

Abstract
Pancreatic cancer is a kind of devastating disease with a high mortality rate. Fentanyl has been widely applied to anesthesia and analgesia in pancreatic cancer therapy, and is also demonstrated to inhibit the growth of some kinds of cancer cells in existed studies. To investigate the functions of fentanyl in pancreatic cancer, we conducted a series of in vivo and in vitro experiments using human pancreatic cancer cells SW1990 and fentanyl treatment. The cells were transplanted to BALB/c nude mice to generate pancreatic tumor for monitoring tumor growth. Viability, apoptosis, migration and invasion, and cell cycle of SW1990 cells were also analyzed. To reveal the functional mechanisms of fentanyl, the expression changes of factors in these cellular activities were detected. Results showed a significant inhibition of pancreatic tumor growth in the fentanyl-treated group. Fentanyl also inhibited viability of SW1990 cells in vitro. Detailed results showed fentanyl led to promoted cell apoptosis via arresting cells in G0/G1 phase. It also suppressed cell migration and invasion. Further proofs indicated that the factors related to cell apoptosis (Bcl-2, p53 and Caspase-3), cell cycle (p21, Cyclin D1 and CDK4) and epithelial-mesenchymal transition (E-cadherin, Vimentin and α-SMA) showed the corresponding expression changes. Fentanyl might execute its functions via the suppressed MAPK pathways, since the key factors, p38, ERK1/2 and JNK were all down-regulated by fentanyl. This study indicated fentanyl could inhibit viability and growth of pancreatic cancer cells, providing a possible strategy for pancreatic cancer treatment.
Keywords: Pancreatic cancer, fentanyl, tumor growth, cell viability, cell apoptosis
Introduction
Pancreatic cancer is the most aggressive cancer with a five-year survival rate of less than 5% [1]. Pancreatic cancer is hard to be diagnosed in early stages, and specific symptoms usually do not arise until the disease has reached advanced stages [2], when it has spread to other organs . This is a main reason generating the poor survival rate of pancreatic cancer. Pancreatic cancer resection, chemotherapy and chemoradiation are frequently-used treatment methods [3,4]. However, postoperative recurrences and metastatic diseases are still major problems causing the high death rates in pancreatic cancer patients [5,6]. Some anesthetics, propofol for example, have been reported to induce the immunosuppression of macrophages [7] and inhibit the proliferation of T cells [8], thus leading to postoperative tumor growth and metastasis.
Fentanyl is a kind of synthetic opioid that has been widely used for anesthesia and analgesia. Recent studies have shown its abilities to affect growth of many kinds of cancer cells. For instance, it inhibits human gastric cell growth, promotes cell apoptosis, and leads to G2/M arrest [9]. It also promotes the apoptosis of colorectal carcinoma cells [10]. In bone cancer, it reduces bone pain symptoms as well as the cancer cell-induced bone lesions [11]. Though fentanyl is a familiar anesthetic in operations and analgesic therapy of pancreatic cancer [12], its impacts on proliferation and apoptosis of pancreatic cancer cells remain unclear.
To investigate the roles of fentanyl in regulating pancreatic cancer cells and reveal the potential mechanisms, both in vivo and in vitro experiments were performed in this study on pancreatic cancer cells under fentanyl treatment of different dosages. The human pancreatic cancer cells SW1990 were transplanted to BALB/c nude mice to generate pancreatic tumor and analyze the impacts of fentanyl on tumor growth. SW1990 cells were also used for analyses of cell viability, apoptosis, migration and invasion, and expression changes of related factors, and factors in mitogen-activated protein kinase (MAPK) pathways. These results will uncover new roles of fentanyl in regulating tumor cells, and provide possible strategies for pancreatic cancer treatment.
Materials and methods
Xenograft in mice
Fifty specific pathogen-free (SPF) grade BALB/c nude mice of 4-week-old were purchased from Vital River Laboratories (Beijing, China). The human pancreatic cancer cells SW1990 (Goybio, Shanghai, China) of 5×106 were suspended in 100 μL phosphate buffer saline (PBS) and subcutaneously injected into the flanks of mice. On the fifth day after inoculation, the 24 mice were randomly groups into four group (12 individuals in each group), and injected into the tumor with fentanyl (Humanwell, Yichang, China) of 0 mg/kg, 0.05 mg/kg, 0.1 mg/kg and 0.2 mg/kg, respectively. The fentanyl injection was conducted every other day and lasted for 3 weeks. The mice were sacrificed for tumor sampling at 5 d, 10 d, 14 d and 21 d post fentanyl injection. The tumors were weighted at 21 d post fentanyl injection, and the tumor volume was estimated at the four sampling points by (π/6) (L×W2), in which L was the length of tumors and W was the width of tumors [13]. All experiments with animals were performed according to the instructions of our institute and approved by a local committee for ethics.
Cell culture
The human pancreatic cancer cell line SW1990 was cultured in Roswell Park Memorial Institute (RPMI)-1640 medium (Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS, Gibco) and 1×105 U/L penicillin-streptomycin (Gibco), and incubated in humidified atmosphere with 5% CO2 at 37°C. The medium was changed every 24 h. Cells were passaged when the confluence reached 70%.
Cell viability assay
Cell viability was detected by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay using MTT Cell Proliferation and Cytotoxicity Assay Kit (Beyotime, Shanghai, China) according to the manuals. Cells of 2×103 in 100 μL medium were transferred to each well of 96-well plates. Fentanyl was added at the concentration of 0 ng/mL, 0.5 ng/mL, 2 ng/mL and 5 ng/mL, respectively. Then 10 μL MTT solution (5 mg/mL) was added and the cells were cultured for 4 h. After adding 100 μL Formanzan solution, the cells were incubated for another 4 h with shakes. The absorbance at 570 nm was detected using a multifunctional microplate reader SpectraMax M5 (Molecular Devices, Silicon Valley, CA, USA) at 24 h, 48 h and 72 h post fentanyl treatment.
Cell apoptosis assay
Cells treated with different dosages of fentanyl for 48 h was labeled with fluorescein isothiocyanate (FITC) and propidium iodide (PI) using Annexin V-FITC Apoptosis Detection Kit I (Univ-bio, Shanghai, China) according to the manuals. Cells were digested by trypsin (Gibco) and washed three times using ice-cold PBS. Then 300 μL 1× Binding Buffer and 5 μL Annexin V-FITC was added to the collected cells. The cells were incubated for 15 min in dark at room temperature. After the incubation, 5 μL PI and 200 μL 1× Binding Buffer were added to the cells, followed by an immediate detection using BD FACSCanto II flow cytometry (BD Biosciences, San Jose, CA, USA).
Cell cycle analysis
The cells were seeded in 24-well plates to the concentration of 1×106 cell/mL. After 48 h of fentanyl treatment, the cells were digested by trypsin (Gibco), centrifuged and collected. The cells were resuspended and washed using ice-cold PBS for two times and fixed in ice-cold 75% alcohol for 4 h at 4°C. After washed with PBS for three times, the cells were incubated in medium with 100 μg/mL Ribonuclease A (Sigma-Aldrich, Shanghai, China) and 50 μg/mL PI (Sigma-Aldrich) for 30 min at 4°C. Then cells were filtered, detected with BD FACSCanto II flow cytometry, and analyzed by Mod Fit LT software (Verity Software House).
Wound-healing assay
Cells treated with fentanyl for 48 h were seeded in 6-well plates with 7×105 cells in each well. After serum-starved for 24 h, the cells were cultured in medium with 10% FBS. A wound was made by a plastic tip sweeping across the monolayer cells. At 0 h and 24 h after the wound was made, the cells were photographed and the wound width was analyzed using Image-Pro Plus program (Media Cybernetics). The covered area of migrated cells (%) were estimated by ((wound area at 0 h-wound area at 24 h)/wound area at 0 h) ×100%.
Transwell analysis
Cell migration and invasion assay was conducted using transwell Millicell Standing Cell Culture 24 well CM (Millipore, Billerica, MA, USA). For cell invasion assay, the upper chamber was pre-coated with diluted Matrigel (50%, BD Biosciences), and incubated at 37°C for gel formation. Cells (1×105) treated with fentanyl for 48 h were seeded to the upper chamber. Culture medium (500 μL) supplemented with 20% FBS was added to the lower chamber. After 24 h of incubation, the cells in the upper chamber were carefully removed and the cells passed through the gel and membrane were stained with 0.1% crystal violet (Sigma-Aldrich). Three visual fields for each well were randomly selected under a microscope to count cells. For cell migration assay, similar procedures were performed but without the Matrigel.
Real-time quantitative PCR (qRT-PCR)
qRT-PCR was conducted to analyze the mRNA expression levels. Total RNAs of cells treated with fentanyl for 48 h were isolated using TRIzol (Invitrogen, Calsbad, CA, USA). DNA contamination was removed by DNase I (Invitrogen) and the quantity and quality of RNA samples were detected using NanoDrop 2000 (Thermo Scientific, Carlbad, CA, USA). The complementary cDNAs were synthesized from 1 μg RNAs using PrimeScript 1st Strand cDNA Synthesis Kit (TaKaRa, Dalian, China). In each reaction system, 20 ng cDNA templates, the corresponding specific primers (Table 1), and SYBR Green I Master (Roche) were included. The reactions were conducted on LightCycler 480 (Roche). GAPDH was used as the internal reference. The data were calculated with the 2-ΔΔCt method.
Table 1.
Primers used in qRT-PCR.
| Primer | Sequence (5’ to 3’) |
|---|---|
| GAPDH-Fw | GAAGGTGAAGGTCGGAGTCAAC |
| GAPDH-Rv | CAGAGTTAAAAGCAGCCCTGGT |
| E-cadherin-Fw | GAACTCAGCCAAGTGTAAAAGCC |
| E-cadherin-Rv | GAGTCTGAACTGACTTCCGC |
| Vimentin-Fw | AAAGTGTGGCTGCCAAGAAC |
| Vimentin-Rv | AGCCTCAGAGAGGTCAGCAA |
| α-SMA-Fw | GGCCGAGATCTCACTGACTAC |
| α-SMA-Rv | TTCATGGATGCCAGCAGA |
| p38-Fw | GAGAACTGCGGTTACTTA |
| p38-Rv | ATGGGTCACCAGATACACAT |
| ERK1/2-Fw | CTCAAGCCTTCCAACCTC |
| ERK1/2-Rv | TTCCACGGCACCTTATTT |
| JNK-Fw | AGTGACAGTAAAAGCGATGG |
| JNK-Rv | TTTAGGAGGACAAGTTCACG |
Western blot
Western blot was performed to detect the protein expression levels. Cells treated with fentanyl for 48 h were lysed in 0.2 mL lysis buffer (0.15 M NaCl, 50 mM Tris-HCl (pH 7.5), 2 mM ethylene diamine tetraacetic acid, 0.5% Triton-100, 5 mM dithiothreitol, 0.2 mM phenylmethanesulfonyl fluoride and 2 mg/mL apoptinin). Cell protein samples of 20 μg were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and transferred to polyvinylidene difluoride membranes (Roche). The blot was blocked in 5% skim milk for 2 h at room temperature and incubated in primary antibodies (Thermo Scientific) overnight at 4°C. After washed three times using Tris-buffered saline Tween-20 (TBST), the blot was incubated in horseradish peroxidase-conjugated secondary antibody (Thermo Scientific) for 2 h. Then the signals were visualized using ECL plus Western Blotting Substrate (Thermo Scientific) and photographed. GAPDH was used as the internal reference.
Statistical analysis
All experiments were performed in triplicate. Data were presented as means ± standard deviation. Analysis of variance or Student’s t-test was conducted by Statistical Product and Service Solutions 19 (IBM, New York, USA). Differences were considered significant if P < 0.05.
Results
Fentanyl inhibits cell viability and tumor growth of pancreatic cancer
In fentanyl-treated human pancreatic cancer cell line SW1990, we observed the decreased cell viability along with the increasing fentanyl dosages (Figure 1A). At each sampling point, the cell viability was significantly inhibited by fentanyl compared to the cells without fentanyl treatment (P < 0.05), and the viability inhibition function of fentanyl was dose-dependent. It seemed that fentanyl was capable of suppress the viability of pancreatic cancer cells. To further testify its functions in regulating tumor growth, we conducted in vivo experiments by transplanting human pancreatic cancer cells into mice for tumor generation, and injecting fentanyl of different dosages. Results showed that as the tumor grew, the tumor size was reduced with fentanyl injection (Figure 1B), which was even obvious at later sampling points. The tumor weight measured at 21 d post treatment was also reduced by fentanyl, which was dose-dependent (Figure 1C). Significant differences were detected when the dosage of injected fentanyl reached 0.10 mg/kg (P < 0.05) and 0.20 mg/kg (P < 0.01). These in vitro and in vivo results indicated fentanyl could inhibit the viability of pancreatic cancer cells, and limit the growth of tumors.
Figure 1.
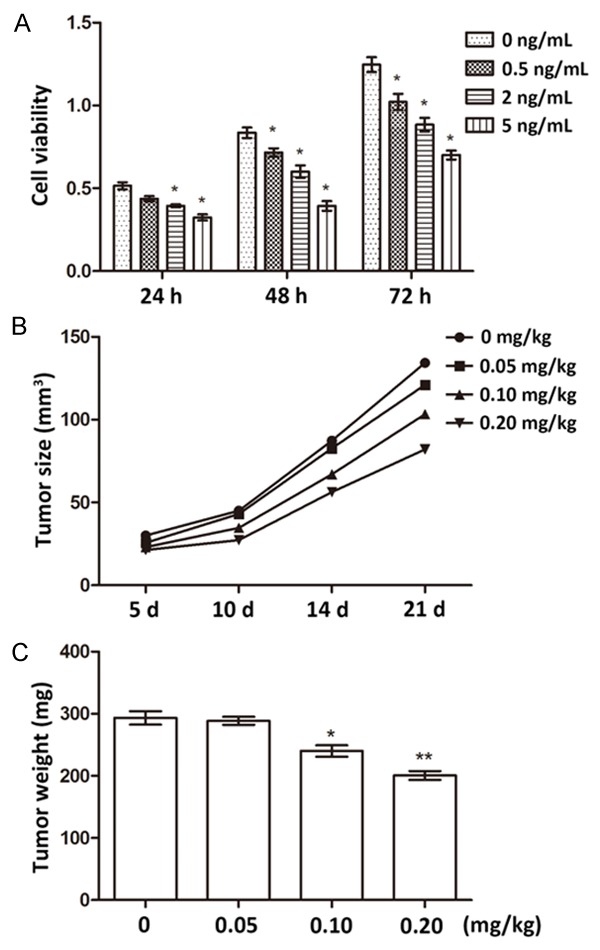
Change of cell viability and tumor growth after fentanyl treatment. A. Viability changes of human pancreatic cancer cells after fentanyl treatment of different dosages (0, 0.5, 2, and 5 ng/mL). Viability is detected at 24, 48, and 72 h post fentanyl treatment. B. Tumor sizes changes after the mice are injected with fentanyl of difference dosages (0, 0.05, 0.10, and 0.20 mg/kg). Samples are taken at 5, 10, 14, and 21 d post fentanyl injection. C. Tumor weight changes after the mice are injected with fentanyl of difference dosages (0, 0.05, 0.10, and 0.20 mg/kg). Samples were taken at 21 d post fentanyl injection. *, significant differences between groups (P < 0.05). **, extremely significant differences between groups (P < 0.01).
Fentanyl induces cell apoptosis and G0/G1 arrest
To reveal the regulatory mechanisms of fentanyl in pancreatic cancer cell growth, we first analyzed the impacts of fentanyl on cell apoptosis and cell cycle. When the cultured human pancreatic cancer cells were treated with fentanyl, the percent of apoptotic cells increased significantly along with the increasing fentanyl dosages (P < 0.05 or P < 0.01, Figure 2A and 2B). Cell cycle analyses showed that with the increase of fentanyl dosages, the percent of cells in G0/G1 phase gradually increased, and the percent of cells in S and G2 phases decreased (Figure 2C and 2D), which implied most of the pancreatic cancer cells were arrested in G0/G1 phase.
Figure 2.
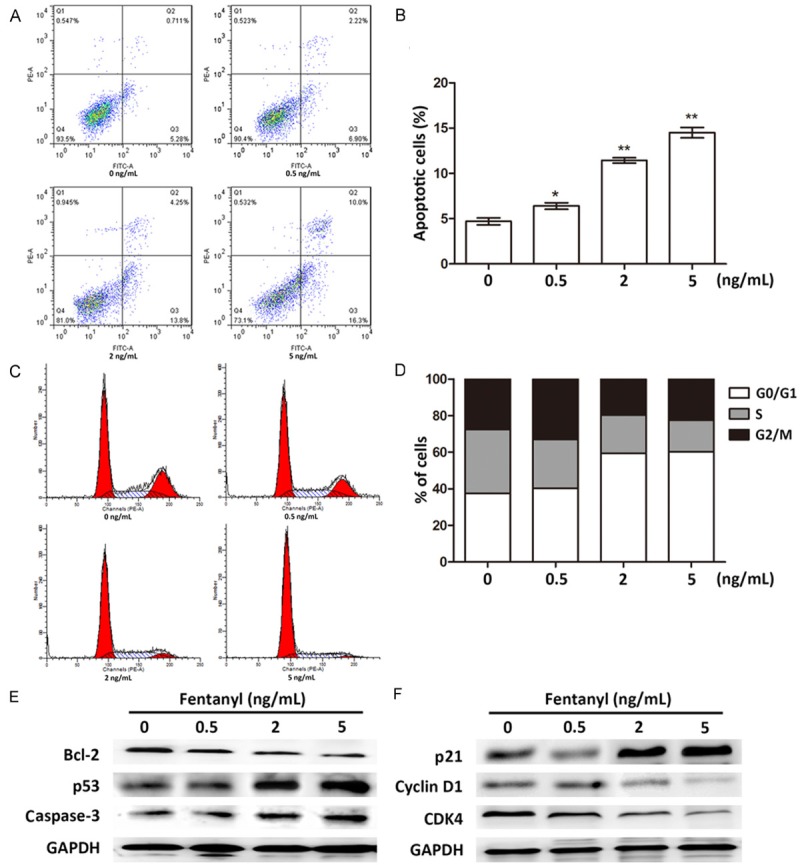
Change of cell apoptosis and cell cycle after fentanyl treatment. A. Flow cytometry results showing cell apoptosis under fentanyl dosage of 0, 0.5, 2, and 5 ng/mL. B. Histogram of the apoptotic cell percent. *, significant differences between groups (P < 0.05). **, extremely significant differences between groups (P < 0.01). C. Flow cytometry results showing the cell cycle distribution under fentanyl dosage of 0, 0.5, 2, and 5 ng/mL. The three peaks indicate cells in G1, S and G2 phases, respectively. D. Histogram showing the percent of cells in different phases. E. Protein expression of apoptosis-related factors in cells treated with fentanyl of 0, 0.5, 2, and 5 ng/mL. Bcl-2, B-Cell CLL/lymphoma 2. F. Protein expression of cell cycle-related factors in cells treated with fentanyl of 0, 0.5, 2, and 5 ng/mL. p21, cyclin-dependent kinase inhibitor 1A. CDK4, cyclin-dependent kinase 4. GAPDH is used as the internal control of western blot.
Then we analyzed the factors functioning in cell apoptosis and cell cycle. B-Cell CLL/lymphoma 2 (Bcl-2), tumor suppressor p53 and cysteine-aspartic acid protease-3 (Caspase-3), three cell apoptosis-related factors, were all influenced by fentanyl (Figure 2E). Specifically, Bcl-2 expression was inhibited, while p53 and Caspase-3 expression were promoted by fentanyl, which agreed with the anti-apoptosis roles of Bcl-2 [14] and the pro-apoptosis roles of p53 and Caspase-3 [15,16], indicating fentanyl promoted apoptosis of pancreatic cancer cells. Previous studies proved that up-regulation of cyclin-dependent kinase inhibitor 1A (p21) and down-regulation of Cyclin D1 and cyclin-dependent kinase 4 (CDK4) were related to G1 arrest [17-19]. So we chose these three cell cycle-related factors to verify the influence of fentanyl on cell cycle. Results showed that with the increase of fentanyl dosages, p21 expression was up-regulated, and Cyclin D1 and CDK4 expression was down-regulated (Figure 2F), implying the fentanyl-treated pancreatic cancer cells were arrested in G0/G1 phase. Taken together, fentanyl could affect the expression of apoptosis and cell cycle-related factors, thus promoting apoptosis of pancreatic cancer cells and leading to the cells arrested at G0/G1 phase.
Fentanyl inhibits cell migration and invasion
We next detected the influences of fentanyl on migration and invasion of pancreatic cancer cells. Wound-healing assay indicated that at 24 h after the wound was made, migration of cells treated with 2 ng/mL or 5 ng/mL fentanyl were significantly suppressed (P < 0.01, Figure 3A and 3B). Similar results were found in the transwell assay without using Matrigel, which showed significant decreases in migration of cells treated with 2 ng/mL or 5 ng/mL fentanyl (P < 0.01, Figure 3C). Cell invasion assay using transwell revealed that the numbers of invasion cells were reduced in cells treated with 2 ng/mL or 5 ng/mL fentanyl (P < 0.01, Figure 3D and 3E). So fentanyl was capable of inhibiting migration and invasion of pancreatic cancer cells.
Figure 3.
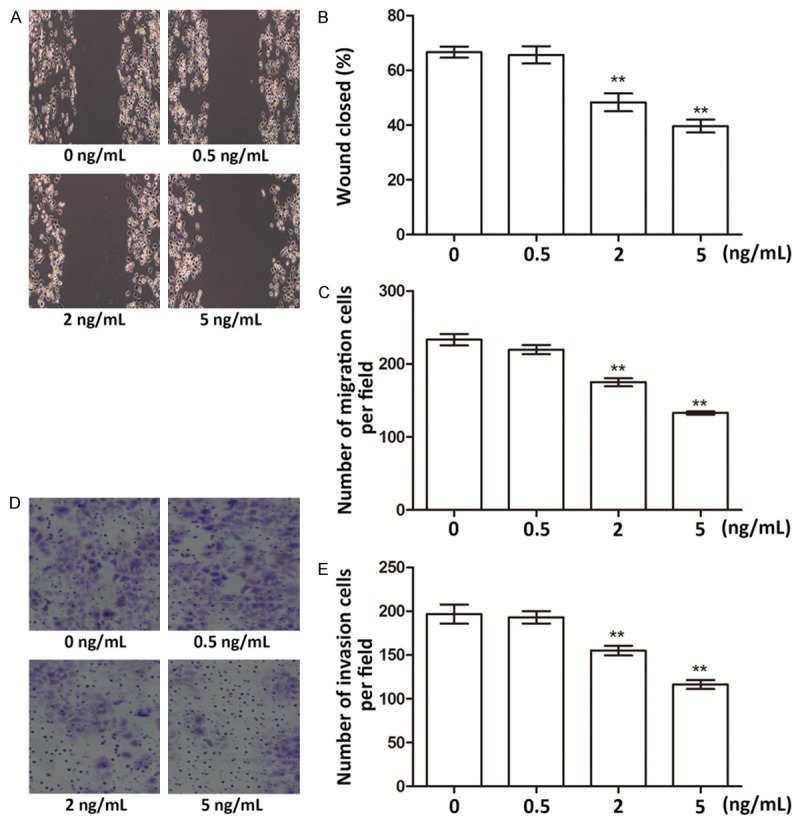
Migration and invasion assays of cells treated with fentanyl of 0, 0.5, 2, and 5 ng/mL. A. Wound-healing photographs of fentanyl-treated cells. B. Histogram showing percent of closed areas. C. Number of migration cells detected by transwell assay. D. Photograph of invasion cells detected by transwell assay. E. Number of invasion cells in the transwell assay. *, significant differences between groups (P < 0.05). **, extremely significant differences between groups (P < 0.01).
We supposed its functions were related to regulating epithelial-mesenchymal transition (EMT) since numerous studies have found various factors regulate cell migration and invasion via EMT [20,21]. So we analyzed the expression changes of some EMT markers, including E-cadherin, Vimentin and α-smooth muscle actin (α-SMA). Results showed the E-cadherin mRNA level was significantly up-regulated (Figure 4A), and the Vimentin and α-SMA mRNA levels were significantly down-regulated (P < 0.01, Figure 4B and 4C), all dependent on the increasing fentanyl dosages. The protein level changes of the three factors were in accordance with their mRNA levels, which were also dose-dependent (Figure 4D). Since studies have proved down-regulated E-cadherin and up-regulated Vimentin and α-SMA were markers of aberrant EMT [22-24]. So their expression changes detected in this study indicated EMT was inhibited in the fentanyl-treated cells. Taken together, fentanyl was capable of inhibiting migration and invasion of pancreatic cancer cells, possibly via suppressing EMT.
Figure 4.
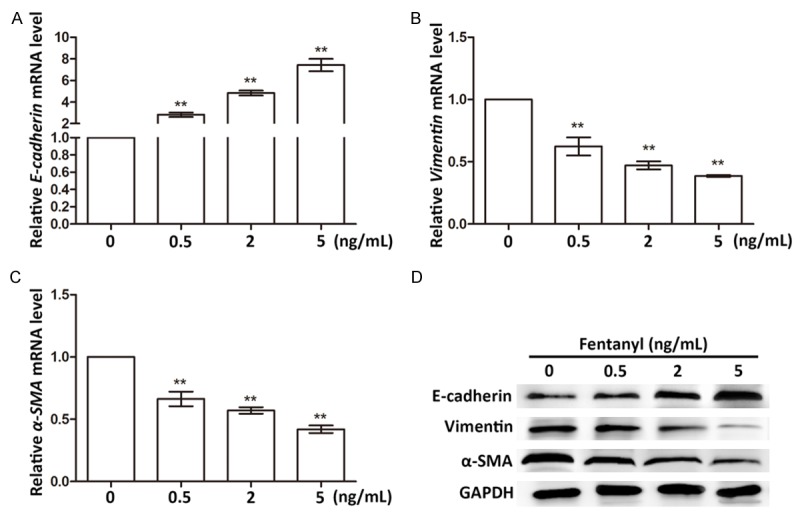
Expression changes of EMT markers in cells treated with fentanyl of 0, 0.5, 2, and 5 ng/mL. A. E-cadherin mRNA levels in different groups. B. Vimentin mRNA levels in different groups. C. α-SMA mRNA levels in different groups. **, extremely significant differences between groups (P < 0.01). D. Protein expression levels of EMT markers in different groups. GAPDH is the internal control in western blot. α-SMA, α-smooth muscle actin.
Fentanyl suppresses MAPK pathways
MAPK pathways are involved in cell growth, proliferation and apoptosis and stress response [25-27]. So we analyzed the influences of fentanyl on MAPK pathways by detecting the expression changes of representative factors of MAPK pathways, namely, p38 (alias MAPK14), extracellular signal-regulated kinase 1/2 (ERK1/2, alias MAPK3/1) and c-Jun N-terminal kinase (JNK, alias MAPK8). Results showed the mRNA expressions of these three factors were all significantly down-regulated in fentanyl-treated cells, exhibiting a dose-dependent pattern (P < 0.05 or P < 0.01, Figure 5A-C). Similarly, the protein expression levels of the three factors were all inhibited when the dosage of fentanyl was over 2 ng/mL (Figure 5D). But it seemed that when the fentanyl dosage was 0.5 ng/mL, all the three factors were slightly up-regulated. So the impacts of fentanyl on MAPK pathways might be dose-dependent. Generally, it could be deduced from these results that higher dosages of fentanyl (over 2 ng/mL) inhibited MAPK pathways, which might result in the suppressed viability and promoted apoptosis of pancreatic cancer cells.
Figure 5.
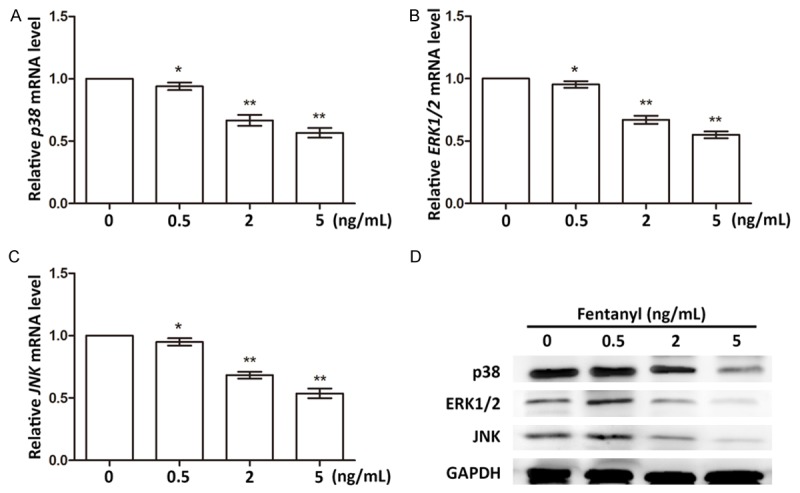
Expression changes of factors in MAPK pathways in cells treated with fentanyl of 0, 0.5, 2, and 5 ng/mL. A. p38 mRNA levels in different groups. B. ERK1/2 mRNA levels in different groups. C. JNK mRNA levels in different groups. *, significant differences between groups (P < 0.05). **, extremely significant differences between groups (P < 0.01). D. Protein expression levels of factors in MAPK pathways. GAPDH is the internal control in western blot. ERK1/2, extracellular signal-regulated kinase 1/2. JNK, c-Jun N-terminal kinase.
Discussion
As a frequently used anesthetic in operation and cancer treatments, fentanyl may influence the viability and apoptosis of cancer cells, like the reported roles of propofol [7,8]. In this study, we investigate the possible roles of fentanyl in regulating pancreatic cancer cells. In vivo and in vitro experiments indicate fentanyl is a potent inhibitor of pancreatic cancer cell viability and tumor growth. Further mechanism studies reveal that fentanyl can induce apoptosis and G0/G1 arrest, and suppress migration and invasion of pancreatic cancer cells, possibly via regulating factors related to apoptosis, cell cycle and EMT. Fentanyl is also a suppressor of MAPK pathways, which may help it to execute the anti-tumor functions.
Fentanyl was proved to influence various factors involved in cell apoptosis, cell cycle, EMT and MAPK pathway in this study, which was to be expected since previous studies have revealed multiple factors are impacted by fentanyl. Fentanyl can induce Bcl-2 and inhibit Bcl-2-associated X protein, thus suppressing cell apoptosis after cerebral vascular spasm [28]. In human breast carcinoma cell line MCF-7, fentanyl promotes the expression of p53 and p21, causing cell arrest in G1 phase [29]. Besides, fentanyl regulates interleukin-1β, tumor necrosis factor-α and nuclear factor κB, which has been reported in global ischemia/reperfusion and gastric cancer [9,30]. So it was reasonable to deduce that fentanyl inhibited cell viability and the growth of pancreatic tumor via regulating factors participating in various cellular activities, like cell apoptosis, cell cycle arrest and EMT.
MAPK pathways include three major pathways in mammals, namely ERK1/2, JNK and p38MAPK, each playing specific roles in cell survival and apoptosis. Furthermore, the crosstalk among these pathways is of great significance for a comprehensive and effective regulation of cell activities. Inactivation of ERK1/2 and p38MAPK is concomitant with suppressed differentiated metastatic of prostate cancer [31]. Increased phosphorylated p38 is observed in human cancer, and inhibition of p38 and the consequent blocked p38/ERK crosstalk can help to induce cell apoptosis in colorectal cancer [32]. In cardiomyocytes, the negative crosstalk between JNK and ERK1/2, p38MAPK pathways may be important to myocardial function improvement [33]. Additionally, there are studies showing the relationship between MAPK pathways and factors related to cell apoptosis and cell cycle, like the above-mentioned Bcl-2, p53 and p21 [34-36]. Together with the findings of this study that fentanyl could inhibit MAPK, it could be inferred that fentanyl impacted apoptosis, migration and invasion of pancreatic cancer cells via suppressing related factors and altering the crosstalk in MAPK pathways.
Taken together, fentanyl is shown in this study to be potent in suppressing viability of pancreatic cancer cell in vitro and postponing the growth of pancreatic tumor in grafted mice. Its functions are achieved possibly by regulating factors related to cell apoptosis, cell cycle, EMT and MAPK pathways. These results present new efficacies of fentanyl in addition to analgesia and anesthesia, and provide a possible strategy for pancreatic cancer treatment.
Disclosure of conflict of interest
None.
References
- 1.Society AC. Cancer Facts & Figures 2010. Atlanta: American Cancer Society; 2010. [Google Scholar]
- 2.Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med. 2014;371:1039–1049. doi: 10.1056/NEJMra1404198. [DOI] [PubMed] [Google Scholar]
- 3.Paulson AS, Tran Cao HS, Tempero MA, Lowy AM. Therapeutic advances in pancreatic cancer. Gastroenterology. 2013;144:1316–1326. doi: 10.1053/j.gastro.2013.01.078. [DOI] [PubMed] [Google Scholar]
- 4.Bond-Smith G, Banga N, Hammond TM, Imber CJ. Pancreatic adenocarcinoma. BMJ. 2012;344:e2476. doi: 10.1136/bmj.e2476. [DOI] [PubMed] [Google Scholar]
- 5.Fujioka S, Misawa T, Okamoto T, Gocho T, Futagawa Y, Yanaga K. Predictors for postoperative liver metastasis in patients with resectable pancreatic cancer. Int Surg. 2008;93:324–330. [PubMed] [Google Scholar]
- 6.Roeder F, Timke C, Uhl M, Habl G, Hensley FW, Buechler MW, Krempien R, Huber PE, Debus J, Werner J. Aggressive local treatment containing intraoperative radiation therapy (IORT) for patients with isolated local recurrences of pancreatic cancer: a retrospective analysis. BMC Cancer. 2012;12:295. doi: 10.1186/1471-2407-12-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu GJ, Tai YT, Chen TL, Lin LL, Ueng YF, Chen RM. Propofol specifically inhibits mitochondrial membrane potential but not complex I NADH dehydrogenase activity, thus reducing cellular ATP biosynthesis and migration of macrophages. Ann N Y Acad Sci. 2005;1042:168–176. doi: 10.1196/annals.1338.019. [DOI] [PubMed] [Google Scholar]
- 8.Yuki K, Soriano SG, Shimaoka M. Sedative drug modulates T-cell and lymphocyte function-associated antigen-1 function. Anesth Analg. 2011;112:830–838. doi: 10.1213/ANE.0b013e31820dcabb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qin Y, Li L, Chen J, Tang X, Liao C, Xie Y, Xiao Q. Fentanyl inhibits progression of human gastric cancer MGC-803 cells by NF-kappaB downregulation and PTEN upregulation in vitro. Oncol Res. 2012;20:61–69. doi: 10.3727/096504012x13473664562501. [DOI] [PubMed] [Google Scholar]
- 10.Zhang XL, Chen ML, Zhou SL. Fentanyl Increases Colorectal Carcinoma Cell Apoptosis by Inhibition of NF-κB in a Sirt1-dependent Manner. Asian Pac J Cancer Prev. 2014;15:10015–10020. doi: 10.7314/apjcp.2014.15.22.10015. [DOI] [PubMed] [Google Scholar]
- 11.El Mouedden M, Meert TF. The impact of the opioids fentanyl and morphine on nociception and bone destruction in a murine model of bone cancer pain. Pharmacol Biochem Behav. 2007;87:30–40. doi: 10.1016/j.pbb.2007.03.017. [DOI] [PubMed] [Google Scholar]
- 12.Matsuyama K, Satomi E, Ueno H, Inoue A, Oike M, Todaka K, Yoshida T, Sekiyama R, Hirotsune H, Sueta K, Miyamoto A, Nakamori S, Tsujinaka T, Komori K. A case of unresectable advanced pancreatic cancer treated by gemcitabine-based chemotherapy following cancer pain alleviation by low-dose matrix-type transdermal fentanyl. Gan To Kagaku Ryoho. 2009;36:1351–1353. [PubMed] [Google Scholar]
- 13.Cao Q, Mani RS, Ateeq B, Dhanasekaran SM, Asangani IA, Prensner JR, Kim JH, Brenner JC, Jing X, Cao X, Wang R, Li Y, Dahiya A, Wang L, Pandhi M, Lonigro RJ, Wu YM, Tomlins SA, Palanisamy N, Qin Z, Yu J, Maher CA, Varambally S, Chinnaiyan AM. Coordinated regulation of polycomb group complexes through microRNAs in cancer. Cancer Cell. 2011;20:187–199. doi: 10.1016/j.ccr.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- 15.Speidel D. Transcription-independent p53 apoptosis: an alternative route to death. Trends Cell Biol. 2010;20:14–24. doi: 10.1016/j.tcb.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 16.Brentnall M, Rodriguez-Menocal L, De Guevara RL, Cepero E, Boise LH. Caspase-9, caspase-3 and caspase-7 have distinct roles during intrinsic apoptosis. BMC Cell Biol. 2013;14:32–40. doi: 10.1186/1471-2121-14-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zurlo D, Leone C, Assante G, Salzano S, Renzone G, Scaloni A, Foresta C, Colantuoni V, Lupo A. Cladosporol a stimulates G1-phase arrest of the cell cycle by up-regulation of p21(waf1/cip1) expression in human colon carcinoma HT-29 cells. Mol Carcinog. 2013;52:1–17. doi: 10.1002/mc.20872. [DOI] [PubMed] [Google Scholar]
- 18.Lin JT, Li HY, Chang NS, Lin CH, Chen YC, Lu PJ. WWOX suppresses prostate cancer cell progression through cyclin D1-mediated cell cycle arrest in the G1 phase. Cell Cycle. 2015;14:408–416. doi: 10.4161/15384101.2014.977103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gulappa T, Reddy RS, Suman S, Nyakeriga AM, Damodaran C. Molecular interplay between cdk4 and p21 dictates G0/G1 cell cycle arrest in prostate cancer cells. Cancer Lett. 2013;337:177–183. doi: 10.1016/j.canlet.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maier HJ, Schmidt-Strassburger U, Huber MA, Wiedemann EM, Beug H, Wirth T. NF-κB promotes epithelial-mesenchymal transition, migration and invasion of pancreatic carcinoma cells. Cancer Lett. 2010;295:214–228. doi: 10.1016/j.canlet.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 21.Hongo K, Tsuno NH, Kawai K, Sasaki K, Kaneko M, Hiyoshi M, Murono K, Tada N, Nirei T, Sunami E, Takahashi K, Nagawa H, Kitayama J, Watanabe T. Hypoxia enhances colon cancer migration and invasion through promotion of epithelial-mesenchymal transition. J Surg Res. 2013;182:75–84. doi: 10.1016/j.jss.2012.08.034. [DOI] [PubMed] [Google Scholar]
- 22.Yori JL, Johnson E, Zhou G, Jain MK, Keri RA. Kruppel-like factor 4 inhibits epithelial-to-mesenchymal transition through regulation of E-cadherin gene expression. J Biol Chem. 2010;285:16854–16863. doi: 10.1074/jbc.M110.114546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mendez MG, Kojima S, Goldman RD. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010;24:1838–1851. doi: 10.1096/fj.09-151639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Valcz G, Sipos F, Krenács T, Molnár J, Patai AV, Leiszter K, Tóth K, Wichmann B, Molnár B, Tulassay Z. Increase of alpha-SMA(+) and CK (+) cells as an early sign of epithelial-mesenchymal transition during colorectal carcinogenesis. Pathol Oncol Res. 2012;18:371–376. doi: 10.1007/s12253-011-9454-z. [DOI] [PubMed] [Google Scholar]
- 25.Leelahavanichkul K, Amornphimoltham P, Molinolo AA, Basile JR, Koontongkaew S, Gutkind JS. A role for p38 MAPK in head and neck cancer cell growth and tumor-induced angiogenesis and lymphangiogenesis. Mol Oncol. 2014;8:105–118. doi: 10.1016/j.molonc.2013.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang PY, Chen MF, Tsai CH, Hu DN, Chang FR, Wu YC. Involvement of caspase and MAPK activities in norcantharidin-induced colorectal cancer cell apoptosis. Toxicol In Vitro. 2010;24:766–775. doi: 10.1016/j.tiv.2009.12.025. [DOI] [PubMed] [Google Scholar]
- 27.Darling NJ, Cook SJ. The role of MAPK signalling pathways in the response to endoplasmic reticulum stress. Biochim Biophys Acta. 2014;1843:2150–2163. doi: 10.1016/j.bbamcr.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 28.Yu-Ming L, Jian-Rong M. Effect of fentanyl on cerebral vasospasm after subarachnoid hemorrhage. Hainan Medical Journal. 2012;23:27–31. [Google Scholar]
- 29.Xian WZ, Han LD, Chuan HZ, Yu KT. The effect of fentanyl on proliferation and cell cycle of human breast carcinoma cell line MCF-7. Journal of Chinese Physician. 2005;7:23–25. [Google Scholar]
- 30.Oh WS. Effect of fentanyl on TNF-alpha and IL-1beta levels during global ischemia/reperfusion in rats. Int J Tissue React. 2002;24:11–21. [PubMed] [Google Scholar]
- 31.Uzgare AR, Kaplan PJ, Greenberg NM. Differential expression and/or activation of P38MAPK, erk1/2, and jnk during the initiation and progression of prostate cancer. Prostate. 2003;55:128–139. doi: 10.1002/pros.10212. [DOI] [PubMed] [Google Scholar]
- 32.Chiacchiera F, Grossi V, Cappellari M, Peserico A, Simonatto M, Germani A, Russo S, Moyer MP, Resta N, Murzilli S, Simone C. Blocking p38/ERK crosstalk affects colorectal cancer growth by inducing apoptosis in vitro and in preclinical mouse models. Cancer Lett. 2012;324:98–108. doi: 10.1016/j.canlet.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 33.Peng T, Zhang T, Lu X, Feng Q. JNK1/c-fos inhibits cardiomyocyte TNF-alpha expression via a negative crosstalk with ERK and p38 MAPK in endotoxaemia. Cardiovasc Res. 2009;81:733–741. doi: 10.1093/cvr/cvn336. [DOI] [PubMed] [Google Scholar]
- 34.De Chiara G, Marcocci ME, Torcia M, Lucibello M, Rosini P, Bonini P, Higashimoto Y, Damonte G, Armirotti A, Amodei S, Palamara AT, Russo T, Garaci E, Cozzolino F. Bcl-2 Phosphorylation by p38 MAPK: identification of target sites and biologic consequences. J Biol Chem. 2006;281:21353–21361. doi: 10.1074/jbc.M511052200. [DOI] [PubMed] [Google Scholar]
- 35.Yu SY, Liao CH, Chien MH, Tsai TY, Lin JK, Weng MS. Induction of p21(Waf1/Cip1) by garcinol via downregulation of p38-MAPK signaling in p53-independent H1299 lung cancer. J Agric Food Chem. 2014;62:2085–2095. doi: 10.1021/jf4037722. [DOI] [PubMed] [Google Scholar]
- 36.Tu Y, Wu W, Wu T, Cao Z, Wilkins R, Toh BH, Cooper ME, Chai Z. Antiproliferative autoantigen CDA1 transcriptionally up-regulates p21(Waf1/Cip1) by activating p53 and MEK/ERK1/2 MAPK pathways. J Biol Chem. 2007;282:11722–11731. doi: 10.1074/jbc.M609623200. [DOI] [PubMed] [Google Scholar]