

Abstract
The understanding of molecular mechanism underlying ischemia/reperfusion-induced neuronal death and neurological dysfunction may provide therapeutic targets for ischemic stroke. In this study, miR-525-5p is clearly reduced in the ischemic brain after oxygen-glucose deprivation (OGD). Using TargetScan, MicroCosm Targets version 5, and microRNA.org databases, we identified miR-525-5p as a possible regulator of the ADAMTS13. We validated that ADAMTS13 is a target for miR-525-5p with a luciferase reporter activity assay. Moreover, adult rats subjected to focal cerebral ischemia exhibited a substantial reduction of miR-525-5p expression, which was inversely upregulated by ADAMTS13 expression. In vivo treatment with miR-525-5p agomir effectively decreased ADAMTS13 mRNA and protein levels in the ischemic region. Furthermore, knockdown of cerebral miR-525-5p reduced cell death and infarct size. In addition, the knockdown of ADAMTS13 by ADAMTS13 siRNA apparently abrogated the protective effect of miR-525-5p antagomir on OGD-induced cell death. Our data demonstrate that miR-525-5p is an endogenous regulator of ADAMTS13 that improves ischemia/reperfusion (I/R)-induced brain injury and dysfunction.
Keywords: miR-525-5p, ADAMTS13, ischemia/reperfusion injury-induced
Introduction
Ischemic brain injury is caused by insufficient blood flow to the brain, characterized by oxidative stress, hypoxia, inflammation, and glutamate excitotoxicity, eventually leading to cell death [1]. Although clinical trials have revealed that there are opportunities to prevent and treat acute stroke [2], ischemic stroke remains one of the leading causes of disability and mortality, and numerous surviving stroke victims suffer from disability for the rest of their lives. However, the precise mechanism of ischemia-induced neuronal death remains poorly understood. Therefore, understanding of the pathological mechanism of ischemia-related cell death is of great importance for developing effective therapies for acute stroke.
MicroRNAs are highly conserved, small, noncoding RNAs that are typically 18-24 nucleotides in length, and these molecules have emerged as important regulators of many biological processes, including differentiation, proliferation, development, migration, and apoptosis. They regulate protein translation through interaction with the 3’-untranslated region (UTR) of messenger RNA (mRNA) of the target gene, leading to destabilization and degradation of mRNA [3]. Compelling evidences have revealed that a variety of miRNAs were involved and functional in cerebral ischemia reperfusion injury [4]. The effects of miR-525-5p in cerebral ischemia reperfusion injury, however, have not yet been well understood.
ADAMTS13 (A Disintegrin And Metalloprotease with Thrombospondin type I repeats-13) plays an important role in preventing microvascular thrombosis by cleaving ultra large von Willebrand factor (ULVWF) multimers, the most thrombogenic form of VWF, into smaller less active multimers, reducing potential thrombotic activity. Early studies have shown that ADAMTS13 deficiency aggravates brain injury in a murine model of acute ischemic stroke [5,6]. The protective effect of ADAMTS13 in this process was hypothesized to occur secondary to reduced platelet-dependent thrombotic potential and concomitant reduction of ischemia/reperfusion injury [5]. A previous study also showed that in the ADAMTS13 deficient mice after a brief focal ischemia the post-ischemic hypoperfusion was significantly amplified partly because of enhanced microvascular plugging by VWF-platelet-leukocyte complex.
In the current study, using the oxygen-glucose deprivation (OGD) model of cell ischemia in vitro, we investigated the role of miR-525-5p in regulating OGD-induced neuron death and the underlying mechanism. We found that miR-525-5p plays an important role in OGD-induced neuron death. Furthermore, miR-525-5p was identified and characterized to interact with 3’-UTR of ADAMTS13; thereby, miR-525-5p antagomir was capable of upregulating ADAMTS13 expression that might protect neurons from ischemia-induced cell death.
Materials and methods
Primary culture of rat cortical neurons
All animal protocols were approved by the second hospital of Jilin University on Animal Research. Cortical neurons were prepared from brains of 18-day-old Sprague-Dawley rat embryos. Cells were plated in neurobasal medium supplemented with 2% B27 (Invitrogen) on plates coated with poly-D-lysine. Neurons were cultured at 37°C in a humidified 5% CO2 atmosphere and used after 10 days in vitro.
To initiate oxygen-glucose deprivation (OGD), cortical neurons were exposed to DMEM without serum or glucose in a humidified atmosphere containing 95% nitrogen and 5% CO2. After 3 h of OGD, neurons were fed with serum and glucose-supplemented original medium, and returned to the incubator under normoxic conditions (95% air, 5% CO2). Cells cultured in growth culture medium under normoxic condition were used as control.
Transfection of microRNA agomir or antagomir into neurons
The cells were seeded at a density of 1.0 × 105 (24-well plates). The dosage of microRNA agomir or antagomir (Ribobio, China) was determined according to the manufacturer’s protocol. MicroRNA agomir (6 nM, final concentration) or antagomir (20 nmol, final concentration) in 50 μl of Opti-MEM was complexed with 1 μl of lipofectamine in 50 μl of Opti-MEM (Ambion, USA). The cells were transfected with these complexes and maintained for 8 h, then neurons were fed with serum and glucose-supplemented original medium and cultured in normoxic conditions for another 24 h prior to OGD insult. In all experiments, an equal concentration of scrambled non-targeting controls (agomir or antagomir-negative control, Ribobio, China) was used as a control for non-sequence-specific effects in microRNA experiments.
RT-qPCR
Total RNA was extracted from TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. For miRNA detection, reverse transcription was performed using the one-step primescript miRNA complementary DNA (cDNA) synthesis kit (TaKaRa, Dalian, China). For mRNA detection, cDNA was generated by using M-MLV reverse transcriptase (Clontech, Palo Alto, CA, USA). To analyze the gene expression, the RT-qPCR mixture system containing cDNA templates, primers, and SYBR Green qPCR Master Mix were subjected to RT-qPCR quantification according to standard methods. β-Actin and U6 were used as internal control of mRNA or miRNA, respectively. Relative gene expression was quantified by 2-ΔΔCt method.
Western blot analysis
A total of 25 μg proteins extracted from cells were loaded and separated by sodium dodecyl sulfatepolyacrylamide gel electrophoresis and then electrotransferred to nitrocellulose membranes (Amersham, Little Chalfont, UK). The membranes were then blocked in 2.5 % nonfat milk for 1 h at 37°C. After washed with Tris-buffered saline with Tween, the membranes were incubated with primary antibodies against ADAMTS13 and GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 4°C overnight. Then, peroxidase-conjugated secondary antibody (Boster Corporation, Wuhan, Hubei, China) diluted in 1:1,000 were added and incubated for 1 h at room temperature. The immunoreactive protein bands were visualized using an enhanced chemiluminescence detection system (Amersham, Little Chalfont, UK).
Dual-luciferase reporter assay
The fragment from the 3’-UTR of ADAMTS13 mRNA containing the predicted miR-525-5p-binding sequences was amplified and subcloned into pGL3 luciferase promoter vector (Promega, Madison, WI, USA). The pGL3 vector containing 3’-UTR of ADAMTS13 mRNA or mutated forms was co-transfected with miR-525-5p agomir or controls into HEK293 cells and incubated for 48 h. Then, cells were harvested and lysed, and the luciferase activity was detected using the dual-luciferase reporter assay kit (Promega, Madison, WI, USA) as per standard protocols. The relative luciferase activities were normalized to that of the control cells.
Measurement of infarct volume and neurological deficit
Infarct volume was measured using 2% TTC (2,3,5-triphenyltetrazolium chloride) (Sangon Biotech, Shanghai, China) staining. Rat brains were removed after 2 h MCAO followed by reperfusion for 24 h and sliced into six 2-mm thick coronal sections on a rat brain matrix. The slices were stained with 2% TTC for 15 min at 37°C. The percent of infarct volume was calculated using a derived formula in which infarct volume as a percentage of the contralateral hemisphere was determined as follows: 100% × (contralateral hemisphere volume-non-infarct ipsilateral hemisphere volume)/contralateral hemisphere volume. Following cerebral ischemia, rats were also tested for neurological deficits and scored on a 5-point scale as follows: 0, no observable neurological deficits (normal); 1, failure to extend right forepaw (mild); 2, circling to the contralateral side (moderate); 3, falling to the right (severe); and 4, mice could not walk spontaneously, depressed level of consciousness (very severe).
TUNEL assays
Coronal brain sections (approximately 2-mm thick) were taken immediately from in front of and behind the optic chiasm after intracardial paraformaldehyde perfusion. The brain specimens were individually immersed in 4% paraformaldehyde fixative for 24 h, dehydrated in alcohol, and embedded in paraffin. They were then cut into slices of 4-μm for TUNEL analysis as described in the commercial kit manual (Roche Applied Science, Mannheim, Germany). Positively labeled nuclei (brown color) were identified from unstained nuclei (blue color). The number of positive nuclei was determined by manually counting (× 400 magnification) all the positively labeled nuclei present in five randomly selected fields under a microscope. The percentage of TUNEL-positive nuclei was used as an apoptotic index. The apoptotic cells had the following characteristics: single cells, no inflammation, cell membrane curling, brown particulate, or fragmented nuclei.
Statistical analysis
The quantitative data were expressed as mean ± SD from 3 independent experiments. Statistical analysis was carried out using one-way analysis of variance (ANOVA) followed by Bonferroni test for multiple groups or Student t test between two groups. Differences with a P value less than 0.05 were regarded as statistically significant.
Results
MiR-525-5p is directly target ADAMTS13
To assess the miRNAs that could target ADAMTS13, a bioinformatic search was performed in TargetScan (http://www.targetscan.org/), MicroCosm Targets Version 5 (http://www.ebi.ac.uk/Enright-srv/microcosm/cgi-bin/targets/v5/search.pl), and microrna.org (http://www.microrna.org/microrna/getGeneForm.do) databases (Figure 1A). To validate a direct interaction between miR-525-5p and ADAMTS13 mRNA, its 3’-UTR target sites was cloned into a dual luciferase reporter vector. We then co-transfected HEK293 cells with luciferase reporter vector containing the 3’-UTR (wild type or mutated type) of ADAMTS13 and miR-525-5p agomir or negative control. Co-transfection of miR-525-5p agomir in HEK 293T cells strongly inhibited luciferase activity produced by the reporter construct containing the wild 3’-UTR segment of ADAMTS13, whereas no effect was observed with a construct containing a mutated segment of ADAMTS13 3’-UTR. This effect was determined to be specific because no change was seen in luciferase reporter activity when negative control was co-transfected with either reporter construct (Figure 1B). Collectively, these data indicate that ADAMTS13 is a potential target of miR-525-5p.
Figure 1.
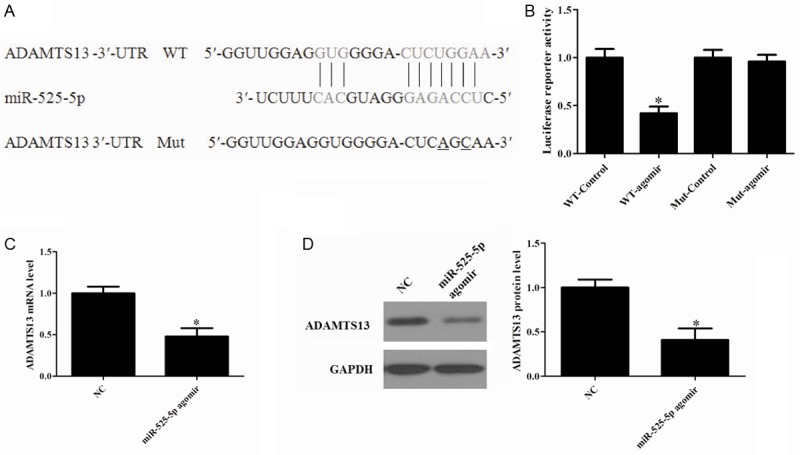
MiR-525-5p agomir attenuates ADAMTS13 expression in OGD neurons. A: ADAMTS13 3’-UTR fragment containing wild-type (WT) or mutated (Mut) miR-525-5p-binding sites was cloned downstream of the luciferase reporter gene. Mutations were generated in the ADAMTS13 3’-UTR sequences complementary to the seed region of miR-525-5p, as indicated. B: Luciferase activity assays using reporters with WT or Mut ADAMTS13 3’-UTRs were performed after co-transfection with miR-525-5p agomir or NC. C: Agomir inhibited most of the endogenous ADAMTS13 expression after OGD. D: The relative expression levels of ADAMTS13 protein were calculated. The data shown were the means ± S.D. from 3 independent experiments. *P<0.05 vs. Control.
MiR-525-5p agomir attenuates ADAMTS13 expression in OGD neurons
To confirm that miR-525-5p acts as a negative regulator of ADAMTS13 translation under ischemic conditions. Neurons cultured in vitro were transfected with agomir or negative controls and maintained for 8 h, then fed with original medium for another 24 h prior to OGD. We observed that ADAMTS13 mRNA and protein expression levels were significantly decreased (Figure 1C and 1D). This is consistent with the idea that an ischemia-induced reduction in miR-525-5p negatively regulates ADAMTS13 expression.
Expression of ADAMTS13 and miR-525-5p in OGD-treated neurons
To determine the expression profiles of miR-525-5p in OGD-treated neurons, we firstly detected the expression levels of miR-525-5p in hippocampal neurons subjected to OGD by RT-qPCR. As shown in Figure 2A, the expression level of miR-525-5p was significantly decreased in neurons after OGD and continued to be down-regulated during 12-48 h after OGD when compared with the control group. In addition, the expression levels of ADAMTS13 OGD-treated neurons were determined by using RT-qPCR (Figure 2B) and Western blot analysis (Figure 2C). The quantitative data exhibited that ADAMTS13 expression was significantly increased following 12-48 h after OGD (Figure 2D). These results suggested a critical role of miR-525-5p and ADAMTS13 in ischemic injury.
Figure 2.
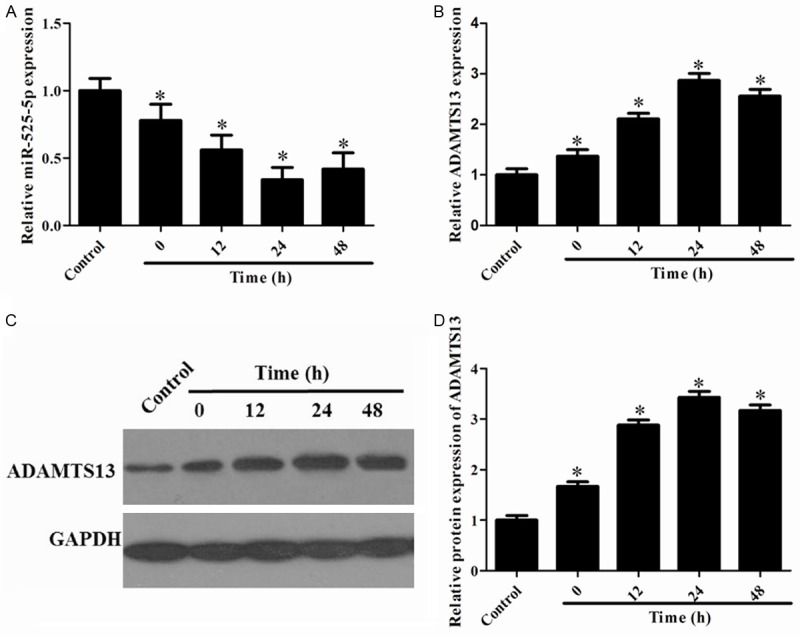
Expression of ADAMTS13 and miR-525-5p in cultured rat neurons after OGD-induced ischemic injury. A: RT-qPCR analysis of miR-525-5p expression levels in neurons during 12-48 h after OGD. B: The level of ADAMTS13 mRNA expression was detected by RT-PCR. C: Western blot analysis of total ADAMTS13 protein expression levels in neurons during 12-48 h after OGD. D: Quantitative analysis of relative ADAMTS13 protein level using Image-Pro Plus 6.0 software and normalized to GAPDH. *P<0.05 vs. Control.
MiR-525-5p antagomir attenuates ischemic brain damage
To evaluate the biological role of miR-525-5p in OGD-induced ischemic injury in vivo, we altered brain levels by intracerebroventricular injection of miR-525-5p antagomir or negative control. As shown in Figure 3A, miR-525-5p antagomir greatly decreased infarct volume relative to the control group. We observed that the infarct volume was reduced in miR-525-5p antagomir-injected rat brains. Given the markedly improved recovery of brain function in miR-525-5p antagomir-injected rats, we next determined the effects of miR-525-5p on postischemic cellular damage. TUNEL staining showed that the proportion of apoptotic cells was much lower in miR-525-5p antagomir-injected rat brains than in negative control (Figure 3B). Taken together, these results demonstrate that miR-525-5p attenuates cerebral I/R damage in vivo.
Figure 3.
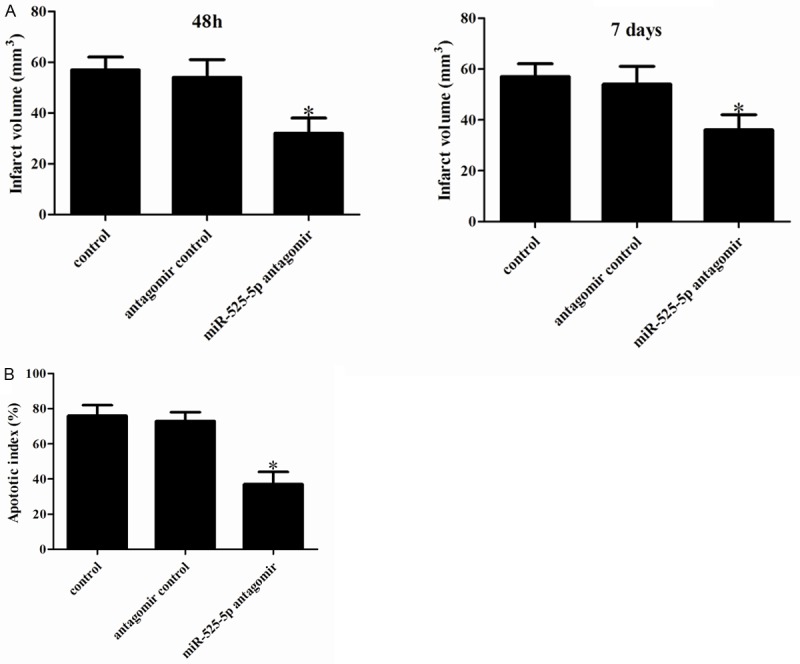
Effect of miR-525-5p on ischemic brain damage. A: Representative TTC-stained coronal sections demonstrate decreased infarct size in a representative miR-525-5p antagomir -transfected brain compared with the brain of negative control-infused rat. B: Neuronal apoptosis in the ipsilateral cortex as detected by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL). The number of TUNEL-positive cells was significantly reduced in miR-525-5p antagomir infused rat brains. *P<0.05 vs. Control.
Knockdown of ADAMTS13 abrogates the neuroprotective effect of miR-525-5p antagomir in OGD-induced ischemic injury
To further confirm the contribution of ADAMTS13 to the biological effects of miR-525-5p in OGD-induced cell injury. Because miRs can theoretically target a large number of different genes, we investigated whether reduction of ADAMTS13 expression by miR-525-5p might be a mechanism contributing to cell death. Cultures were also treated with small interfering RNA to ADAMTS13 or control to reduce ADAMTS13 mRNA expression. Western blot results showed that the stimulation effects of miR-525-5p antagomir on the expression levels of ADAMTS13 were apparently blocked by ADAMTS13 siRNA (Figure 4A). MiR-525-5p antagomir decreased cell death from this injury paradigm (Figure 4B). Also, ADAMTS13 knockdown abolished the protective effect of miR-525-5p antagomir, confirming a role for ADAMTS13 in the mechanism of miR-525-5p-mediated cell death.
Figure 4.
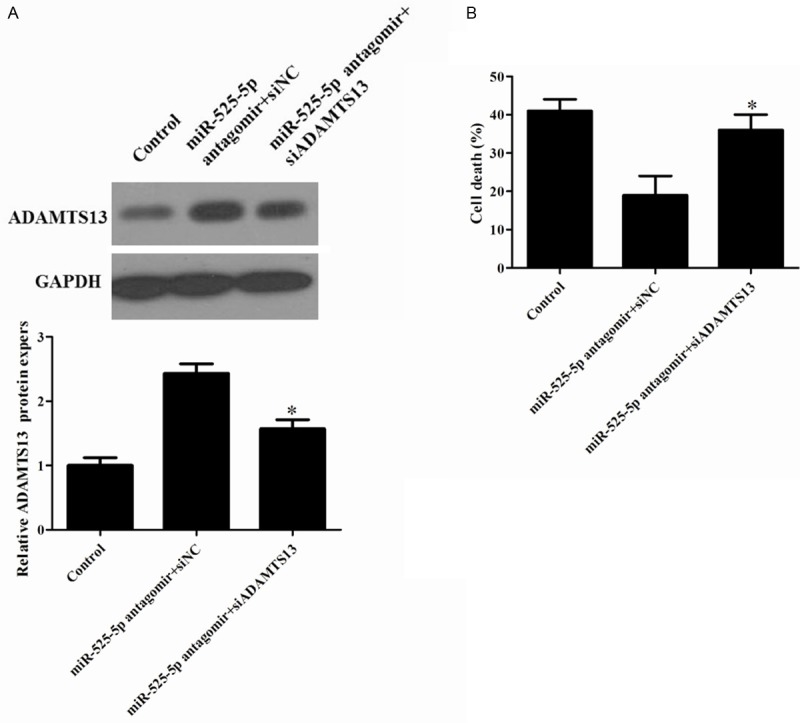
Effect of ADAMTS13 siRNA on miR-525-5p neuroprotective effect in OGD-treated neurons. A: Western blot analysis of ADAMTS13 protein expression levels in different groups. B: Detection the effect of co-transfection of miR-525-5p antagomir and ADAMTS13 siRNA on cell death by LDH. *P<0.05 vs. Control.
Discussion
miRNAs are a class of sophisticated gene expression regulators that inhibit translation and/or degrade target mRNAs by recognizing them through base pairing with short regions near 3’-UTRs. A subset of miRNAs are abundantly expressed in the human brain [7], and play important roles in numerous brain diseases [8-10]. Currently, increasing studies have demonstrated that miRNAs play a critical role in the pathophysiology of brain seizures, ischemia, and trauma [10,11]. Downregulation of miR-30a inhibited cerebral ischemic injury through upregulation of Beclin-1-mediated autophagy [12]. It has been reported that inhibition of miR-18b provides a protective effect against ischemia injury by directly regulating the expression of ubiquitin carboxyl-terminal hydrolase isozyme L1 and heat shock protein A5 [13]. miR-124, another reported brain-specific miRNA, has been demonstrated to be downregulated in rats subjected to focal cerebral [14]. In addition, Liu et al. have reported that the inhibition of miR-124 is capable of enhancing the expression of apoptosis-stimulating proteins of the p53 family and reduces neuronal apoptosis and infarction in mouse focal cerebral ischemia [15]. In the present study, for the first time, we show that miR-525-5p expression was downregulated in cortical neurons subjected to OGD, and that the expression pattern of miR-525-5p correlates inversely with the expression pattern of ADAMTS13.
ADAMTS-13 is a VWF-cleaving protease that inhibits thrombus formation. ADAMTS-13 has been described as a key protein in linking thrombosis with inflammation. In experimental ischemic stroke models, ADAMTS13 deficiency resulted in aggravation, and administration of ADAMTS13 in amelioration, of neuronal injury [5,6,16]. Although not directly investigated, the mechanisms underlying these observations were thought to involve alterations in post-ischemic thrombotic potential; either decreased thrombosis in the case of VWF deficiency or enhanced post-ischemic thrombosis in the case of ADAMTS13 deficiency. In this study, according to the prediction of miRNA targets in human and in vertebrates, miR-525-5p may regulate the expression of ADAMTS13 at the post-transcriptional level by pairing with partially complementary sites in the 3’-UTR of ADAMTS13 mRNA. In addition, we also demonstrated that miR-525-5p could directly bind with the 3’-UTR of ADAMTS13 mRNA and negatively regulate ADAMTS13 gene expression. miR-525-5p agomir decreased the expression of ADAMTS13 after OGD-induced ischemic injury. Furthermore, knockdown of endogenous miR-525-5p expression reduced brain infarct size after focal ischemia. Taken together, these results provide strong evidence that miR-525-5p contributes to the cerebral ischemic injury through negatively regulating ADAMTS13 level. Our study further confirmed the importance of miR-525-5p in regulating cerebral ischemic injury that might contribute to the development of intervention therapy for cerebral ischemic stroke.
It has been reported that microRNAs are endogenous molecules with stable and high tissue compatibility characteristics [17]. MicroRNA agomirs and antagomirs used in the present study are chemically modified small riboneucleotides with specific sequences to mimic or rivalry miR-525-5p. The specificity and stability of the agomirs and antagomirs have been proved in several recent researches [18]. In the present study, we find that miR-525-5p antagomir remains at a high level 7 days after injection in vivo, and attenuates brain damage 7 days, suggesting it might exert long-term efficacy. However, profound study in longer periods is needed to examine its prospective outcome.
In summary, we have provided evidence that the downregulation of miR-525-5p in OGD-treated neurons could alleviate ischemic injury through enhancing ADAMTS13. The findings point out a novel mechanism for the regulation of ischemic neural cell death through miR-525-5p, and suggest that regulation of miR-525-5p and/or ADAMTS13 in the brain could be potential therapeutic targets for ischemic stroke. Further work is needed to explore the stroke-related function of this particular miRNA in more time points of brain ischemia and reperfusion injury.
Disclosure of conflict of interest
None.
References
- 1.Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4:399–414. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- 2.Marsh JD, Keyrouz SG. Stroke prevention and treatment. J Am Coll Cardiol. 2010;56:683–691. doi: 10.1016/j.jacc.2009.12.072. [DOI] [PubMed] [Google Scholar]
- 3.Mendell JT, Olson EN. MicroRNAs in stress signaling and human disease. Cell. 2012;148:1172–1187. doi: 10.1016/j.cell.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Di Y, Lei Y, Yu F, Changfeng F, Song W, Xuming M. MicroRNAs expression and function in cerebral ischemia reperfusion injury. J Mol Neurosci. 2014;53:242–250. doi: 10.1007/s12031-014-0293-8. [DOI] [PubMed] [Google Scholar]
- 5.Fujioka M, Hayakawa K, Mishima K, Kunizawa A, Irie K, Higuchi S, Nakano T, Muroi C, Fukushima H, Sugimoto M. ADAMTS13 gene deletion aggravates ischemic brain damage: a possible neuroprotective role of ADAMTS13 by ameliorating postischemic hypoperfusion. Blood. 2010;115:1650–1653. doi: 10.1182/blood-2009-06-230110. [DOI] [PubMed] [Google Scholar]
- 6.Zhao BQ, Chauhan AK, Canault M, Patten IS, Yang JJ, Dockal M, Scheiflinger F, Wagner DD. von Willebrand factor-cleaving protease ADAMTS13 reduces ischemic brain injury in experimental stroke. Blood. 2009;114:3329–3334. doi: 10.1182/blood-2009-03-213264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bak M, Silahtaroglu A, Møller M, Christensen M, Rath MF, Skryabin B, Tommerup N, Kauppinen S. MicroRNA expression in the adult mouse central nervous system. Rna. 2008;14:432–444. doi: 10.1261/rna.783108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hébert SS, De Strooper B. Alterations of the microRNA network cause neurodegenerative disease. Trends Neurosci. 2009;32:199–206. doi: 10.1016/j.tins.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 9.Teplyuk NM, Mollenhauer B, Gabriely G, Giese A, Kim E, Smolsky M, Kim RY, Saria MG, Pastorino S, Kesari S. MicroRNAs in cerebrospinal fluid identify glioblastoma and metastatic brain cancers and reflect disease activity. Neuro Oncol. 2012;14:689–700. doi: 10.1093/neuonc/nos074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu DZ, Tian Y, Ander BP, Xu H, Stamova BS, Zhan X, Turner RJ, Jickling G, Sharp FR. Brain and blood microRNA expression profiling of ischemic stroke, intracerebral hemorrhage, and kainate seizures. J Cereb Blood Flow Metab. 2010;30:92–101. doi: 10.1038/jcbfm.2009.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ziu M, Fletcher L, Rana S, Jimenez DF, Digicaylioglu M. Temporal differences in microRNA expression patterns in astrocytes and neurons after ischemic injury. PLoS One. 2011;6:e14724. doi: 10.1371/journal.pone.0014724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang P, Liang J, Li Y, Li J, Yang X, Zhang X, Han S, Li S, Li J. Down-regulation of miRNA-30a alleviates cerebral ischemic injury through enhancing beclin 1-mediated autophagy. Neurochem Res. 2014;39:1279–1291. doi: 10.1007/s11064-014-1310-6. [DOI] [PubMed] [Google Scholar]
- 13.Peng Z, Li J, Li Y, Yang X, Feng S, Han S, Li J. Downregulation of miR-181b in mouse brain following ischemic stroke induces neuroprotection against ischemic injury through targeting heat shock protein A5 and ubiquitin carboxyl-terminal hydrolase isozyme L1. J Neurosci Res. 2013;91:1349–1362. doi: 10.1002/jnr.23255. [DOI] [PubMed] [Google Scholar]
- 14.Zhu F, Liu JL, Li JP, Xiao F, Zhang ZX, Zhang L. MicroRNA-124 (miR-124) regulates Ku70 expression and is correlated with neuronal death induced by ischemia/reperfusion. J Mol Neurosci. 2014;52:148–155. doi: 10.1007/s12031-013-0155-9. [DOI] [PubMed] [Google Scholar]
- 15.Liu X, Li F, Zhao S, Luo Y, Kang J, Zhao H, Yan F, Li S, Ji X. MicroRNA-124–Mediated Regulation of Inhibitory Member of Apoptosis-Stimulating Protein of p53 Family in Experimental Stroke. Stroke. 2013;44:1973–1980. doi: 10.1161/STROKEAHA.111.000613. [DOI] [PubMed] [Google Scholar]
- 16.Fujioka M, Nakano T, Hayakawa K, Irie K, Akitake Y, Sakamoto Y, Mishima K, Muroi C, Yonekawa Y, Banno F. ADAMTS13 gene deletion enhances plasma high-mobility group box1 elevation and neuroinflammation in brain ischemia–reperfusion injury. Neurol Sci. 2012;33:1107–1115. doi: 10.1007/s10072-011-0913-9. [DOI] [PubMed] [Google Scholar]
- 17.Sethupathy P, Collins FS. MicroRNA target site polymorphisms and human disease. Trends Genet. 2008;24:489–497. doi: 10.1016/j.tig.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 18.Duan Q, Wang X, Gong W, Ni L, Chen C, He X, Chen F, Yang L, Wang P, Wang DW. ER stress negatively modulates the expression of the miR-199a/214 cluster to regulates tumor survival and progression in human hepatocellular cancer. PLoS One. 2012;7:e31518. doi: 10.1371/journal.pone.0031518. [DOI] [PMC free article] [PubMed] [Google Scholar]