

Abstract
The deletion of chromosomal region 6q was commonly found in several types of human cancers, although the tumor suppressor genes (TSGs) located within this genomic region are not well established. Our recent work detected recurrent chromosomal truncation at the Na+/K+ transporting ATPase interacting 2 (NKAIN2) gene in prostate cancer, which was also found to be truncated in leukemia and lymphoma, suggesting that NKAIN2 is potentially one of the TSGs located in the 6q commonly deleted region in human cancers. NKAIN2 gene consists of eight coding exons that span approximately 1 Mb of genomic DNA on chromosome 6q and there are four main splice variants. The function of this gene is not well investigated and the limited knowledge of this gene pointed to nervous system development. The chromosomal translocations in nervous development disorders usually lead to inactivation of this gene. In human tumors, both chromosomal deletion and translocation may also inactivate this gene and consequently contribute to tumorigenesis. Further genetic and cellular functional studies are required to establish its tumor suppressor role.
Keywords: Chromosomal deletion, genomic rearrangement, tumor suppressor gene, NKAIN2 (TCBA1)
Introduction
Deletions of 6q are frequently found in human cancers, including prostate cancer [1-3], breast cancer [4], pancreatic cancer [5], renal cell carcinoma [6], lung adenocarcinoma [7], malignant melanoma [8], esophageal squamous cell cancer [9], lymphoblastic leukemia [10], and non-Hodgkin’s B-cell lymphomas [11]. Several studies on the loss of heterozygosity (LOH) have also suggested that chromosome 6q is involved in the pathogenesis of various human malignancies, including prostate cancer [12], acute lymphoblastic leukemia [13], non-Hodgkin’s B-cell lymphoma [13], ovarian carcinoma [14], breast carcinoma [15], malignant melanoma [16], renal cell carcinoma [17], hepatocellular carcinoma [18], salivary gland adenocarcinoma [19], pancreatic cancer [20], and parathyroid adenoma [21]. Re-introducing a normal chromosome 6 into melanoma cells suppresses tumorigenesis [3] and a potential tumor metastasis suppressor locus has been functionally linked to 6q16.3-q23 [3]. These studies suggest that chromosome 6 may harbor one or more genes that suppress tumorigenesis and metastasis. However, while several candidate TSGs have been proposed, such as MAP3K7 [22], PLAGL1 [23], and LATS1 [24], no TSGs have been well established. In our recent study of prostate cancer genomic alterations, we found that in prostate cancer the majority of genomic truncations occurred at TSGs rather than oncogenes, indicating that genomic truncation is also a major mechanism causing TSG inactivation in prostate cancer [25]. Na+/K+ transporting ATPase interacting 2 (NKAIN2) is one of the genes recurrently truncated by chromosomal rearrangement [25] and our unpublished data showed that NKAIN2 was generally under-expressed in prostate cancer cell compare to adjacent non-malignant prostate tissues, suggesting that NKAIN2 is potentially one of the TSGs located on 6q. In the present article, we review the biological function of NKAIN2 and existing evidence to support its role in tumorigenesis. We speculate that NKAIN2 could be a novel tumor suppressor on the 6q commonly deleted chromosomal region in human cancer and propose further researches required to investigate its potential tumor suppresser role. With the deepening of its involvement in human cancer and its cellular functions, the role of NKAIN2 in tumorigenesis will be uncovered, which may impact the treatment of human malignancy.
Basic structure and cellular function of NKAIN2
NKAIN2 was originally named T-cell lymphoma breakpoint associated target 1 (TCBA1), because it was involved in chromosome 6q aberrations in T-cell lymphoma and leukemia cell lines [26]. Potential functions of NKAIN2 can be predicted based on its genetic structure. Searching the Sanger Center Pfam (Protein Families) database (http://www.sanger.ac.uk/resources/databases/pfam.html) for the product of NKAIN2 identifies a family of proteins (Pfam05640/DUF798) with several members that are phylogenetically well conserved. The common feature shared by these proteins is the presence of the DUF798 domain, the function of which is not known. Most of the members of this family are classified as hypothetical proteins and no information is available about their function. In human, NKAIN2 belongs to a superfamily of transmembrane proteins that interact with the b1 subunits of Na+/K+-ATPase and that are encoded by four genes, NKAIN1, NKAIN2, NKAIN3 and NKAIN4 [27] (Table 1).
Table 1.
Basic information of NKAINs family members
| Gene | Location | Expression | Interaction | Involvement in diseases |
|---|---|---|---|---|
| NKAIN1 | 1p35.2 | Neuron-specific | Interact with the b1 subunit of the Na, K-ATPase | alcohol dependence. |
| NKAIN2 | 6q22.31 | Neuron-specific | genital herpes, alcohol dependence, lymphoma, neuroblastoma, prostate cancer, type 2 diabets mellitus. | |
| NKAIN3 | 8q12.13 | Neuron-specific | ||
| NKAIN4 | 20q13.33 | Ubiquitous |
NKAIN genes do not show any similarities to any other known genes, but they do share striking evolutionary conservation among species, there is a striking amino acid conservation in the first two transmembrane domains from Drosophila to human, which is considered a good indicator of their functional significance [27]. Preliminary data concerning the putative C. elegans homolog gene (T13H5.6) suggest that its expression pattern is also conserved among phylogenetically diverse species [28]. The NKAIN2 gene consists of eight coding exons that span approximately 1 Mb of genomic DNA on chromosome 6q [28]. Four main splice variants have been identified, including two short isoforms that contain four exons and two long isoforms containing seven exons [26,28] (Figure 1). A PSI-BLAST analysis, which allows the identification of distantly related proteins, has revealed a string of 70 residues that perfectly match a mouse protein similar to B-RAF [29]. In addition to the matching 70-residue sequence, this protein mainly consists of a serine/threonine kinase domain homologous to mouse B-RAF. Raf proteins, which are encoded by known proto-oncogenes, belong to the family of serine/threonine kinases and are intracellular signal transducers that are associated with membrane receptors [30-32].
Figure 1.
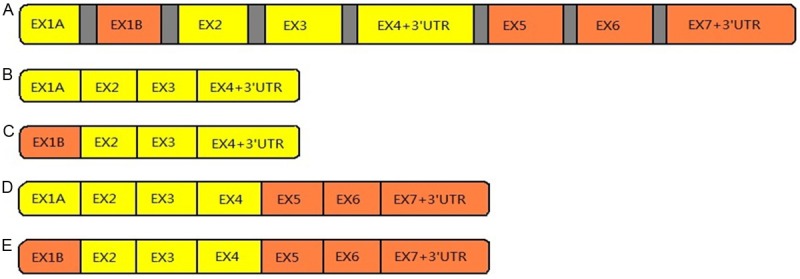
(A) Schematic representation of the NKAIN2 (TCBA1) gene. The gene consists of eight coding exons spanning about 1 Mb of genomic DNA on chromosome 6q (proportions are not respected in this picture). (B-E) Four main splice variants of NKAIN2, including two short isoforms (B and C) that contain four exons and two long isoforms (D and E) containing seven exons.
Involvement of NKAIN2 in human malignance
Chromosomal rearrangements, including translocations, inversions, deletions, and duplications, are the hallmarks of human cancer [33,34]. Those chromosomal rearrangements can either activate oncogenes by deregulating them and even generate novel tumorigenic fusion product or inactivate TSGs by deleting the whole or part of the genes. As mentioned above, NKAIN2 is located within the 6q commonly deleted region in human cancers, thus reducing expression by loss of genomic copies and potentially causing haploid insufficiency. In addition, chromosome translocations affecting NKAIN2 genomic region can also lead to loss of function of this gene, both in developmental diseases and human malignancies [26,28,35].
It is well established that translocation and genomic fusion can deregulate oncogenes or generate gain of function tumorigenic genes. The generation of fusion proteins is a well-known mechanism capable of promoting oncogene activation, because the fusion proteins often have novel functional features such as the unscheduled expression or constitutive activation of an intrinsic function [36,37]. Nowell and Hungerford’s discovery of the Philadelphia chromosome in chronic myeloid leukemia (CML) [38], the first consistent chromosome change seen in human malignancy, clearly supports the notion that chromosomal abnormalities, especially translocations and their corresponding gene fusions, play an important role in the initiation of carcinogenesis. Fusion genes and the accompanying deregulation of oncogenes associated with chromosomal rearrangements have been extensively studied in hematological malignancies and soft tissue sarcomas [33], and can frequently be used to define tumor subtypes and predict prognosis. In last 10 years, the commonness and importance of fusion genes in carcinomas have also been revealed [39]. TMPRSS2:ERG, for instance, is the most frequently found fusion gene in human malignancies and occurs in about 50% of prostate cancer cases [2,33]. However, in the case of chromosomal translocation and genomic fusion involving NKAIN2, this gene is inactivated rather than gain of function both in malignant and non-malignant diseases.
Translocation involving NKAIN2 has been identified in both T-cell lymphoma and leukemia cell lines [26]. In a T-cell lymphoblastic lymphoma cell line, HT-1, NKAIN2 was found to be fused to SUSP1 (SUMO-1-specific protease), creating a SUSP1-NKAIN2 chimeric gene. However, this fusion was not in frame, which led to the production of a abnormal SUPS1 protein and loss of NKAIN2 protein from this chromosome [26]. In an adult T-cell leukemia cell line, ATN-1, aberrant NKAIN2 transcripts were produced and no chimeric gene was detected. Therefore, in both cases, the chromosomal rearrangements affecting this genomic region led to loss of function of NKAIN2 [26]. Translocations of t(1;6)(q32.3;q22.3) and t(2;6)(q24.3;q22.31) with breakpoint at NKAIN2 have been reported in developmental delay [35] and neurological disorders [28] respectively, and in both cases, the consequence is constitutional inactivation of the NKAIN2 gene.
In our SNP array analysis of 71 samples from patients with prostate cancer, NKAIN2 was truncated in 5 cases and deleted in 10 cases [25]. Importantly, in two of the five cases where NKAIN2 was truncated, a small genomic deletion event occurred that covered nearly the entire gene but very little of the adjacent genomic regions [25]. This finding suggests that NKAIN2 is also inactivated by not only chromosome deletion but also genomic truncation in prostate cancer. Further supporting this proposition, Kanishka Sircar et al. [40] have reported that NKAIN2 is downregulated and deleted in castration-resistant prostate cancer (CRPC), which appears at the early stage of the development of castration resistance. These data support the hypothesis that NKAIN2 is an important prostate cancer TSG in the frequently deleted 6q region.
In addition to chromosome loss, mutation and promoter methylation can also result in inactivation of TSG. The expression of NKAIN2 is downregulated in human brain and CNS cancer (Data from Oncomine database http://www.oncomine.org/resource/login). In addition, 80 mutations affecting the NKIAN2 coding region have been characterized and NKAIN2 gene mutation recurrence has been found in breast carcinoma (4/1233 samples), endometrioid carcinoma (4/494 samples), clear cell renal cell carcinoma (2/878 samples), lung adenocarcinoma (7/639 samples), lung squamous cell carcinoma (4/531 samples), oesophagus adenocarcinoma (2/151 samples), skin malignant melanoma (13/526 samples), stomachadenocarcinoma (6/338 samples), bladder carcinoma (2/327 samples) (Data from COSMIC database http://cancer.sanger.ac.uk/cosmic and International Genome Cancer Consortium database http://dcc.icgc.org/). All these data reveals that NKAIN2 maybe a TSG inactivated in many types of human cancers.
However, based on the tissue type involved, NKAIN2 may not only act as a TSG, but also promotes tumorigenesis in tissue types where it is required for cell proliferation, such as the neurons. In fact, Romania et al. revealed that NKAIN2 expressed at high mRNA levels in affected siblings in peripheral blood and tumor samples, in a study involving an Italian family with three neuroblastoma patients [41]. Elevated NKAIN2 was also detected in MYCN-amplified neuroblastoma cell lines, in the most aggressive neuroblastoma lesions and on the cell membranes of neuroblasts in the peripheral blood of a large cohort of neuroblastoma patients. These data indicate that NKAIN2 contribute to neuroblastoma development and/or progression, which is consistent with its role in neuron cell growth.
Biological function of NKAIN2 and its involvement in human diseases
The biological function of NKAIN2 remains unclear. NKAIN2 is transcribed in different splice variants and abundantly expressed in the brain tissues [27]. Several studies have suggested that NKAIN2 is highly specific to the central nervous system and is necessary for nervous system health and development [28,35,42-44]. Bocciardi et al. described the characterization of a de novo balanced translocation, t(2;6)(q24.3;q22.31), in a patient with a severe neurologic phenotype that included epileptic encephalopathy with spastic tetraparesis, severe psychomotor retardation associated with cerebral atrophy and involvement of the periventricular white matter [28]. Calboli et al. analyzed 430,000 autosomal SNPs together with an additional 1.2 million SNPs with high estimated quality from the International Hap Map Project’s CEU samples and demonstrated that neuroticism is one of the primary effects associated with SNPs in NKAIN2 [42]. Yue et al. found that NKAIN2 is disrupted in intron 4 by a de novo balanced translocation, t(1;6)(q32.2;q22.3), in a child with developmental delay and recurrent infections [35]. In an Australian twin-family based association analysis of alcohol dependence in the Collaborative Study on the Genetics of Alcoholism (COGA) study of two cohorts of Australian twins and their spouses, Wang et al. revealed that rs637547 in NKAIN2 at 6q21 showed a strong association with alcohol dependence [43]. Lind et al. performed a meta-analysis of Australian and Dutch Data and found that rs594664 in the NKAIN2 intron was the thirty most significant association with nicotine dependence, although it failed to reach genome-wide significance (P = 2.63 × 10-5) [44].
In addition to the strong expression in brain tissues [26,35], NKAIN2 was also expression in thymus [26], skeletal muscle [28], spinalcord, and ovary (Data from GeneCards http://www.genecards.org). However, the role of NKAIN2 in those tissues is unknown. NKAIN2 expression was detected in hematological malignancy cell lines but not those T cell derived malignant cell lines with NKAIN2 truncation, suggesting its potential involvement in hematological cell differentiation, which is also supported by the recurrent infection in the case with constitutional t(1;6)(q32.2;q22.3) [35].
NKAIN2 in prostate cancer and the difference between Chinese and western populations
Despite numerous investigations into the molecular mechanisms underlying the pathogenesis of the disease, the genetic changes driving prostate cancer development and progression are incompletely characterized [45]. The chromosomal regions that are most commonly lost in prostate cancer are 1p, 6q, 8p21.2, 10p15, 10q21, 10q23.31, 12p12-13, 13q21, 16q22, 21q22.2 [2,46]. Prostate cancer has been shown commonly to involve very complex chromosome rearrangements, which lead to many chromosome breakpoints and gene fusions, including the most common fusion gene in human malignancies, TMPRSS2:ERG [47,48]. The fusions frequently involve the ETS family of transcription factors, which are placed under the control of genes highly active in prostate epithelial cells, predominately as TMPRSS2, but also including SLC45A3, HERV-K_22q11.23, C15orf21, and HNRPA2B1 [49-52]. However, in our recent work, we found that chromosome breakpoints in prostate cancer may more frequently lead to TSG inactivation than oncogene activation [25].
By analyzing 71 clinical prostate cancer cases and 6 prostate cancer cell lines using Affymetrix array 6.0 and 500K SNP microarrays, we identified many recurrent breakpoints (n ≥ 2) and 41 located at known TSGs, oncogenes and genes previously identified as partner genes in gene fusion events. While the ERG and TMPRSS2 genes were affected by the frequent breakpoints (identified in 18/77 and 15/77 cases, respectively), there was a preferential involvement of TSGs (n = 27) compared to oncogenes (n = 6) at the breakpoints [25]. NKAIN2 was also recurrently truncated by chromosomal rearrangement [25]. Together with our unpublished data that NKAIN2 was generally under-expressed in prostate cancer cell compare to adjacent non-malignant prostate tissues, it suggests that NKAIN2 is potentially a TSG in prostate cancer.
Interestingly, NKAIN2 was truncated in four of the 39 Chinese prostate cancer cases but not in any of the 32 cases from UK. Remarkable disparities of prostate cancer incidence and mortality exist among different racial groups and the prevalence of prostate cancer in Asian countries is much lower than that observed in Western countries [53-56]. In our previous study, we have revealed that deletions of chromosomes 21 (causing TMPRSS2:ERG and consequently ERG overexpression) and 10q (inactivating PTEN) in Chinese patients are far less common than those reported in Western populations [53,57]. Magi-Galluzzi et al. confirmed the difference in ERG rearrangement frequency between Western and Asian patients [58]. We have also found that different ERG proteins are expressed in prostate cancer from UK or Chinese patients and that this difference was detected in prostate cancer precursor lesion, high-grade prostatic intraepithelial neoplasia [59]. PTEN is a well-characterized TSG, recurrently deleted as a result of chromosome rearrangements [60]. AKT pathway activation resulting from PTEN inactivity is a commonly detected phenomenon in prostate cancers from Western countries, particularly during the progression of cancer from localized tumors to metastatic cancer lesions [53]. However, the activation of the AKT pathway through the inactivation of PTEN may have limited contribution to Chinese prostate cancer [53]. While TMPRSS2:ERG gene fusion and PTEN loss were detected much less frequently in Chinese than Western prostate cancer, recently we also found a higher frequency of BRAF and RAF1 alterations in Chinese patients with prostate cancer than was observed in their Western counterparts [61]. All those observations suggest that UK and Chinese prostate cancer tumorigenesis may result from different genetic mechanisms and loss of function of NKAIN2 may contribute more significantly to prostate carcinogenesis in Chinese than the Western population. In fact, deletion of 6q was also more frequent in prostate cancers in Chinese than Western men, although 6q deletion occurred at a high frequency in the prostate cancer in the Western population.
Future directions
Clearly, there are limited studies on NKAIN2. It has been found to be involved in the nervous system developmental and genetic alterations affect its function lead to developmental diseases and mental disorders [28,35,42-44]. There are several lines of evidence suggest that NKAIN2 is potentially a TSG [25,26,40], but much more extensive investigations are required to confirm its tumor suppressor role. It is necessary to continue to study the chromosomal alterations of NKAIN2 gene and the functional consequence of those genomic alterations in prostate cancer and other human cancers. NKAIN2 is currently known as a transmembrane protein, which interacts with Na+/K+-ATPase. Apart from this, little is known of its cellular functions; therefore it is unclear how it may play the role to suppress tumor growth. Extensive biological study of the cellular function of NAKIN2 is urgently required, in particular its involvement in tumorigenesis pathways. Until the cellular and biological functions of NKAIN2 revealed, we will not be able to use it for the diagnosis and treatment of related diseases.
Acknowledgements
This work was supported by the Joint Research Fund for Overseas Chinese Scholars and Scholars in Hong Kong and Macao, the National Natural Science Foundation of China (81328017), Orchid and the Science and Technology Planning Project of Guangdong Province, China (2013B051000050, 2014A020212538).
Disclosure of conflict of interest
None.
References
- 1.Boström PJ, Bjartell AS, Catto JW, Eggener SE, Lilja H, Loeb S, Schalken J, Schlomm T, Cooperberg MR. Genomic Predictors of Outcome in Prostate Cancer. Eur Urol. 2015;68:1033–44. doi: 10.1016/j.eururo.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 2.Boyd LK, Mao X, Lu YJ. The complexity of prostate cancer: genomic alterations and heterogeneity. Nat Rev Urol. 2012;9:652–664. doi: 10.1038/nrurol.2012.185. [DOI] [PubMed] [Google Scholar]
- 3.Sun M, Srikantan V, Ma L, Li J, Zhang W, Petrovics G, Makarem M, Strovel JW, Horrigan SG, Augustus M, Sesterhenn IA, Moul JW, Chandrasekharappa S, Zou Z, Srivastava S. Characterization of frequently deleted 6q locus in prostate cancer. Dna Cell Biol. 2006;25:597–607. doi: 10.1089/dna.2006.25.597. [DOI] [PubMed] [Google Scholar]
- 4.Fabris VT. From chromosomal abnormalities to the identification of target genes in mouse models of breast cancer. Cancer Genet. 2014;207:233–246. doi: 10.1016/j.cancergen.2014.06.025. [DOI] [PubMed] [Google Scholar]
- 5.Capurso G, Festa S, Valente R, Piciucchi M, Panzuto F, Jensen RT, Delle Fave G. Molecular pathology and genetics of pancreatic endocrine tumours. J Mol Endocrinol. 2012;49:R37–R50. doi: 10.1530/JME-12-0069. [DOI] [PubMed] [Google Scholar]
- 6.Maher ER. Genomics and epigenomics of renal cell carcinoma. Semin Cancer Biol. 2013;23:10–17. doi: 10.1016/j.semcancer.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 7.Yen C, Liang S, Jong Y, Chen Y, Lin C, Chen Y, Wu Y, Su W, Huang CF, Tseng S, Whang-Peng J. Chromosomal aberrations of malignant pleural effusions of lung adenocarcinoma: Different cytogenetic changes are correlated with genders and smoking habits. Lung Cancer. 2007;57:292–301. doi: 10.1016/j.lungcan.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 8.van den Hurk K, Niessen HE, Veeck J, van den Oord JJ, van Steensel MA, Zur Hausen A, van Engeland M, Winnepenninckx VJ. Genetics and epigenetics of cutaneous malignant melanoma: A concert out of tune. Biochim Biophys Acta. 2012;1826:89–102. doi: 10.1016/j.bbcan.2012.03.011. [DOI] [PubMed] [Google Scholar]
- 9.Bellini MF, Silva AE, Varella-Garcia M. Genomic imbalances in esophageal squamous cell carcinoma identified by molecular cytogenetic techniques. Genet Mol Biol. 2010;33:205–213. doi: 10.1590/S1415-47572010005000028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borchers CH, Kast J, Foster LJ, Siu KW, Overall CM, Binkowski TA, Hildebrand WH, Scherer A, Mansoor M, Keown PA. The Human Proteome Organization Chromosome 6 Consortium: Integrating chromosome-centric and biology/disease driven strategies. J Proteomics. 2014;100:60–67. doi: 10.1016/j.jprot.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Honma K, Tsuzuki S, Nakagawa M, Tagawa H, Nakamura S, Morishima Y, Seto M. TNFAIP3/A20 functions as a novel tumor suppressor gene in several subtypes of non-Hodgkin lymphomas. Blood. 2009;114:2467–2475. doi: 10.1182/blood-2008-12-194852. [DOI] [PubMed] [Google Scholar]
- 12.Konishi N, Shimada K, Ishida E, Nakamura M. Molecular pathology of prostate cancer. Pathol Int. 2005;55:531–539. doi: 10.1111/j.1440-1827.2005.01865.x. [DOI] [PubMed] [Google Scholar]
- 13.Nakamura M, Kishi M, Sakaki T, Hashimoto H, Nakase H, Shimada K, Ishida E, Konishi N. Novel tumor suppressor loci on 6q22-23 in primary central nervous system lymphomas. Cancer Res. 2003;63:737–741. [PubMed] [Google Scholar]
- 14.Orphanos V, McGown G, Hey Y, Thorncroft M, Santibanez-Koref M, Russell SE, Hickey I, Atkinson RJ, Boyle JM. Allelic imbalance of chromosome 6q in ovarian tumours. Br J Cancer. 1995;71:666–9. doi: 10.1038/bjc.1995.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smeds J, Wärnberg F, Norberg T, Nordgren H, Holmberg L, Bergh J. Ductal carcinoma in situ of the breast with different histopathological grades and corresponding new breast tumour events: Analysis of loss of heterozygosity. Acta Oncol. 2005;44:41–49. doi: 10.1080/02841860410002842. [DOI] [PubMed] [Google Scholar]
- 16.Stark M, Hayward N. Genome-wide loss of heterozygosity and copy number analysis in melanoma using high-density single-nucleotide polymorphism arrays. Cancer Res. 2007;67:2632–2642. doi: 10.1158/0008-5472.CAN-06-4152. [DOI] [PubMed] [Google Scholar]
- 17.Chen M, Ye Y, Yang H, Tamboli P, Matin S, Tannir NM, Wood CG, Gu J, Wu X. Genome-wide profiling of chromosomal alterations in renal cell carcinoma using high-density single nucleotide polymorphism arrays. Int J Cancer. 2009;125:2342–2348. doi: 10.1002/ijc.24642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maximin S, Ganeshan DM, Shanbhogue AK, Dighe MK, Yeh MM, Kolokythas O, Bhargava P, Lalwani N. Current update on combined hepatocellular-cholangiocarcinoma. European Journal of Radiology Open. 2014;1:40–48. doi: 10.1016/j.ejro.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Namboodiripad PC. A review: Immunological markers for malignant salivary gland tumors. J Oral Biol Craniofac Res. 2014;4:127–134. doi: 10.1016/j.jobcr.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sunamura M, Takeda K, Matsuno S, Yatsuoka T, Motoi F, Duda DG, Kimura M, Abe T, Yokoyama T, Inoue H, Oonuma M. Gene therapy for pancreatic cancer based on genetic characterization of the disease. J Hepatobiliary Pancreat Surg. 2002;9:32–38. doi: 10.1007/s005340200002. [DOI] [PubMed] [Google Scholar]
- 21.Correa P, Juhlin C, Rastad J, Akerström G, Westin G, Carling T. Allelic loss in clinically and screening-detected primary hyperparathyroidism. Clin Endocrinol. 2002;56:113–117. doi: 10.1046/j.0300-0664.2001.01436.x. [DOI] [PubMed] [Google Scholar]
- 22.Roh YS, Song J, Seki E. TAK1 regulates hepatic cell survival and carcinogenesis. J Gastroenterol. 2014;49:185–194. doi: 10.1007/s00535-013-0931-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou Y, Zhang X, Klibanski A. Genetic and epigenetic mutations of tumor suppressive genes in sporadic pituitary adenoma. Mol Cell Endocrinol. 2014;386:16–33. doi: 10.1016/j.mce.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Visser S, Yang X. LATS tumor suppressor: A new governor of cellular homeostasis. Cell Cycle. 2010;9:3892–3903. doi: 10.4161/cc.9.19.13386. [DOI] [PubMed] [Google Scholar]
- 25.Mao X, Boyd LK, Yanez-Munoz RJ, Chaplin T, Xue L, Lin D, Shan L, Berney DM, Young BD, Lu Y. Chromosome rearrangement associated inactivation of tumour suppressor genes in prostate cancer. Am J Cancer Res. 2011;1:604–617. [PMC free article] [PubMed] [Google Scholar]
- 26.Tagawa H, Miura I, Suzuki R, Suzuki H, Hosokawa Y, Seto M. Molecular cytogenetic analysis of the breakpoint region at 6q21-22 in T-cell lymphoma/leukemia cell lines. Genes Chromosomes Cancer. 2002;34:175–185. doi: 10.1002/gcc.10057. [DOI] [PubMed] [Google Scholar]
- 27.Gorokhova S, Bibert S, Geering K, Heintz N. A novel family of transmembrane proteins interacting with subunits of the Na, K-ATPase. Hum Mol Genet. 2007;16:2394–2410. doi: 10.1093/hmg/ddm167. [DOI] [PubMed] [Google Scholar]
- 28.Bocciardi R, Giorda R, Marigo V, Zordan P, Montanaro D, Gimelli S, Seri M, Lerone M, Ravazzolo R, Gimelli G. Molecular characterization of a t(2;6) balanced translocation that is associated with a complex phenotype and leads to truncation of the. Hum Mutat. 2005;26:426–436. doi: 10.1002/humu.20235. [DOI] [PubMed] [Google Scholar]
- 29.Barnier JV, Papin C, Eychène A, Lecoq O, Calothy G. The mouse B-raf gene encodes multiple protein isoforms with tissue-specific expression. J Biol Chem. 1995;270:23381–23389. doi: 10.1074/jbc.270.40.23381. [DOI] [PubMed] [Google Scholar]
- 30.Hindley A, Kolch W. Extracellular signal regulated kinase (ERK)/mitogen activated protein kinase (MAPK)-independent functions of Raf kinases. J Cell Sci. 2002;115:1575–1581. doi: 10.1242/jcs.115.8.1575. [DOI] [PubMed] [Google Scholar]
- 31.Mercer KE, Pritchard CA. Raf proteins and cancer: B-Raf is identified as a mutational target. Biochim Biophys Acta. 2003;1653:25–40. doi: 10.1016/s0304-419x(03)00016-7. [DOI] [PubMed] [Google Scholar]
- 32.Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5:875–885. doi: 10.1038/nrm1498. [DOI] [PubMed] [Google Scholar]
- 33.Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer. 2007;7:233–245. doi: 10.1038/nrc2091. [DOI] [PubMed] [Google Scholar]
- 34.Zhang C, Leibowitz ML, Pellman D. Chromothripsis and beyond: rapid genome evolution from complex chromosomal rearrangements. Gene Dev. 2013;27:2513–2530. doi: 10.1101/gad.229559.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yue Y, Stout K, Grossmann B, Zechner U, Brinckmann A, White C, Pilz DT, Haaf T. Disruption of TCBA1 associated with a de novo t(1;6)(q32.2;q22.3) presenting in a child with developmental delay and recurrent infections. J Med Genet. 2006;43:143–147. doi: 10.1136/jmg.2004.029660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alberti L, Carniti C, Miranda C, Roccato E, Pierotti MA. RET and NTRK1 proto-oncogenes in human diseases. J Cell Physiol. 2003;195:168–186. doi: 10.1002/jcp.10252. [DOI] [PubMed] [Google Scholar]
- 37.Bystritskiy AA, Razin SV. Breakpoint clusters: Reason or consequence? Crit Rev Eukaryot Gene Expr. 2004;14:65–77. [PubMed] [Google Scholar]
- 38.Nowell PC. A minute chromosome in human chronic granulocytic leukemia. Science. 1960;132:1497. [Google Scholar]
- 39.Qi M, Li Y, Liu J, Yang X, Wang L, Zhou Z, Han B. Morphologic features of carcinomas with recurrent gene fusions. Adv Anat Pathol. 2012;19:417. doi: 10.1097/PAP.0b013e318273baae. [DOI] [PubMed] [Google Scholar]
- 40.Sircar K, Huang H, Hu L, Cogdell D, Dhillon J, Tzelepi V, Efstathiou E, Koumakpayi IH, Saad F, Luo D, Bismar TA, Aparicio A, Troncoso P, Navone N, Zhang W. Integrative Molecular Profiling Reveals Asparagine Synthetase Is a Target in Castration-Resistant Prostate Cancer. Am J Pathol. 2012;180:895–903. doi: 10.1016/j.ajpath.2011.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Romania P, Castellano A, Surace C, Citti A, De Ioris MA, Sirleto P, De Mariano M, Longo L, Boldrini R, Angioni A, Locatelli F, Fruci D. High-Resolution Array CGH Profiling Identifies Na/K Transporting ATPase Interacting 2 (NKAIN2) as a Predisposing Candidate Gene in Neuroblastoma. PLoS One. 2013;8:e78481. doi: 10.1371/journal.pone.0078481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Calboli FCF, Tozzi F, Galwey NW, Antoniades A, Mooser V, Preisig M, Vollenweider P, Waterworth D, Waeber G, Johnson MR, Muglia P, Balding DJ. A Genome-Wide Association Study of Neuroticism in a Population-Based Sample. PLoS One. 2010;5:e11504. doi: 10.1371/journal.pone.0011504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang K, Liu X, Aragam N, Jian X, Mullersman JE, Liu Y, Pan Y. Family-based association analysis of alcohol dependence in the COGA sample and replication in the Australian twin-family study. J Neural Transm. 2011;118:1293–1299. doi: 10.1007/s00702-011-0628-3. [DOI] [PubMed] [Google Scholar]
- 44.Lind PA, Macgregor S, Vink JM, Pergadia ML, Hansell NK, de Moor MH, Smit AB, Hottenga JJ, Richter MM, Heath AC, Martin NG, Willemsen G, de Geus EJ, Vogelzangs N, Penninx BW, Whitfield JB, Montgomery GW, Boomsma DI, Madden PA. A Genomewide Association Study of Nicotine and Alcohol Dependence in Australian and Dutch Populations. Twin Res Hum Genet. 2010;13:10–29. doi: 10.1375/twin.13.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Sivachenko AY, Sboner A, Esgueva R, Pflueger D, Sougnez C, Onofrio R, Carter SL, Park K, Habegger L, Ambrogio L, Fennell T, Parkin M, Saksena G, Voet D, Ramos AH, Pugh TJ, Wilkinson J, Fisher S, Winckler W, Mahan S, Ardlie K, Baldwin J, Simons JW, Kitabayashi N, MacDonald TY, Kantoff PW, Chin L, Gabriel SB, Gerstein MB, Golub TR, Meyerson M, Tewari A, Lander ES, Getz G, Rubin MA, Garraway LA. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–220. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barbieri CE, Bangma CH, Bjartell A, Catto JWF, Culig Z, Grönberg H, Luo J, Visakorpi T, Rubin MA. The Mutational Landscape of Prostate Cancer. Eur Urol. 2013;64:567–576. doi: 10.1016/j.eururo.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mao X, James SY, Yanez-Munoz RJ, Chaplin T, Molloy G, Oliver RTD, Young BD, Lu Y. Rapid high-resolution karyotyping with precise identification of chromosome breakpoints. Genes Chromosomes Cancer. 2007;46:675–683. doi: 10.1002/gcc.20452. [DOI] [PubMed] [Google Scholar]
- 48.van Bokhoven A, Caires A, Maria MD, Schulte AP, Lucia MS, Nordeen SK, Miller GJ, Varella-Garcia M. Spectral karyotype (SKY) analysis of human prostate carcinoma cell lines. Prostate. 2003;57:226–244. doi: 10.1002/pros.10291. [DOI] [PubMed] [Google Scholar]
- 49.Maher CA, Kumar-Sinha C, Cao X, Kalyana-Sundaram S, Han B, Jing X, Sam L, Barrette T, Palanisamy N, Chinnaiyan AM. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009;458:97–99. doi: 10.1038/nature07638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Esgueva R, Perner S, LaFargue CJ, Scheble V, Stephan C, Lein M, Fritzsche FR, Dietel M, Kristiansen G, Rubin MA. Prevalence of TMPRSS2-ERG and SLC45A3-ERG gene fusions in a large prostatectomy cohort. Mod Pathol. 2010;23:539–546. doi: 10.1038/modpathol.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pflueger D, Rickman DS, Sboner A, Perner S, LaFargue CJ, Svensson MA, Moss BJ, Kitabayashi N, Pan Y, de la Taille A, Kuefer R, Tewari AK, Demichelis F, Chee MS, Gerstein MB, Rubin MA. N-myc Downstream Regulated Gene 1 (NDRG1) Is Fused to ERG in Prostate Cancer. Neoplasia. 2009;11:804–11. doi: 10.1593/neo.09572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pflueger D, Terry S, Sboner A, Habegger L, Esgueva R, Lin P, Svensson MA, Kitabayashi N, Moss BJ, MacDonald TY, Cao X, Barrette T, Tewari AK, Chee MS, Chinnaiyan AM, Rickman DS, Demichelis F, Gerstein MB, Rubin MA. Discovery of non-ETS gene fusions in human prostate cancer using next-generation RNA sequencing. Genome Res. 2011;21:56–67. doi: 10.1101/gr.110684.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mao X, Yu Y, Boyd LK, Ren G, Lin D, Chaplin T, Kudahetti SC, Stankiewicz E, Xue L, Beltran L, Gupta M, Oliver RTD, Lemoine NR, Berney DM, Young BD, Lu Y. Distinct Genomic Alterations in Prostate Cancers in Chinese and Western Populations Suggest Alternative Pathways of Prostate Carcinogenesis. Cancer Res. 2010;70:5207–5212. doi: 10.1158/0008-5472.CAN-09-4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mahal BA, Aizer AA, Ziehr DR, Hyatt AS, Choueiri TK, Hu JC, Hoffman KE, Sweeney CJ, Beard CJ, D’Amico AV, Martin NE, Kim SP, Quoc-Dien T, Nguyen PL. Racial Disparities in Prostate Cancere-Specific Mortality in Men With Low-Risk Prostate Cancer. Clin Genitourin Cancer. 2014;12:E189–E195. doi: 10.1016/j.clgc.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 55.Sim HG, Cheng C. Changing demography of prostate cancer in Asia. Eur J Cancer. 2005;41:834–845. doi: 10.1016/j.ejca.2004.12.033. [DOI] [PubMed] [Google Scholar]
- 56.Gronberg H. Prostate cancer epidemiology. Lancet. 2003;361:859–864. doi: 10.1016/S0140-6736(03)12713-4. [DOI] [PubMed] [Google Scholar]
- 57.Mani RS, Chinnaiyan AM. Triggers for genomic rearrangements: insights into genomic, cellular and environmental influences. Nat Rev Genet. 2010;11:819–829. doi: 10.1038/nrg2883. [DOI] [PubMed] [Google Scholar]
- 58.Magi-Galluzzi C, Tsusuki T, Elson P, Simmerman K, LaFargue C, Esgueva R, Klein E, Rubin MA, Zhou M. TMPRSS2-ERG Gene Fusion Prevalence and Class Are Significantly Different in Prostate Cancer of Caucasian, African-American and Japanese Patients. Prostate. 2011;71:489–497. doi: 10.1002/pros.21265. [DOI] [PubMed] [Google Scholar]
- 59.Xue LY, Mao XY, Ren GP, Stankiewicz E, Kudahetti SC, Lin DM, Beltran L, Berney DM, Lu YJ. Chinese and Western prostate cancers show alternate pathogenetic pathways in association with ERG status. Am J Cancer Res. 2012;2:736–744. [PMC free article] [PubMed] [Google Scholar]
- 60.Hollander MC, Blumenthal GM, Dennis PA. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat Rev Cancer. 2011;11:289–301. doi: 10.1038/nrc3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ren G, Liu X, Mao X, Zhang Y, Stankiewicz E, Hylands L, Song R, Berney DM, Clark J, Cooper C, Lu Y. Identification of frequent BRAF copy number gain and alterations of RAF genes in chinese prostate cancer. Genes Chromosomes Cancer. 2012;51:1014–1023. doi: 10.1002/gcc.21984. [DOI] [PubMed] [Google Scholar]