weak structural similarity to MltA from Escherichia coli and no activity on wild‐type PG, was shown to preferentially degrade PG devoid of peptide stems, contributing to daughter cell separation. Under normal growth conditions, the mutant was reported to have a wild‐type phenotype. Bacterial strains and plasmids used in this study are listed in Table 1. Pseudomonas aeruginosa mutant strains were created using either a Flp‐FRT gene disruption system or unmarked gene deletion (Hoang et al. 1998). Genes were disrupted by FRT insertion following previously described methods (Cavallari et al. 2013). To create unmarked gene deletions, appropriate pEX18Gm constructs were transferred to P. aeruginosa strains of interest by conjugation with donor strain E. coli SM10, and mating mixtures were plated on Pseudomonas isolation agar containing 100 μg/mL gentamicin (Gm) to counter‐select the donor. Gm‐resistant colonies were plated on modified Luria–Bertani agar (LBA), no salt, 5% sucrose to select for double recombinants that lost the sacB‐expressing pEX18Gm suicide vector. Gm‐sensitive mutants with insertion or deletion in the gene of interest were confirmed by PCR using gene‐specific and flanking primers (see Table S1).
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Description | Reference or source | Notes |
|---|---|---|---|
| Strains | |||
| PAO1 | Pseudomonas aeruginosa wild‐type strain | Li et al. (1998), Lamers et al. (2013) | |
| PAO1 mltA | WT with mltA deletion (PA1222) | This study | Family 2 |
| PAO1 mltB | WT with mltB deletion (PA4444) | This study | Family 3 |
| PAO1 mltD | WT with mltD deletion (PA1812) | This study | Family 1D |
| PAO1 mltF | WT with mltF deletion (PA3764) | This study | Family 1E |
| PAO1 mltF2 | WT with mltF2 deletion (PA2865) | This study | Family 1E |
| PAO1 slt | WT with slt deletion (PA3020) | This study | Family 1A |
| PAO1 sltB1 | WT with FRT scar at nucleotide 577 of sltB1 (PA4001) | Cavallari et al. (2013) | Family 3 |
| PAO1 sltG | WT with sltG deletion (PA1171) | This study | Family 3 |
| PAO1 sltH | WT with sltH deletion (PA3992) | This study | Family 3 |
| PAO1 sltB1/slt | sltB1 mutant with slt deletion | This study | |
| PAO1 sltB1/G | sltB1 mutant with sltG deletion | This study | |
| PAO1 sltB1/H | sltB1 mutant with sltH deletion | This study | |
| PAO1 sltB1/mltB | sltB1 mutant with slt deletion | This study | |
| PAO1 sltB1/G/mltB | sltB1/mltB mutant with sltG deletion | This study | |
| PAO1 sltB1/G/H | sltB1/H mutant with sltG deletion | This study | |
| PAO1 sltB1/G/H/slt | sltB1/G/H mutant with slt deletion | This study | Strain lacking all soluble LTs |
| PAO1 sltB1/G/H/mltB | sltB1/G/H mutant with mltB deletion | This study | Strain lacking all Family 3 LTs |
| PAO1 slt/mltF | slt mutant with mltF deletion | This study | |
| PAO1 slt/mltB | slt mutant with mltB deletion | This study | |
| PAO1 mltB/F | mltB mutant with mltF deletion | This study | |
| PAO1 mltD/F | mltD mutant with mltF deletion | This study | |
| PAO1 mltB/D/F | mltD/F mutant with mltB deletion | This study | |
| PAO1 mltB/F/F2 | mltB/F mutant with mltF2 deletion | This study | |
| PAO1 mltD/F/F2 | mltD/F mutant with mltF2 deletion | This study | |
| PAO1 mltA/B/F | mltB/F mutant with mltA deletion | This study | |
| PAO1 mltB/D/F/F2 | mltD/F/F2 deletion with mltB deletion | This study | |
| PAO1 mltD/F/F2/slt | mltD/F/F2 deletion with slt deletion | This study | Strain lacking all Family 1 LTs |
| PAO1 mltA/B/D/F/F2 | mltB/D/F/F2 deletion with mltA deletion | This study | Strain lacking all membrane‐bound LTs |
| PAO1 ampC | WT with FRT scar at nucleotide 1070 of ampC (PA4110) | Cavallari et al. (2013) | |
| PAO1 sltB1/ampC | sltB1 mutant with FRT scar at nucleotide 1070 of ampC | Cavallari et al. (2013) | |
| PAO1 sltH/ampC | sltH mutant with FRT scar at nucleotide 1070 of ampC | This study | |
| PAO1 mltB/ampC | mltB mutant with FRT scar at nucleotide 1070 of ampC | This study | |
| PAO1 mltD/ampC | mltD mutant with FRT scar at nucleotide 1070 of ampC | This study | |
| PAO1 mltF2/ampC | mltF2 mutant with FRT scar at nucleotide 1070 of ampC | This study | |
| PAO1 dacB | WT with FRT scar at nucleotide 168 of dacB (PA3047) | Lamers et al. (2013) | |
| PAO1 dacB/slt | dacB mutant with slt deletion | This study | |
| PAO1 sltB1/dacB | sltB1 mutant with FRT scar at nucleotide 168 of dacB | Cavallari et al. (2013) | |
| PAO1 sltB1/G/H/mltB/dacB | sltB1/G/H/mltB mutant with FRT scar at nucleotide 168 of dacB | This study | |
| PAO1 sltG/H/mltB/dacB | sltB1/G/H/mltB/dacB mutant with replacement of sltB1::FRT with wild‐type sltB1 | This study | |
| PAO1 sltB1/G/H/slt/dacB | sltB1/G/H/slt mutant with FRT scar at nucleotide 168 of dacB | This study | |
| PAO1 sltG/H/slt/dacB | sltB1/G/H/slt/dacB mutant with replacement of sltB1::FRT with wild‐type sltB1 | This study | |
| Plasmids | |||
| pPS856 | Source of FRT‐flanked gentamicin cassette; Gmr | Hoang et al. (1998) | |
| pEX18Ap | Suicide vector used for gene disruption; Apr | Hoang et al. (1998) | |
| pEX18Gm | Suicide vector used for gene deletion; Gmr | Hoang et al. (1998) | |
| pFLP2 | Suicide vector encoding Flp recombinase; Apr | Hoang et al. (1998) | |
| pBADGr | pMLBAD backbone with dhfr replaced with aacC1; arabinose inducible; Gmr | Asikyan et al. (2008), Cavallari et al. 2013) | |
| pEX18Ap‐sltB1::FRTGmFRT | Suicide vector containing sltB1 disrupted at nucleotide position 577 with FRT‐flanked gentamicin resistance cassette; Apr Gmr | Cavallari et al. (2013) | |
| pEX18Ap‐ampC::FRTGmFRT | Suicide vector containing ampC disrupted at nucleotide position 1070 with FRT‐flanked gentamicin resistance cassette; Apr Gmr | Cavallari et al. (2013) | |
| pEX18Ap‐dacB::FRTGmFRT | Suicide vector containing dacB disrupted at nucleotide position 168 with FRT‐flanked gentamicin resistance cassette; Apr Gmr | Lamers et al. (2013) | |
| pEX18Gm‐mltA | Suicide vector containing 500 nucleotides flanking each side of mltA for recombination; Gmr | This study | |
| pEX18Gm‐mltB | Suicide vector containing 500 nucleotides flanking each side of mltB for recombination; Gmr | This study | |
| pEX18Gm‐mltD | Suicide vector containing 500 nucleotides flanking each side of mltD for recombination; Gmr | This study | |
| pEX18Gm‐mltF | Suicide vector containing 500 nucleotides flanking each side of mltF for recombination; Gmr | This study | |
| pEX18Gm‐mltF2 | Suicide vector containing 500 nucleotides flanking each side of mltF2 for recombination; Gmr | This study | |
| pEX18Gm‐sltB1 WT | Suicide vector containing wild‐type sltB1; Gmr | This study | |
| pEX18Gm‐slt | Suicide vector containing 500 nucleotides flanking each side of slt for recombination; Gmr | This study | |
| pEX18Gm‐sltG | Suicide vector containing 500 nucleotides flanking each side of sltG for recombination; Gmr | This study | |
| pEX18Gm‐sltH | Suicide vector containing 500 nucleotides flanking each side of sltH for recombination; Gmr | This study | |
| pBADGr‐OprI | pBADGr derivative containing OprI (Braun's lipoprotein, Lpp; PA2853) on an EcoRI to HindIII fragment; Gmr | This study | |
| pBADGr‐OprL | pBADGr derivative containing OprL (peptidoglycan‐associated lipoprotein, Pal; PA0973) on an EcoRI to HindIII fragment; Gmr | This study | |
Antibiotic sensitivity assays
Antibiotic sensitivity assays were performed using Etest strips (BioMérieux Canada, Inc., St. Laurent, Quebec, Canada) as previously described (Cavallari et al. 2013), and broth microdilution. For Etest assays, overnight bacterial cultures were subcultured 1:50 in Mueller–Hinton Broth (MHB; Becton, Dickinson and Company, Mississauga, Ontario, Canada) and grown to logarithmic phase at 37°C, with shaking at 200 rpm. Cultures were standardized to an optical density at 600 nm (OD600 nm) of 0.25 in MHB, and 100 μL was evenly spread on Mueller–Hinton agar (MHA) and allowed to dry. Etest strips were overlaid and plates were incubated for 18 h at 37°C. As per the manufacturer's recommendations, MICs were determined to be the concentration at which the zone of inhibition intersected the Etest strip. Etest assays were performed three times independently and the modal MIC value was listed in Table 2. Variations in MICs between replicates were never more than 1.5‐fold; and thus, only mutants with MIC differences twofold or greater than wild type were tested further for reproducibility using the broth microdilution method.
Table 2.
MICs for LT mutants of β‐lactam, fluoroquinolone, and glycopeptide antibiotics
| Strain | Minimum inhibitory concentration (μg/mL)a , b | ||||||
|---|---|---|---|---|---|---|---|
| PP | CT | TZ | IP | CI | VA | PMB | |
| PAO1 | 6 (4) | 12 (8) | 1 (2) | 1 | 0.19 | >256 (2048) | (1) |
| ampC | 2 (1) | 4 (4) | – (0.5) | 0.5 | nd | nd | nd |
| dacB | 64 (32) | >256 (128) | 16 (16) | – | nd | nd | nd |
| dacB/slt | 32 (8) | 128 (64) | 8 (8) | – | nd | nd | nd |
| slt | 3 (1) | 6 (4) | 0.5 (1) | – | – | – | nd |
| slt (E503A) | 4 (1) | 6 (4) | 0.5 (1) | – | nd | nd | nd |
| sltB1 | 16 (8) | 24 (16) | – (–) | – | – | – | nd |
| sltB1/ampC | 4 (2) | 4 (4) | – (1) | 0.5 | nd | nd | nd |
| sltH | 12 (8) | 32 (16) | – (–) | 1.5 | – | – | nd |
| sltH/ampC | 4 (2) | 16 (4) | – (1) | 0.25 | nd | nd | nd |
| sltB1/mltB | 32 (16) | 96 (32) | 1.5 (–) | 0.75 | – | – | nd |
| mltB | 12 (–) | 32 (16) | 1.5 (–) | 1.5 | – | – | nd |
| mltB (E162A) | 8 (–) | 16 (–) | – (–) | – | nd | nd | nd |
| mltB/ampC | 8 (2) | 16 (4) | – (–) | 0.5 | nd | nd | nd |
| mltD | 12 (–) | 24 (16) | – (–) | – | – | – (–) | (–) |
| mltD (E144A) | – (–) | 16 (–) | – (–) | – | nd | nd | nd |
| mltD/ampC | – (2) | 16 (4) | – (–) | 0.5 | nd | nd | nd |
| mltF2 | 12 (–) | 32 (16) | – (–) | – | – | – | nd |
| mltF2/ampC | – (–) | 8 (4) | – | 0.5 | nd | nd | nd |
| mltB/D/F | 4 (2) | 6 (4) | 0.75 (1) | – | – | – | nd |
| mltB/F/F2 | 4 (2) | 6 (4) | 0.75 (1) | – | – | – | nd |
| mltD/F/F2 | 3 (1) | 6 (4) | – (1) | 0.75 | – | – (512) | (–) |
| mltB/D/F/F2 | 3 (1) | 4 (4) | – (1) | – | – | – (256) | (0.5) |
| mltD/F/F2/slt | 1.5 (0.5) | 2 (2) | 0.5 (0.5) | 0.5 | 0.125 | 128 (256) | (0.5) |
| mltA/B/D/F/F2 | 2 (0.5) | 4 (4) | 0.75 (0.5) | 0.5 | 0.125 | 128 (128) | (0.5) |
PP, piperacillin; CT, cefotaxime; TZ, ceftazidime; IP, imipenem; CI, ciprofloxacin; VA, vancomycin; PMB, Polymyxin B; nd, not done;–, MIC is same as wild type.
MICs in blue are ≥twofold higher than wild type, while those in red are ≥twofold lower than wild type, , as confirmed by Etest and broth microdilution methods.
MIC values in parentheses were determined using broth microdilution, while all other MIC values were determined by Etest.
Strains with Etest MIC differences twofold or greater than control – and their respective control strains – were tested further using broth microdilution following Clinical and Laboratory Standards Institute (CLSI) guidelines. For these assays, antibiotics were serially diluted twofold and 1 μL of the desired concentration was added to the appropriate wells of a 96‐well microtitre plate. Overnight bacterial cultures were subcultured 1:50 in MHB and grown to logarithmic phase at 37°C, with shaking at 200 rpm. Cultures were standardized to ~4.0 × 105 colony forming units per mL (CFU/mL) in MHB, and a 99 μL inoculum was combined with the desired antibiotic in the appropriate wells of a 96‐well microtitre plate. Plates were covered with Mylar seals (Thermo Scientific, Mississauga, Ontario, Canada) and incubated at 37°C. MICs were determined as the concentration at which no visible growth was observed after 18 h incubation. Broth microdilution MIC assays were performed three times independently with three technical replicates each. Only reproducible MIC changes (i.e., confirmed by Etest and broth microdilution methods both) were listed as different than wild type in Table 2. We note that MIC differences of twofold are generally not regarded as clinically significant; however, modest changes in MIC may contribute to stepwise changes to resistance (Juan et al. 2006; Alvarez‐Ortega et al. 2010; Kumari et al. 2014), and therefore, we listed all reproducible MIC changes of twofold or greater in Table 2.
AmpC β‐lactamase western blot analyses
Overnight bacterial cultures were subcultured 1:20 in 5 mL LB and grown to an OD600 nm = 0.6 at 37°C and 200 rpm. For assays using chemical induction of AmpC, cultures were split 1:1 in 5 mL LB with or without 50 μg/mL cefoxitin (inducer) and incubated for an additional 2 h at 37°C and 200 rpm. Cell cultures were then standardized to an OD600 nm = 0.6 and 1 mL was centrifuged at 16,100g for 1 min. For assays using genetic induction of AmpC (i.e., via disruption of dacB), logarithmic‐phase cell cultures were standardized to an OD600 nm = 0.6 and 1 mL was centrifuged at 16,100g for 1 min. Cell pellets were resuspended in 100 mL of sodium dodecyl sulfate (SDS) sample buffer (0.3 mol/L Tris‐HCl, pH 6.8, SDS [6.7% w/v], glycerol [10% v/v], 2‐mercaptoethanol [5.3% v/v] and bromophenol blue [0.2% w/v]), boiled for 10 min and stored at −20°C until immunoblotting.
For polyclonal antibody production, P. aeruginosa ampC (PA4110) lacking the first 78 nucleotides (amino acids 1–26) was cloned into pET151/D‐TOPO vector (Life Technologies, Mississauga, Ontario, Canada) and expressed following the manufacturer's instructions in E. coli BL21 cells at 37°C without induction. Cells were lysed by sonication and AmpCΔ26 was purified by nickel affinity chromatography, followed by Tobacco Etch Virus (TEV) protease cleavage and a second nickel affinity purification step to isolate untagged AmpCΔ26. Purified AmpCΔ26 was dialyzed (using Slide‐A‐Lyzer Dialysis Cassette, 10 kDa molecular weight cutoff; Thermo Scientific, Mississauga, Ontario, Canada) with phosphate‐buffered saline (PBS; pH 7.0), the protein concentration was adjusted to 1 mg/mL, and sample was submitted to Cedarlane Laboratories (Burlington, Ontario, Canada) for rabbit immunization.
Immunoblotting was performed by first separating samples on 12.5% SDS‐PAGE (polyacrylamide gel electrophoresis) gels at 150 V for 90 min followed by transfer to nitrocellulose membranes at 225 mA for 60 min. Membranes were blocked using 5% skim milk in PBS at 37°C for 60 min, followed by washing with PBS and incubation with α‐AmpC primary antibody (1/2000 dilution in PBS) overnight at 4°C. Membranes were washed with PBS and incubated with α‐rabbit secondary antibody conjugated to alkaline phosphatase (1/3000 dilution in PBS) for 1 h at 37°C followed by washing again with PBS. Membranes were developed in a solution containing 100 μL nitro‐blue tetrazolium and 100 μL 5‐bromo‐4‐chloro‐3‐indolyl phosphate (BCIP) in 10 mL of alkaline phosphatase buffer (100 mmol/L NaCl, 5 mmol/L MgCl2, 100 mmol/L Tris pH 9.5).
Cell envelope integrity assays
Cell envelope integrity was tested by measuring bacterial survival on bile salts containing or high‐osmolarity media (Wang 2002; Korsak et al. 2005). Overnight bacterial cultures were subcultured 1:50 in LB and grown at 37°C and 200 rpm to an OD600 nm = 0.6. For bile salt assays, cultures were serially diluted and 50 μL spread on LBA, with or without 0.15% No. 3 bile salts (Sigma‐Aldrich, Oakville, ON, Canada). Plates were incubated overnight at 37°C. Percent survival was determined by enumerating CFUs from samples treated with bile salts compared to matched, untreated samples. For high‐osmolarity assays, logarithmic‐phase cultures were standardized to OD600 nm = 0.1 in LB containing either 85.5 mmol/L or 2.5 mol/L NaCl and incubated for 2 h at room temperature. Cultures were then serially diluted followed by spreading 50 μL on LBA and incubated overnight at 37°C. The percent survival of strains to high‐osmolarity conditions was determined by enumerating CFUs from samples treated with 2.5 mol/L NaCl compared to matched samples treated with 85.5 mmol/L NaCl. Cell envelope integrity assays were performed three times independently and statistical significance was assessed using a two‐tailed Student's t‐test. A P ≤ 0.05 was considered statistically significant.
Biofilm assays
Biofilm assays were performed as previously described (Wenderska et al. 2011). Briefly, overnight cultures grown in PBS (137 mmol/L NaCl, 2.7 mmol/L KCl, 10.1 mmol/L Na2HPO4, 1.8 mmol/L KH2PO4, pH 7.4) containing 0.2% (w/v) LB broth mix were diluted 1:25 in the same medium and grown to an OD600 nm = 0.1. Subcultures were diluted 1:500 in PBS/LB medium and 150 μL was added to triplicate wells of a flat‐bottom 96‐well polystyrene plate. Plates were covered with 96‐well pin lids (Nalge Nunc International, Rochester, NY) and sealed with parafilm. Following 19 h incubation at 37°C and 200 rpm, pin lids were submerged in PBS without shaking for 10 min to remove loosely adhering bacteria, then stained for 15 min with 200 μL 0.1% (w/v) crystal violet (CV) in 96‐well plates. Following CV staining, pin lids were washed 5 × 15 min in sterile water. CV was solubilized in 200 μL of 33.3% acetic acid per well for 5 min in 96‐well plates and quantified by measuring absorbance at 600 nm. To rule out growth rate‐related effects on biofilm production, planktonic growth was also measured at the end of the 19 h assay at OD600 nm and all strains reached the same terminal OD. Assays were performed three times independently and statistical significance was calculated using a two‐tailed Student's t‐test. A P ≤ 0.05 was considered statistically significant.
Time–kill assays
Overnight bacterial cultures in MHB were subcultured 1:50 in the same medium and grown to logarithmic phase at 37°C, with shaking at 200 rpm. Cultures were standardized to an OD600 nm = 0.1 and diluted 1:200 in MHB. Aliquots of 1 mL were treated with and without piperacillin (12 μg/mL final concentration) and incubated at 37°C for up to 24 h. Cultures were then serially diluted in MHB followed by spreading 50 μL on MHA and incubated overnight at 37°C. The survival of strains treated with piperacillin was determined by enumerating CFUs from antibiotic‐treated samples compared to matched, untreated samples. Data were plotted as a log10 reduction in CFUs. Time–kill assays were performed three times independently and statistical significance was assessed using a two‐tailed Student's t‐test. A P ≤ 0.05 was considered statistically significant.
Results
Loss of specific LTs decreases β‐lactam sensitivity
To understand the potential of LTs to modulate β‐lactam sensitivity in P. aeruginosa, single or multiple LT genes were deleted. An antibiotic sensitivity screen of single mutants revealed six with altered sensitivities to piperacillin, cefotaxime, and ceftazidime (Table 2). Loss of sltB1 increased piperacillin and cefotaxime MICs ~twofold in Etest and broth microdilution assays. Loss of mltB increased piperacillin MICs by twofold using Etest; however, there was no increase in MIC using broth microdilution. Loss of mltB increased cefotaxime MICs by ≥twofold as confirmed by both methods. Loss of sltH resulted in a ≥twofold increase in piperacillin and cefotaxime MICs by Etest and broth microdilution. Mutants lacking mltD and mltF2 had piperacillin MICs twofold higher than wild type as determined by Etest; however, the broth microdilution method showed no increase. Loss of mltD and mltF2 also increased cefotaxime MICs by ≥twofold by both methods. In contrast, loss of slt resulted in a two to fourfold decrease in MICs of piperacillin, cefotaxime, and ceftazidime using Etest and broth microdilution methods, consistent with a previous report (Cavallari et al. 2013). Loss of sltG, mltA, or mltF had no effect on β‐lactam sensitivity (refer to Table S2 for a summary of MICs). Single mutants had wild‐type sensitivities to non‐β‐lactam antibiotics ciprofloxacin and vancomycin (Table 2), suggesting that MIC changes were not due to differences in efflux or membrane permeability. There were no differences between single LT mutants and the wild type in morphology (not shown) or growth rates over 48 h (Fig. S1), ruling out growth rate‐related changes in sensitivity.
To relate the mutants' β‐lactam MICs to AmpC expression, AmpC levels in uninduced and cefoxitin‐induced cultures were examined. No differences in AmpC expression or induction were detected for single LT mutants (Fig. S2). The MICs of double mutants lacking LTs and ampC were also determined (Table 2). While the loss of AmpC in LT mutant backgrounds reduced piperacillin and cefotaxime MICs, resistance levels remained above those of the ampC control. Thus, the data suggest that additional mechanism(s) – beyond AmpC expression – contributed to the increased MICs.
We showed previously by complementation with active‐site point mutants that loss of SltB1 activity – rather than potential disruption of protein complexes containing SltB1 – increased β‐lactam MICs (Cavallari et al. 2013). To determine whether this was true for other LT mutants with altered MICs, the mltB, mltD, and slt genes were replaced at their native loci with versions encoding putative active‐site mutants. The catalytic Glu162 of MltB (van Asselt et al. 1999; Blackburn and Clarke 2001), Glu144 of MltD (Blackburn and Clarke 2001), and Glu503 of Slt (Thunnissen et al. 1994; Blackburn and Clarke 2001) were each replaced with Ala. The mltB and mltD point mutants had wild‐type MICs (Table 2), suggesting a structural rather than enzymatic contribution of their protein products to β‐lactam sensitivity. In contrast, the Slt active‐site mutant failed to restore wild‐type β‐lactam MICs, suggesting that loss of Slt activity underlies enhanced sensitivity of the deletion mutant.
The phenotypes of LT combination mutants correlate with loss of specific enzymes
Similar to the way in which β‐lactams can target multiple PBPs (Kocaoglu and Carlson 2015; Kocaoglu et al. 2015), a small molecule inhibitor of LTs could simultaneously inactivate multiple enzymes due to the conservation of the LT active site (Scheurwater et al. 2008). To examine the effect of coinactivating multiple LTs on β‐lactam sensitivity, combination mutants lacking up to five LTs were generated (Table 1). The strategy for generation of combination mutants was based on a previous report in which LTs were divided into four families (Blackburn and Clarke 2001). We made combination mutants lacking all mLTs (i.e., mltA, mltB, mltD, mltF, and mltF2), all Family 1 LTs (i.e., mltD, mltF, mltF2, and slt), all sLTs (i.e., sltB1, sltG, sltH, and slt), or all Family 3 LTs (i.e., sltB1, sltG, sltH, and mltB) (Table 1). With the exception of Slt, all Family 1 LTs are membrane‐bound enzymes, and with the exception of MltB, all Family 3 LTs are soluble enzymes. Two different phenotypes resulted, depending on the nature of the combinations. The sltB1 mltB double mutant had the highest β‐lactam MICs among combination mutants, with a ≥fourfold increase in piperacillin and cefotaxime MIC compared to wild type (Table 2). We confirmed by anti‐AmpC western blot (data not shown) that this increase in MIC was not due to increased AmpC levels in the sltB1 mltB mutant (Cavallari et al. 2013). Other combination mutants lacking sLTs or Family 3 LTs had increased β‐lactam MICs compared to wild type; however, none were as high as the sltB1 mltB double mutant. Changes to β‐lactam sensitivities were independent of changes in AmpC levels, which remained similar to wild type (Fig. 1) (Cavallari et al. 2013). No differences in ciprofloxacin or vancomycin MICs for any of the sLT or Family 3 combination mutants were observed. To test whether loss of Slt in the β‐lactam‐resistant dacB background could restore β‐lactam sensitivity, a dacB slt double mutant was generated. β‐lactam MICs were reduced at least twofold in the dacB slt double mutant compared to its dacB parent (Table 2).
Figure 1.

Only loss of sltB1 further increases AmpC β‐lactamase expression in the dacB background. Loss of dacB in strains lacking all sLTs or Family 3 LTs increased AmpC levels, but not to the same level as the dacB sltB1 double mutant. AmpC expression was reverted to that of the dacB single mutant when sltB1 is present. Shown is a representative AmpC β‐lactamase immunoblot. Values represent average AmpC levels (N = 3), relative to the dacB mutant. sLTs, soluble lytic transglycosylases.
The loss of all mLTs (i.e., mltA/B/D/F/F2) or Family 1 LTs (i.e., mltD/F/F2/slt) reduced β‐lactam MICs below wild‐type levels (Table 2). The mltB/D/F and mltB/F/F2 triple mutants had cefotaxime MICs twofold below wild type. These strains lack mltB – which alone, increased cefotaxime MICs. The mltD/F/F2 triple mutant and the mltB/D/F/F2 quadruple mutant had twofold reductions in piperacillin and cefotaxime MICs compared to wild type, as confirmed by Etest and broth microdilution methods. The Family 1 LT mutant lacking mltD/F/F2/slt was highly sensitive to β‐lactams, with ≥fourfold decreases in piperacillin and cefotaxime MICs, respectively. This mutant was also ≥twofold more sensitive to ceftazidime and imipenem. The mltA/B/D/F/F2 mutant lacking all mLTs was ≥three and ≥twofold more sensitive to piperacillin and cefotaxime, respectively, as confirmed by Etest and broth microdilution methods. Surprisingly, the mltD/F/F2, mltB/D/F/F2, mltD/F/F2/slt and mltA/B/D/F/F2 mutants had vancomycin MICs that were 4‐, 8‐, 8‐, and 16‐fold lower than wild type, respectively, as determined by broth microdilution (Table 2). Vancomycin is normally incapable of crossing the OM, suggesting that increased drug sensitivities of combination mLT mutants were due in part to cell envelope perturbations.
Only the loss of SltB1 – but not other sLTs or Family 3 LTs – increases AmpC expression in the dacB background
PBP4 (dacB) mutants of P. aeruginosa express AmpC at high levels, even in the absence of antibiotics (Moya et al. 2009; Cavallari et al. 2013). We showed previously that the combined loss of sltB1 and dacB further increases AmpC expression, even though an sltB1 mutant had wild‐type AmpC expression (Cavallari et al. 2013). Thus, we asked whether AmpC expression in the dacB background was altered by the loss of other sLTs or Family 3 LTs. A strain lacking all Family 3 LTs plus dacB (i.e., the sltB1/G/H/mltB/dacB mutant) had higher levels of AmpC than the dacB mutant alone (P < 0.01), but lower than the sltB1 dacB double mutant (P < 0.01; Fig. 1, compare lanes 5 and 7). To test whether the elevated AmpC levels in the sltB1/G/H/mltB/dacB mutant were due to the loss of SltB1, the disrupted sltB1 gene was replaced with the wild‐type sltB1 allele, yielding a sltG/H/mltB/dacB mutant. This mutant's AmpC expression was similar to that of the dacB single mutant (P = 0.97) (Fig. 1, compare lanes 7 and 8). Similarly, a strain lacking all sLTs (sltB1/G/H/slt) and dacB expressed more AmpC than the dacB mutant (P = 0.02), but less than the sltB1 dacB double mutant (P < 0.01; Fig. 1, compare lanes 5 and 10). Knock‐in of the wild‐type sltB1 allele in this mutant – yielding the sltG/H/slt/dacB mutant – reversed AmpC levels to those of a dacB single mutant (P = 0.2) (Fig. 1, compare lanes 10 and 11). These data suggest that only loss of SltB1 increased AmpC expression in the dacB background. Furthermore, the loss of additional LTs in a strain lacking both PBP4 and SltB1 reduces its very high AmpC levels; however, the cause(s) of the attenuated AmpC levels in these strains is unknown.
Previous studies suggest that the loss of lytic enzymes slows β‐lactam‐induced cell death due to decreased cell lysis upon the inhibition of synthetic enzymes (Tomasz et al. 1970; Tomasz 1979; Heidrich et al. 2002; Uehara et al. 2009). This phenomenon has been referred to as penicillin tolerance (Tomasz et al. 1970; Tomasz 1979). To test whether slower rates of death could explain why mutants lacking Family 3 LTs had higher β‐lactam MICs despite wild‐type AmpC levels (Fig. 1, Fig. S2; Cavallari et al. 2013), piperacillin time–kill assays were performed (Fig. 2). Within 15 min, there was a ~0.4 log reduction in wild‐type CFU versus ~0.07 log (P = 0.02) for sltB1, mltB, and sltB1 mltB mutants (Fig. 2A). The reduced rate of killing by piperacillin in the LT mutants continued throughout the first 3 h, with a ~4.5‐log reduction in wild‐type CFU compared to ~3.5‐log and ~3‐log reductions for sltB1 (P < 0.01) and sltB1 mltB (P < 0.001) mutants, respectively. After 6 h, cell death leveled off in all strains (Fig. 2B). Marginal regrowth was observed in wild‐type and single mutants between 6 and 24 h, whereas the sltB1 mltB mutant regrew to nearly the same concentration as the untreated cells. The wild‐type and sltB1 mltB strains had indistinguishable growth curves under the assay conditions, ruling out growth rate‐related differences in MIC (Fig. 2C). These data suggest that the elevated β‐lactam MICs for some LT mutants is due in part to a reduced rate of autolysis.
Figure 2.
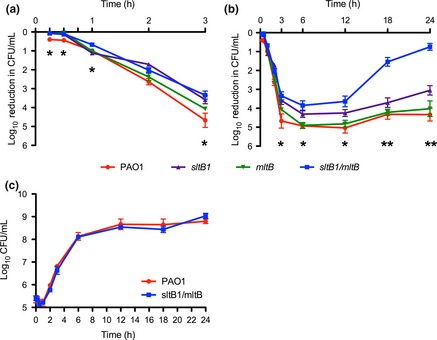
Strains lacking SltB1 and MltB die more slowly than wild‐type cells when treated with piperacillin. LT mutant cells were treated with piperacillin and their viability over 24 h was compared to untreated cells. Loss of sltB1/mltB slows piperacillin‐induced death over the first 3 h (A) with the remaining cells regrowing over 24 h (B). The growth rates between PAO1 and sltB1/mltB are the same (C), ruling out growth‐related differences in MIC. Asterisks indicate statistical differences between sltB1/mltB and PAO1. N = 3. Bars represent the mean ± SEM. *P < 0.05; **P < 0.01. LT, lytic transglycosylase; MIC, minimum inhibitory concentration.
Combination mLT mutants have compromised cell envelopes
Gram‐negative bacteria with compromised OMs are sensitive to detergent‐like compounds, including bile salts (Hancock 1984; Lamers et al. 2013). Due to their marked increase in vancomycin susceptibility, we further tested mutants lacking Family 1 and mLT enzymes for increased OM permeability using bile salt sensitivity assays (Fig. 3). Of the single LT mutants, only mltA (P = 0.03) and mltD (P = 0.02) were more sensitive than wild type (Fig. 3A). Chromosomal replacement of mltA and mltD with versions encoding active‐site mutants MltA D305A (van Straaten et al. 2005; Powell et al. 2006) and MltD E144A (Blackburn and Clarke 2001) restored bile salt resistance to near‐wild‐type levels (Fig. 3A). Mutants lacking Family 3 LTs or all sLTs were unaffected by bile salts, with the exception of the sltB1/G/H/mltB quadruple mutant (~55% of wild‐type viability; P = 0.018) (Fig. 3B). Conversely, mutants lacking all Family 1 LTs or all mLTs were more sensitive than wild type. Those mutants most sensitive to β‐lactams – mltD/F/F2, mltD/F/F2/slt, mltB/D/F/F2, and mltA/B/D/F/F2 – had ~15–30% survival rates on bile salts compared to matched, untreated cells (Fig. 3A). Triple mutants mltB/D/F and mltB/F/F2 had survival rates of ~50% (P = 0.01 and 0.02, respectively).
Figure 3.
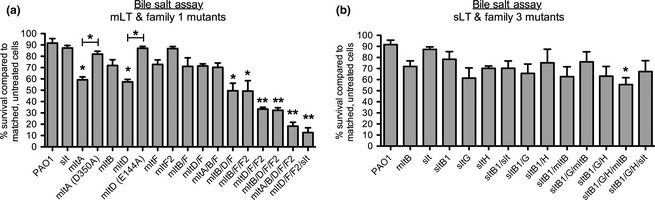
Combination mLT and Family 1 LT mutants have increased OM permeability. Viability of LT mutants grown in the presence/absence of bile salts was compared. (A) mLT and Family 1 mutant strains were sensitive to bile salts while (B) sLT and Family 3 mutants were not. N = 3. Bars represent the mean ± SEM. *P < 0.05; **P < 0.01. mLT, membrane‐bound lytic transglycosylase; OM, outer membrane; sLT, soluble lytic transglycosylase.
Loss of lipopolysaccharide (LPS) integrity can increase OM permeability (Hancock 1984; Nikaido 2003; Sutterlin et al. 2014); however, supplementation with 1 mmol/L MgCl2 (Plesiat et al. 1997; Nikaido 2003; Lamers et al. 2013) had no effect on the sensitivity of mltD/F/F2/slt or mltA/B/D/F/F2 mutants to bile salts (Fig. S3), suggesting that increased OM permeability was independent of LPS stability. To further establish whether OM permeability was related to LPS instability, mLT mutants were assessed for sensitivity to the antimicrobial peptide polymyxin B (PMB). The potency of PMB is directly affected by LPS integrity and requires the negative charge to access the periplasm (Delcour 2009). PMB sensitivity was generally unaffected by the loss of mLTs except for a twofold reduction in MIC for mltB/D/F/F2, mltD/F/F2/slt, or mltA/B/D/F/F2 mutants (0.5 μg/mL) compared to wild type (1 μg/mL) (Table 2) – further suggesting that the increase in OM permeability is largely independent of LPS stability.
The integrity of the PG layer in cells lacking hydrolases can be investigated by osmotic stress challenge, by growth on media containing 2.5 mol/L NaCl (Heidrich et al. 2002; Korsak et al. 2005). Single mLT mutants lacking mltA, mltB, mltD, mltF, or mltF2 (Fig. 4A) and single sLT mutants lacking slt, sltB1, sltG, or sltH (Fig. 4B) had wild‐type survival on high‐salt medium. While combination sLT and Family 3 LT mutants had wild‐type levels of survival on high salt (Fig. 4B), combination mLT and Family 1 LT mutants lacking two or more enzymes had statistically significant reductions in viability (Fig. 4A). The sensitivity of combination mLT mutants – but not combination sLT mutants – to osmotic stress supports a role for mLTs in cell wall integrity.
Figure 4.
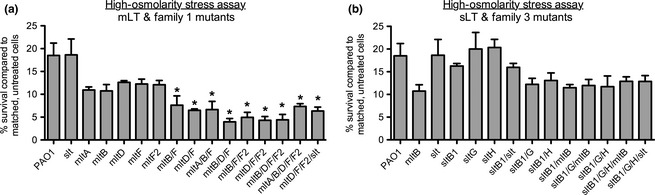
Combination mLT and Family 1 LT mutants are sensitive to osmotic stress. Viability of LT mutants was compared between cells treated with 2.5 mol/L NaCl and untreated, control cells. (A) mLT and Family 1 mutant strains were more sensitive to osmotic stress than wild type while (B) sLT and Family 3 mutants were not. N = 3. Bars represent the mean ± SEM. *P < 0.05. mLT, membrane‐bound lytic transglycosylase; sLT, soluble lytic transglycosylase.
Complementation of mLT mutants with Lpp reduces sensitivity to bile salts
Based on the phenotypes of multi‐mLT mutants, we hypothesized that these lipoproteins might help to tether the OM to PG, similar to Lpp (called OprI or PA2853 in P. aeruginosa) (Mizuno and Kageyama 1979; Cornelis et al. 1989; Cascales et al. 2002; Ni and Chen 2004, 2005; Wessel et al. 2013) and PG‐associated lipoprotein, Pal (called OprL or PA0973) (Mizuno 1979; Mizuno and Kageyama 1979; Hancock et al. 1981; Cascales et al. 2002). To test whether increased OM–PG interactions might improve the mLT mutants' cell envelope integrity, OprI or OprL was expressed in trans in strains sensitive to bile salts (Fig. 5). Low‐level expression of OprI from the vector's leaky promoter in the absence of induction (Giltner et al. 2010) increased survival on bile salts from ~34% to ~73% for the mltD/F/F2 mutant and for strains lacking all mLTs (mltA/B/D/F/F2), or all Family 1 LTs (mltD/F/F2/slt), from ~14% to ~32% (Fig. 5, left panel). Using 0.1% l‐arabinose to increase OprI expression did not further improve bile salt survival (Fig. 5, right panel). Complementation with OprL, with or without l‐arabinose induction, had no significant effect on bile salt sensitivity of any mutants tested.
Figure 5.
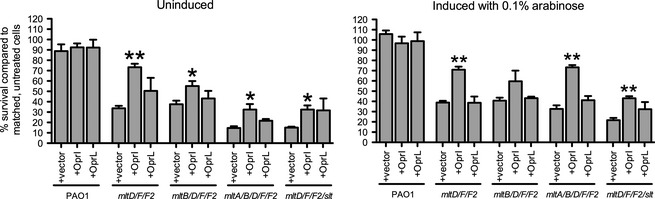
Overexpression of Braun's lipoprotein in trans reduces sensitivity to bile salts in membrane‐bound lytic transglycosylase (mLT) mutants. mLT mutants sensitive to bile salts were complemented with OprI (Braun's lipoprotein; Lpp) or OprL (peptidoglycan‐associated lipoprotein; Pal) and their viability in the presence of bile salts was assessed. Left panel, uninduced; right panel, induced with 0.1% arabinose. N = 3. Bars represent the mean ± SEM. *P < 0.05; **P < 0.01.
Loss of mLTs increases biofilm formation
LT activity, cell wall turnover, and cell envelope damage have previously been linked to increased biofilm formation in both Gram‐negative (Monteiro et al. 2011) and Gram‐positive bacteria (Sailer et al. 2003; Kolar et al. 2011). Moreover, exposure to sub‐MIC β‐lactams can induce biofilm formation in an autolysin‐dependent manner (Kaplan et al. 2012). Cells in a biofilm are protected from exposure to many antibiotics by the surrounding extracellular polymeric substance matrix and are heterogeneous with respect to growth rates, with reduced sensitivity in slow‐growing or persister subpopulations (Kostakioti et al. 2013). Factors affecting cell wall metabolism can modulate bacterial surface properties (Reid et al. 2004; Liu et al. 2012) and biofilm formation (Monteiro et al. 2011; Payne et al. 2013; Brambilla et al. 2014), altering adhesion to surfaces or the ability to form the protective matrix (Monteiro et al. 2011; Liu et al. 2014). Therefore, we asked whether the ability of LT mutants to form biofilms was altered. Only the mltD mutant made less biofilm than wild type (~70% of wild type; P = 0.03), while four mutants – the mltD/F/F2 mutant and its derivatives mltD/F/F2/slt, mltB/D/F/F2, and mltA/B/D/F/F2, all of which have compromised cell envelopes based on the data presented above – showed significantly enhanced biofilm formation (Fig. 6). The parent strain of mltD/F/F2, mltD/F, made wild‐type levels of biofilm. The mltA/B/F and mltB/D/F triple mutants had elevated biofilm production compared with their mltB/F parent strain, which resembled wild type (Fig. 6).
Figure 6.
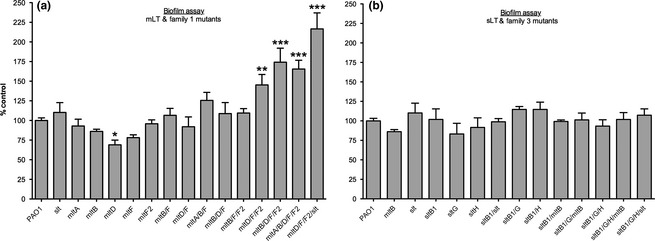
Combination mutants lacking mLTs or Family 1 LTs are hyperbiofilm formers. Biofilms of LT mutants grown for 24 h were compared to those formed by wild type. (A) Loss of multiple mLTs increased biofilm formation while (B) the loss of sLTs did not. N = 3. Bars represent the mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001. mLT, membrane‐bound lytic transglycosylase; sLT, soluble lytic transglycosylase.
Discussion
Resistance to antibiotics has increased dramatically, and the need to identify novel targets is urgent (World Health Organization, 2014). PG is essential for bacterial survival and absent in eukaryotes, making it a favored target for antibacterial agents. Previous profiling of the β‐lactam sensitivities of P. aeruginosa mutants lacking PG‐active enzymes showed that loss of specific LTs – which have been proposed as potential drug targets (Blackburn and Clarke 2001) – had varying effects on resistance (Cavallari et al. 2013). Our work, and that of others (Kraft et al. 1999; Korsak et al. 2005), have questioned the degree of redundancy among LTs, and prompted this more comprehensive study.
We identified five LT deletion mutants with increased β‐lactam MICs (i.e., those lacking SltB1, SltH, MltB, MltD, and MltF2), and one with decreased MICs (i.e., that lacking Slt; summarized in Table S3). Consistent with a previous study of transposon mutants (Cavallari et al. 2013), loss of SltB1 or MltB increased MICs, while loss of Slt decreased MICs. In contrast, loss of MltF had no effect on sensitivity in the current study. These differences may relate to the use of transposon mutants for the previous study versus clean gene deletions for this work. We have now identified three additional LTs – SltH, MltD, and MltF2 – whose absence led to elevated β‐lactam MICs. Loss of AmpC in these strains reduced β‐lactam MICs, but they remained higher than the ampC control. These data suggest that AmpC‐independent mechanisms contribute to the reduced sensitivity of LT mutants.
As has been observed for cells lacking multiple PG hydrolases (Tomasz et al. 1970; Tomasz 1979; Heidrich et al. 2002; Uehara et al. 2009), the loss of LTs could be protective to cells challenged with β‐lactams, by limiting autolysis under conditions where PG biosynthesis is inhibited – the antisuicide hypothesis (Cavallari et al. 2013). Supporting this idea, cells lacking SltB1 and MltB had slower rates of cell death compared to wild‐type cells when treated with piperacillin (Fig. 2). Loss of additional LTs did not exacerbate the phenotype, possibly because only a subset of LTs is active at certain times during the cell cycle, or because different LTs make specific contributions to PG turnover (Blackburn and Clarke 2002). Blocking LT activity with the inhibitor NAG‐thiazoline (Reid et al. 2004) or deleting a combination of specific LTs and amidases (Heidrich et al. 2002) significantly reduced levels of ampicillin‐induced E. coli cell lysis, providing further evidence that loss of lytic activities can prolong cell survival in the presence of PG‐synthesis inhibitors.
Slt is a unique exception to this phenomenon. In E. coli, loss of Slt reduces β‐lactam resistance and prevents induction of AmpC from a plasmid carrying Enterobacter cloacae ampR and ampC (Templin et al. 1992; Kraft et al. 1999; Cho et al. 2014). In P. aeruginosa, Slt was the only LT whose loss increased β‐lactam sensitivity (Table S3), although AmpC levels were unchanged (this study; Cavallari et al. 2013). The critical contribution of E. coli Slt to β‐lactam susceptibility was recently attributed to its role in a futile cycle of nascent PG synthesis and degradation upon β‐lactam challenge (Cho et al. 2014). The simultaneous loss of Slt and β‐lactam challenge was hypothesized to result in the accumulation of aberrantly cross‐linked glycan strands in the mature PG matrix that interfered with the function of downstream PG‐remodeling machineries (Cho et al. 2014). Whether this paradigm holds true for P. aeruginosa and other Gram‐negatives will be an important future consideration, particularly given the differences between species in the number of LTs and phenotypes of deletion mutants.
While the loss of Family 3 LTs or sLTs increased β‐lactam MICs, the loss of mLTs or all Family 1 LTs caused extreme β‐lactam sensitivity, as well as sensitivity to detergent‐like bile salts and to vancomycin, consistent with a compromised OM (compare sltB1/G/H/mltB and sltB1/G/H/slt strains to mltA/B/D/F/F2 and mltD/F/F2/slt in summary Table S3). Strains lacking multiple sLTs had wild‐type sensitivity to bile salts – except for the mutant lacking all Family 3 LTs, which includes MltB – suggesting that only mLTs contribute to OM stability. Combination mLT mutants were also sensitive to osmotic stress – suggestive of perturbations to the PG layer – implying that they suffer from general cell envelope defects (refer to Table S3, strains mltA/B/D/F/F2 and mltD/F/F2/slt). Membrane defects were unlikely to be due to LPS perturbation, since PMB sensitivity was largely unchanged in mLT mutants and Mg2+ supplementation failed to reverse bile salt sensitivity. Furthermore, complementation with MltA and MltD active‐site point mutants restored wild‐type sensitivity to bile salts (Fig. 3A), pointing to a structural role for these proteins – possibly by facilitating the formation or functioning of multiprotein complexes or by stabilizing OM–PG interactions. In contrast, Slt (this study) and SltB1 (Cavallari et al. 2013) active‐site mutants phenocopied their respective deletion mutants (Table S3), suggesting that catalytic activity of sLTs is essential. The specific roles of most LTs in P. aeruginosa remain unknown (Lee et al. 2015), and future studies to further delineate functional differences between sLTs and mLTs are necessary.
mLTs are OM lipoproteins (Derouaux et al. 2014) that could contribute to stabilizing OM–PG interactions. There are multiple examples in Gram‐negative bacteria of PG‐binding lipoproteins that are critical for OM barrier function. These include Lpp (Cascales et al. 2002; Ni and Chen 2004, 2005), called OprI in P. aeruginosa (Mizuno and Kageyama 1979; Wessel et al. 2013), and Pal (Cascales et al. 2002), called OprL (Mizuno 1979; Mizuno and Kageyama 1979; Hancock et al. 1981). Lpp is the most abundant Pal in E. coli, with ~33% covalently bound to PG through the DAP moiety of PG stem peptides (Neidhardt et al. 1990; Ni and Chen 2005). OprI is also abundant in P. aeruginosa, accounting for ~20% of total OM proteins (Mizuno and Kageyama 1979; Cornelis et al. 1989), but exhibits strain‐specific variations in the level of covalent PG attachment (Mizuno and Kageyama 1979; Hancock et al. 1981; Duchene et al. 1989; Wessel et al. 2013). Pal is less abundant in E. coli than Lpp – 2000–3000 molecules per μm2 versus ~100,000 molecules per μm2 for Lpp (Nikaido 1996; Cascales et al. 2002) – but its absence causes similar perturbations of the cell envelope, suggesting that factors in addition to protein density determine the relative contributions of these proteins to cell envelope integrity. Similarly, loss of covalent OM–PG interactions is not solely responsible for cell envelope instability in lpp mutants, since overexpression of Pal in that background restored membrane integrity, while the reverse was not true (Cascales et al. 2002).
Reversal of bile salt sensitivity in multi‐mLT mutants complemented with OprI suggested that in addition to any enzymatic roles they might have, mLTs might help to stabilize OM–PG interactions. The inability of OprL to reverse bile salt sensitivity was unexpected, given Pal's ability to stabilize the OM in Lpp‐deficient E. coli; however, these data are consistent with a recent study (Wessel et al. 2013) investigating P. aeruginosa OM vesicle (OMV) production and OM stability. Loss of OprI increased OMV production in P. aeruginosa – presumably due to decreased OM–PG interactions – while loss of OprL had no effect (Wessel et al. 2013). Thus, the roles of P. aeruginosa Lpp and Pal orthologs in OM stability appear different from those in E. coli. However, we cannot rule out the possibility that overexpression of OprI suppressed the effects of the mLT mutants by stabilizing a cell envelope perturbed via a different mechanism.
Establishing the mechanisms underlying changes in biofilm formation in PG remodeling enzyme mutants has been challenging due to the lack of a consistent phenotype when such enzymes are lost (Gallant et al. 2005; Monteiro et al. 2011; Payne et al. 2013; Brambilla et al. 2014). Only the mltD mutant in this study was defective for biofilm formation, while four strains lacking mLTs and Family 1 LTs produced significantly more biofilm. These four strains have compromised OMs and increased sensitivity to osmotic stress (Table S3). Loss of OM integrity in these mutants may promote biofilm formation by invoking a response to perceived cell envelope damage. Such a response is reminiscent of the triggering of AmpC expression that occurs – even in the absence of antibiotics – upon loss of PBP4 (Moya et al. 2009).
Our previous study (Cavallari et al. 2013) showed that loss of LTs from Family 1 reduced β‐lactam MICs while loss of LTs from Family 3 increased MICs; however, in light of more comprehensive data, it is clear that differences in β‐lactam MICs are more accurately related to the loss of soluble versus membrane‐bound enzymes (Table S3). Our data agree with previous studies (Korsak et al. 2005; Lee et al. 2013) that there is a degree of functional redundancy between LTs, in that (1) multiple enzymes can be deleted without lethality and (2) with the exception of MltA and MltD, multiple enzymes must be deleted before changes to cell wall integrity are observed (Table S3). Furthermore, AmpC induction was unaffected by the loss of multiple LTs, suggesting redundancy in generating activating anhMP fragments. In contrast, inactivation of E. coli Slt, MltA or MltB attenuated AmpC induction (Kraft et al. 1999), implying that those enzymes make unique contributions to the pool of activating anhMPs. Mecillinam treatment of an E. coli slt mutant resulted in a different PG profile than that of wild type challenged with the same drug (Cho et al. 2014), suggesting that Slt has a specific role in muropeptide turnover upon β‐lactam challenge. In P. aeruginosa, only a subset of LTs affects β‐lactam sensitivity, and only the absence of sltB1 – but not other sLTs or mltB – uniquely contributes to further increases in AmpC levels when PBP4 is inactivated. Thus, the repertoire of LTs with unique activities differs between species.
A screen of the P. aeruginosa strain PA14 transposon mutant library identified several genes whose disruption altered β‐lactam MICs; however, none encoded LTs (Alvarez‐Ortega et al. 2010). The use of clean deletion mutants – or the PAO1 background strain – in this study may underlie the differences in our results. Furthermore, combination mutants often reveal phenotypes that are not obvious from single mutant screens, a concept widely exploited in synthetic lethality studies (Bender and Pringle 1991; Bernhardt and de Boer 2004; Butland et al. 2008). Here, the combined loss of multiple mLTs was critical for uncovering their potential roles in cell envelope stability.
A thorough understanding of the roles of sLTs versus mLTs, and of the unique functions of specific LTs, is necessary for their further development as potential therapeutic targets. We showed that increased β‐lactam sensitivity of a mutant lacking all mLTs or all Family 1 LTs corresponds to loss of cell envelope integrity, while decreased β‐lactam sensitivity of sLT mutants is due – at least in part – to slower death. These findings suggest that pan‐inhibitors of LT activity could have the potential to increase β‐lactam resistance in P. aeruginosa, as – except for Slt – the activity of sLTs contributes to sensitivity, while mLT activity is dispensable. However, the collective inhibition of all LTs may yield unexpected results. As the sole LT in both P. aeruginosa and E. coli whose loss leads to increased β‐lactam sensitivity (Templin et al. 1992; Cavallari et al. 2013; Cho et al. 2014), Slt is emerging as the preferred LT target for development of antibiotic adjuvants.
Conflict of Interest
None declared.
Supporting information
Table S1. Primer details for PCR confirmation of LT mutant strains.
Table S2. MICs for all LT mutants of β‐lactam, fluoroquinolone, and glycopeptide antibiotics.
Table S3. Summary of phenotypes associated with LT mutants.
Figure S1. Loss of LTs does not cause growth defects. Bacterial growth (OD600 nm) was monitored every hour for 48 h using a plate‐based assay, described below. Curves are average of three biological replicates with three technical replicates each. Following the strain names in each legend are the growth rates per minute (min−1) and lag times (in min), respectively.
Figure S2. Loss of LTs does not affect AmpC expression. Strains lacking the indicated LT were grown under basal (top panel) or AmpC‐inducing (bottom panel) conditions and immunoblotted using α‐AmpC antibodies, as described in the Materials and Methods section. Loss of LTs does not prevent either basal AmpC expression or AmpC induction.
Figure S3. Magnesium supplementation does not affect bile salt sensitivity in mLT mutants. Bile salt assays were performed as described in the Materials and Methods section with and without MgCl2 (1 mmol/L final) supplementation. Bile salt sensitivity of the mltA/B/D/F/F2 and mltD/F/F2/slt mutants was unaffected by the addition of magnesium. Bars representing non‐MgCl2‐treated samples are the same as those used in Figure 3A. N = 3. Bars represent the mean ± SEM.
Acknowledgments
This study was supported by a Canada–UK Partnership on Antibiotic Resistance award from the Canadian Institutes of Health Research (CIHR; 114045) and a Natural Sciences and Engineering Research Council of Canada (NSERC) award (RGPIN 227817‐2011 to L. L. B.). R. P. L. held the Michael G. DeGroote Post‐Doctoral Fellowship in Basic Biomedical Science from McMaster University and Post‐Doctoral Fellowships from CIHR and Cystic Fibrosis Canada.
MicrobiologyOpen 2015; 4(6): 879–895
References
- Alvarez‐Ortega, C. , Wiegand I., Olivares J., Hancock R. E., and Martinez J. L.. 2010. Genetic determinants involved in the susceptibility of Pseudomonas aeruginosa to beta‐lactam antibiotics. Antimicrob. Agents Chemother. 54:4159–4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asikyan, M. L. , Kus J. V., and Burrows L. L.. 2008. Novel proteins that modulate type IV pilus retraction dynamics in Pseudomonas aeruginosa . J. Bacteriol. 190:7022–7034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Asselt, E. J. , Dijkstra A. J., Kalk K. H., Takacs B., Keck W., and Dijkstra B. W.. 1999. Crystal structure of Escherichia coli lytic transglycosylase Slt35 reveals a lysozyme‐like catalytic domain with an EF‐hand. Structure 7:1167–1180. [DOI] [PubMed] [Google Scholar]
- Bender, A. , and Pringle J. R.. 1991. Use of a screen for synthetic lethal and multicopy suppressee mutants to identify two new genes involved in morphogenesis in Saccharomyces cerevisiae . Mol. Cell. Biol. 11:1295–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernal, P. , Molina‐Santiago C., Daddaoua A., and Llamas M. A.. 2013. Antibiotic adjuvants: identification and clinical use. Microb. Biotechnol. 6:445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhardt, T. G. , and de Boer P. A.. 2004. Screening for synthetic lethal mutants in Escherichia coli and identification of EnvC (YibP) as a periplasmic septal ring factor with murein hydrolase activity. Mol. Microbiol. 52:1255–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn, N. T. , and Clarke A. J.. 2001. Identification of four families of peptidoglycan lytic transglycosylases. J. Mol. Evol. 52:78–84. [DOI] [PubMed] [Google Scholar]
- Blackburn, N. T. , and Clarke A. J.. 2002. Characterization of soluble and membrane‐bound family 3 lytic transglycosylases from Pseudomonas aeruginosa . Biochemistry 41:1001–1013. [DOI] [PubMed] [Google Scholar]
- Boucher, H. W. , Talbot G. H., Bradley J. S., Edwards J. E., Gilbert D., Rice L. B., et al. 2009. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 48:1–12. [DOI] [PubMed] [Google Scholar]
- Brambilla, L. , Moran‐Barrio J., and Viale A. M.. 2014. Low‐molecular‐mass penicillin binding protein 6b (DacD) is required for efficient GOB‐18 metallo‐beta‐lactamase biogenesis in Salmonella enterica and Escherichia coli . Antimicrob. Agents Chemother. 58:205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breidenstein, E. B. , De la Fuente‐Nunez C., and Hancock R. E.. 2011. Pseudomonas aeruginosa: all roads lead to resistance. Trends Microbiol. 19:419–426. [DOI] [PubMed] [Google Scholar]
- Butland, G. , Babu M., Diaz‐Mejia J. J., Bohdana F., Phanse S., Gold B., et al. 2008. eSGA: E. coli synthetic genetic array analysis. Nat. Methods 5:789–795. [DOI] [PubMed] [Google Scholar]
- Cascales, E. , Bernadac A., Gavioli M., Lazzaroni J. C., and Lloubes R.. 2002. Pal lipoprotein of Escherichia coli plays a major role in outer membrane integrity. J. Bacteriol. 184:754–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallari, J. F. , Lamers R. P., Scheurwater E. M., Matos A. L., and Burrows L. L.. 2013. Changes to its peptidoglycan‐remodeling enzyme repertoire modulate beta‐lactam resistance in Pseudomonas aeruginosa . Antimicrob. Agents Chemother. 57:3078–3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho, H. , Uehara T., and Bernhardt T. G.. 2014. Beta‐lactam antibiotics induce a lethal malfunctioning of the bacterial cell wall synthesis machinery. Cell 159:1300–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelis, P. , Bouia A., Belarbi A., Guyonvarch A., Kammerer B., Hannaert V., et al. 1989. Cloning and analysis of the gene for the major outer membrane lipoprotein from Pseudomonas aeruginosa . Mol. Microbiol. 3:421–428. [DOI] [PubMed] [Google Scholar]
- Davies, J. , and Davies D.. 2010. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 74:417–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcour, A. H. 2009. Outer membrane permeability and antibiotic resistance. Biochim. Biophys. Acta 1794:808–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derouaux, A. , Terrak M., Den blaauwen T., and Vollmer W.. 2014. Bacterial cell wall growth, shape and division Pp. 3–54 in Remaut H. and Fronzes R., eds. Bacterial membranes: structural and molecular biology. Caister Academic Press, Norfolk, U.K. [Google Scholar]
- Duchene, M. , Barron C., Schweizer A., von Specht B. U., and Domdey H.. 1989. Pseudomonas aeruginosa outer membrane lipoprotein I gene: molecular cloning, sequence, and expression in Escherichia coli . J. Bacteriol. 171:4130–4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez, L. , Breidenstein E. B., and Hancock R. E.. 2011. Creeping baselines and adaptive resistance to antibiotics. Drug Resist. Updates 14:1–21. [DOI] [PubMed] [Google Scholar]
- Gallant, C. V. , Daniels C., Leung J. M., Ghosh A. S., Young K. D., Kotra L. P., et al. 2005. Common beta‐lactamases inhibit bacterial biofilm formation. Mol. Microbiol. 58:1012–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giltner, C. L. , Habash M., and Burrows L. L.. 2010. Pseudomonas aeruginosa minor pilins are incorporated into type IV pili. J. Mol. Biol. 398:444–461. [DOI] [PubMed] [Google Scholar]
- Hancock, R. E. 1984. Alterations in outer membrane permeability. Annu. Rev. Microbiol. 38:237–264. [DOI] [PubMed] [Google Scholar]
- Hancock, R. E. , Irvin R. T., Costerton J. W., and Carey A. M.. 1981. Pseudomonas aeruginosa outer membrane: peptidoglycan‐associated proteins. J. Bacteriol. 145:628–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidrich, C. , Ursinus A., Berger J., Schwarz H., and Holtje J. V.. 2002. Effects of multiple deletions of murein hydrolases on viability, septum cleavage, and sensitivity to large toxic molecules in Escherichia coli . J. Bacteriol. 184:6093–6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang, T. T. , Karkhoff‐Schweizer R. R., Kutchma A. J., and Schweizer H. P.. 1998. A broad‐host‐range Flp‐FRT recombination system for site‐specific excision of chromosomally‐located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86. [DOI] [PubMed] [Google Scholar]
- Jacobs, C. , Huang L. J., Bartowsky E., Normark S., and Park J. T.. 1994. Bacterial cell wall recycling provides cytosolic muropeptides as effectors for beta‐lactamase induction. EMBO J. 13:4684–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, J. W. , Fisher J. F., and Mobashery S.. 2013. Bacterial cell‐wall recycling. Ann. N. Y. Acad. Sci. 1277:54–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgenson, M. A. , Chen Y., Yahashiri A., Popham D. L., and Weiss D. S.. 2014. The bacterial septal ring protein RlpA is a lytic transglycosylase that contributes to rod shape and daughter cell separation in Pseudomonas aeruginosa . Mol. Microbiol. 93:113–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juan, C. , Moya B., Perez J. L., and Oliver A.. 2006. Stepwise upregulation of the Pseudomonas aeruginosa chromosomal cephalosporinase conferring high‐level beta‐lactam resistance involves three AmpD homologues. Antimicrob. Agents Chemother. 50:1780–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan, J. B. , Izano E. A., Gopal P., Karwacki M. T., Kim S., Bose J. L., et al. 2012. Low levels of beta‐lactam antibiotics induce extracellular DNA release and biofilm formation in Staphylococcus aureus . MBio 3:e00198‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocaoglu, O. , and Carlson E. E.. 2015. Profiling of beta‐lactam selectivity for penicillin‐binding proteins in Escherichia coli strain DC2. Antimicrob. Agents Chemother. 59:2785–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocaoglu, O. , Tsui H. C., Winkler M. E., and Carlson E. E.. 2015. Profiling of beta‐lactam selectivity for penicillin‐binding proteins in Streptococcus pneumoniae D39. Antimicrob. Agents Chemother. 59:3548–3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolar, S. L. , Nagarajan V., Oszmiana A., Rivera F. E., Miller H. K., Davenport J. E., et al. 2011. NsaRS is a cell‐envelope‐stress‐sensing two‐component system of Staphylococcus aureus . Microbiology 157:2206–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsak, D. , Liebscher S., and Vollmer W.. 2005. Susceptibility to antibiotics and beta‐lactamase induction in murein hydrolase mutants of Escherichia coli . Antimicrob. Agents Chemother. 49:1404–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostakioti, M. , Hadjifrangiskou M., and Hultgren S. J.. 2013. Bacterial biofilms: development, dispersal, and therapeutic strategies in the dawn of the postantibiotic era. Cold Spring Harb. Perspect. Med. 3:a010306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft, A. R. , Prabhu J., Ursinus A., and Holtje J. V.. 1999. Interference with murein turnover has no effect on growth but reduces beta‐lactamase induction in Escherichia coli . J. Bacteriol. 181:7192–7198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari, H. , Balasubramanian D., Zincke D., and Mathee K.. 2014. Role of Pseudomonas aeruginosa AmpR on beta‐lactam and non‐beta‐lactam transient cross‐resistance upon pre‐exposure to subinhibitory concentrations of antibiotics. J. Med. Microbiol. 63:544–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamers, R. P. , Cavallari J. F., and Burrows L. L.. 2013. The efflux inhibitor phenylalanine‐arginine beta‐naphthylamide (PAbetaN) permeabilizes the outer membrane of gram‐negative bacteria. PLoS One 8:e60666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, M. , Hesek D., Llarrull L. I., Lastochkin E., Pi H., Boggess B., et al. 2013. Reactions of all Escherichia coli lytic transglycosylases with bacterial cell wall. J. Am. Chem. Soc. 135:3311–3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, M. , Hesek D., Blazquez B., Lastochkin E., Boggess B., Fisher J. F., et al. 2015. Catalytic spectrum of the penicillin‐binding protein 4 of Pseudomonas aeruginosa, a nexus for the induction of beta‐lactam antibiotic resistance. J. Am. Chem. Soc. 137:190–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legaree, B. A. , and Clarke A. J.. 2008. Interaction of penicillin‐binding protein 2 with soluble lytic transglycosylase B1 in Pseudomonas aeruginosa . J. Bacteriol. 190:6922–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X. Z. , Zhang L., Srikumar R., and Poole K.. 1998. Beta‐lactamase inhibitors are substrates for the multidrug efflux pumps of Pseudomonas aeruginosa . Antimicrob. Agents Chemother. 42:399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister, P. D. , Wolter D. J., and Hanson N. D.. 2009. Antibacterial‐resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin. Microbiol. Rev. 22:582–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, W. , Dong N., and Zhang X. H.. 2012. Overexpression of mltA in Edwardsiella tarda reduces resistance to antibiotics and enhances lethality in zebra fish. J. Appl. Microbiol. 112:1075–1085. [DOI] [PubMed] [Google Scholar]
- Liu, Z. , Niu H., Wu S., and Huang R.. 2014. CsgD regulatory network in a bacterial trait‐altering biofilm formation. Emerg. Microbes Infect. 3:e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark, B. L. , Vocadlo D. J., and Oliver A.. 2011. Providing beta‐lactams a helping hand: targeting the AmpC beta‐lactamase induction pathway. Future Microbiol. 6:1415–1427. [DOI] [PubMed] [Google Scholar]
- Mizuno, T. 1979. A novel peptidoglycan‐associated lipoprotein found in the cell envelope of Pseudomonas aeruginosa and Escherichia coli . J. Biochem. 86:991–1000. [DOI] [PubMed] [Google Scholar]
- Mizuno, T. , and Kageyama M.. 1979. Isolation and characterization of a major outer membrane protein of Pseudomonas aeruginosa. Evidence for the occurrence of a lipoprotein. J. Biochem. 85:115–122. [DOI] [PubMed] [Google Scholar]
- Monteiro, C. , Fang X., Ahmad I., Gomelsky M., and Romling U.. 2011. Regulation of biofilm components in Salmonella enterica serovar Typhimurium by lytic transglycosylases involved in cell wall turnover. J. Bacteriol. 193:6443–6451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moya, B. , Dotsch A., Juan C., Blazquez J., Zamorano L., Haussler S., et al. 2009. Beta‐lactam resistance response triggered by inactivation of a nonessential penicillin‐binding protein. PLoS Pathog. 5:e1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neidhardt, F. C. , Ingraham J., and Schaechter M.. 1990. Physiology of the bacterial cell: a molecular approach. Sinauer Associates, Sunderland, MA. [Google Scholar]
- Ni, Y. , and Chen R. R.. 2004. Accelerating whole‐cell biocatalysis by reducing outer membrane permeability barrier. Biotechnol. Bioeng. 87:804–811. [DOI] [PubMed] [Google Scholar]
- Ni, Y. , and Chen R. R.. 2005. Lipoprotein mutation accelerates substrate permeability‐limited toluene dioxygenase‐catalyzed reaction. Biotechnol. Prog. 21:799–805. [DOI] [PubMed] [Google Scholar]
- Nikaido H., ed. 1996. Outer membrane. ASM Press, Washington, DC. [Google Scholar]
- Nikaido, H. 2003. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 67:593–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido, H. 2005. Restoring permeability barrier function to outer membrane. Chem. Biol. 12:507–509. [DOI] [PubMed] [Google Scholar]
- Payne, D. E. , Martin N. R., Parzych K. R., Rickard A. H., Underwood A., and Boles B. R.. 2013. Tannic acid inhibits Staphylococcus aureus surface colonization in an IsaA‐dependent manner. Infect. Immun. 81:496–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plesiat, P. , Aires J. R., Godard C., and Kohler T.. 1997. Use of steroids to monitor alterations in the outer membrane of Pseudomonas aeruginosa . J. Bacteriol. 179:7004–7010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell, A. J. , Liu Z. J., Nicholas R. A., and Davies C.. 2006. Crystal structures of the lytic transglycosylase MltA from N. gonorrhoeae and E. coli: insights into interdomain movements and substrate binding. J. Mol. Biol. 359:122–136. [DOI] [PubMed] [Google Scholar]
- Priyadarshini, R. , Popham D. L., and Young K. D.. 2006. Daughter cell separation by penicillin‐binding proteins and peptidoglycan amidases in Escherichia coli . J. Bacteriol. 188:5345–5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid, C. W. , Blackburn N. T., and Clarke A. J.. 2004. The effect of NAG‐thiazoline on morphology and surface hydrophobicity of Escherichia coli . FEMS Microbiol. Lett. 234:343–348. [DOI] [PubMed] [Google Scholar]
- Rice, L. B. 2008. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J. Infect. Dis. 197:1079–1081. [DOI] [PubMed] [Google Scholar]
- Ruiz, N. , Falcone B., Kahne D., and Silhavy T. J.. 2005. Chemical conditionality: a genetic strategy to probe organelle assembly. Cell 121:307–317. [DOI] [PubMed] [Google Scholar]
- Sailer, F. C. , Meberg B. M., and Young K. D.. 2003. Beta‐lactam induction of colanic acid gene expression in Escherichia coli . FEMS Microbiol. Lett. 226:245–249. [DOI] [PubMed] [Google Scholar]
- Scheurwater, E. , Reid C. W., and Clarke A. J.. 2008. Lytic transglycosylases: bacterial space‐making autolysins. Int. J. Biochem. Cell Biol. 40:586–591. [DOI] [PubMed] [Google Scholar]
- Silhavy, T. J. , Kahne D., and Walker S.. 2010. The bacterial cell envelope. Cold Spring Harb. Perspect. Biol. 2:a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Straaten, K. E. , Dijkstra B. W., Vollmer W., and Thunnissen A. M.. 2005. Crystal structure of MltA from Escherichia coli reveals a unique lytic transglycosylase fold. J. Mol. Biol. 352:1068–1080. [DOI] [PubMed] [Google Scholar]
- Sutterlin, H. A. , Zhang S., and Silhavy T. J.. 2014. Accumulation of phosphatidic acid increases vancomycin resistance in Escherichia coli . J. Bacteriol. 196:3214–3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Templin, M. F. , Edwards D. H., and Holtje J. V.. 1992. A murein hydrolase is the specific target of bulgecin in Escherichia coli . J. Biol. Chem. 267:20039–20043. [PubMed] [Google Scholar]
- Thunnissen, A. M. , Dijkstra A. J., Kalk K. H., Rozeboom H. J., Engel H., Keck W., et al. 1994. Doughnut‐shaped structure of a bacterial muramidase revealed by X‐ray crystallography. Nature 367:750–753. [DOI] [PubMed] [Google Scholar]
- Tomasz, A. 1979. The mechanism of the irreversible antimicrobial effects of penicillins: how the beta‐lactam antibiotics kill and lyse bacteria. Annu. Rev. Microbiol. 33:113–137. [DOI] [PubMed] [Google Scholar]
- Tomasz, A. , Albino A., and Zanati E.. 1970. Multiple antibiotic resistance in a bacterium with suppressed autolytic system. Nature 227:138–140. [DOI] [PubMed] [Google Scholar]
- Uehara, T. , Dinh T., and Bernhardt T. G.. 2009. LytM‐domain factors are required for daughter cell separation and rapid ampicillin‐induced lysis in Escherichia coli . J. Bacteriol. 191:5094–5107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. 2002. The function of OmpA in Escherichia coli . Biochem. Biophys. Res. Commun. 292:396–401. [DOI] [PubMed] [Google Scholar]
- Wenderska, I. B. , Chong M., McNulty J., Wright G. D., and Burrows L. L.. 2011. Palmitoyl‐DL‐carnitine is a multitarget inhibitor of Pseudomonas aeruginosa biofilm development. Chembiochem 12:2759–2766. [DOI] [PubMed] [Google Scholar]
- Wessel, A. K. , Liew J., Kwon T., Marcotte E. M., and Whiteley M.. 2013. Role of Pseudomonas aeruginosa peptidoglycan‐associated outer membrane proteins in vesicle formation. J. Bacteriol. 195:213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization . 2014. Antimicrobial resistance: global report on surveillance.
- Zamorano, L. , Reeve T. M., Deng L., Juan C., Moya B., Cabot G., et al. 2010. NagZ inactivation prevents and reverts beta‐lactam resistance, driven by AmpD and PBP 4 mutations, in Pseudomonas aeruginosa . Antimicrob. Agents Chemother. 54:3557–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamorano, L. , Reeve T. M., Juan C., Moya B., Cabot G., Vocadlo D. J., et al. 2011. AmpG inactivation restores susceptibility of pan‐beta‐lactam‐resistant Pseudomonas aeruginosa clinical strains. Antimicrob. Agents Chemother. 55:1990–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Bao Q., Gagnon L. A., Huletsky A., Oliver A., Jin S., et al. 2010. ampG gene of Pseudomonas aeruginosa and its role in beta‐lactamase expression. Antimicrob. Agents Chemother. 54:4772–4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primer details for PCR confirmation of LT mutant strains.
Table S2. MICs for all LT mutants of β‐lactam, fluoroquinolone, and glycopeptide antibiotics.
Table S3. Summary of phenotypes associated with LT mutants.
Figure S1. Loss of LTs does not cause growth defects. Bacterial growth (OD600 nm) was monitored every hour for 48 h using a plate‐based assay, described below. Curves are average of three biological replicates with three technical replicates each. Following the strain names in each legend are the growth rates per minute (min−1) and lag times (in min), respectively.
Figure S2. Loss of LTs does not affect AmpC expression. Strains lacking the indicated LT were grown under basal (top panel) or AmpC‐inducing (bottom panel) conditions and immunoblotted using α‐AmpC antibodies, as described in the Materials and Methods section. Loss of LTs does not prevent either basal AmpC expression or AmpC induction.
Figure S3. Magnesium supplementation does not affect bile salt sensitivity in mLT mutants. Bile salt assays were performed as described in the Materials and Methods section with and without MgCl2 (1 mmol/L final) supplementation. Bile salt sensitivity of the mltA/B/D/F/F2 and mltD/F/F2/slt mutants was unaffected by the addition of magnesium. Bars representing non‐MgCl2‐treated samples are the same as those used in Figure 3A. N = 3. Bars represent the mean ± SEM.