

Abstract
Background: Colorectal carcinoma (CRC) and non-alcoholic fatty liver disease (NAFLD) share common risk factors. Insulin resistance (IR) has an important role in both diseases. It has been speculated that the prevalence of colorectal neoplasms might be increased in patients with NAFLD. However, It is unclear whether NAFLD is an actual risk factor or any association is incidental coexistance due to the role of IR in both disease. We aimed to assess the risk for CRC in patients with NAFLD in relation to IR. Method: This study was designed prospectively and cross-sectionally. We determined NAFLD by ultrasonography and measured IR by the homeostatic model of assessment-insulin resistance model. Results: The prevalences of CRC and adenoma were shown to be significantly higher in patients with IR (respectively; P: 0.005, P: 0.008). But prevalence of CRC was found to be significantly lower in subjects with NAFLD (P: 0.001). On multivariate logistic regression analysis, the risks of colorectal adenoma and carcinoma were significantly associated with the presence of IR (respectively; OR: 2.338, 95% CI: 1.080-4.993, P: 0.003 and : 5.023, 95% CI: 1.789-9.789, P: 0.001). The risk for CRC was significantly associated with the absence of NAFLD (OR: 7.380, 95% CI: 3.069-7.961, P: 0.010). The absence of NAFLD in the presence of IR was associated with significantly high risk for CRC (OR: 5.218, 95% CI: 1.538-7.448, P: 0.017). Conclusion: The risk of CRC can increased in subjects with IR but without NAFLD. The absence of NAFLD in the presence of IR may predict the CRC.
Keywords: Colonoscopy, colorectal neoplasm screening, hepatosteatosis, cancer pathogenesis, insulin resistance
Introduction
Colorectal carcinoma (CRC) is one of the most common cancers in the world, and is the second leading cause of cancer-related deaths world wide [1]. Insulin resistance (IR), defined as a subnormal glycemic response to endogenous insulin, is characterized by compensatory hyperinsulinemia [2], and has been shown positively associated with CRC [3,4].
It has been proposed that insulin may be involved in colon carcinogenesis [4]. Moreover, increased levels of plasma insulin and glucose, glucose intolerance, obesity and physical inactivity have been showed to be positively associated with CRC [4,5].
Non-alcoholic fatty liver disease (NAFLD) is also a common disease and is strongly associated with IR, obesity, and dyslipidemia. It is also regarded as a liver manifestation of metabolic syndrome (MS) [6]. It has been showed that the prevalence of MS in CRC patients was relatively high [7]. Recently it has been suggested that NAFLD might be a risk factor for colorectal neoplasms [8-10]. However, it is unclear whether NAFLD is a risk factors for colorectal neoplastic lessions, or this association is an incidental coexistance because of both are accompaniying with IR.
We aimed to investigate NAFLD as one of the potential risk factors for CRC in relation to IR.
Method
Study population
This study was a prospective, observational study investigating NAFLD and IR as risk factor for CRC. This study was conducted by using a registry of participants who underwent colonoscopy between February 2014 and July 2014 at a single center in Turkey. This study was approved by the Institutional Ethical Board. The written informed consent about the study and a standard questionnaire regarding their personal medical history, present medications, family history, and life style habits were obtained from all participants. Physical examinations, laboratory assays, imaging studies were performed after a fasting period of at least 12 hours on the day of the colonoscopy.
During the study period, a consecutive series of 450 subjects, 26-85 years of age, were evaluated. We excluded subjects with a history of chronic alchocol consumption (n: 15), chronic liver disease (n: 18), and seropositivity of hepatitis B virus (n: 60) and hepatitis C virus (n: 22). We also excluded subjects who had colonoscopies in which the cecum was not reached (n: 22), poor bowel preparation (n: 108), inflammatory bowel disease (n: 66), active gastrointestinal bleeding (n: 10), history of coloretal surgery (n: 7), CRC diagnosed beforehand (n: 4) and hereditary cancer syndrome (n: 1). After exclusion of above-mentioned conditions, 127 patients were included to the study.
Participants were classified into three groups according to colonoscopic findings and histologic subtypes of polyps, respectively adenoma group who have tubuler, villous and tubulovillous adenomatous polyps; control group who have normal colonoscopic findings, and carcinoma group who have adenocarcinomas.
Measurements, definitions and laboratory assays
Anthropometry, blood pressure, and laboratory tests were measured after a fasting period of at least 12 hours on the day of the colonoscopy.
Trained nurses measured the height and weight of the participants, wearing light weight hospital gowns and no shoes, using a calibrated stadiometer and a balance beamscale. Blood pressure (BP) was measured in seated subjects after a 5-min rest with a standard mercury sphyngomanometer. The presence of hypertension (HT) was defined according to the 2013 guidelines on hypertension of the European Society of Hypertension and the European Society of Cardiology [11], or the use of antihypertensive medication. Waist circumference (WC) was measured at the umbilicus level. Increased WC was based on the definition according to the Regional Office for the Western Pacific Region of World Health Organization criteria [12]. The body mass index (BMI) was calculated as kg/m2. Diabetes mellitus (DM) was determined by American Diabetes Association guidelines, 2003 [13]. MS was defined as having at least 3 of the criteria set by the Adult Treatment Panel III criteria, as updated by the American Heart Association [14].
A venous blood sample was drawn from an antecubital vein. The serum levels of liver enzymes, lipids, glucose and other biochemical markers were measured. Homeostatic model of assessment-insulin resistance (HOMA-IR), as an index of IR, was calculated with the fasting serum insulin and glucose values of an individual [15]. If HOMA-IR score was 2.5 or more, the patient was defined to have IR.
Hepatic ultrasonography (USG) scanning (a 3.5-MHz transducer [Logiq P5; GE, USA]) was performed for all participants by two trained gastroenterologist blindly and independently. The diagnosis of NAFLD was made on the basis of four known criteria, namely, hepatorenal echogenic contrast, liver brightness, deep attenuation and vascular blurring [16].
Statistical analysis
The statistical analysis was performed with the Windows SPSS program ver. 15.0 (SPSS, Chicago, IL, USA). The statistical results are presented as the mean ± standard deviation or percentages. Statistical analyses included an independent sample One way ANOVA test. The concordance between two USG measurements was also measured with Mc Nemar test. Risk estimation and comparisions of categorical data were made by the chi square-test and multinominal logistic regression analysis. Stepwise linear regression analysis with both backward elimination and forward selection was employed to evaluate any association between outcome variables. Adjusted factors were determined from factors which had p value < 0.05 by linear regression analysis indicating to have independent effect on the dependent variables. These factors were considered covariates and included into multivariate regresion analysis. Each odds ratio (OR) is presented together with its 95% confidence interval (CI). P < 0.05 was considered statistically significant.
Results
Baseline characteristics of the subjects
A total of 450 subjects were eligible for the study. Of these individuals, 127 subjects were included in the final analysis, after excluding 323 subjects according to exclusion criteria.
The baseline characteristics are summarized in Table 1. The mean age of the enrolled subjects was 57.3 ± 12.8 and there were 59 men (46%). A total of 44 patients (35%) had 1 or more colorectal neoplastic lessions, of which 18 subjects (40%) had colorectal carcinoma. The subjects with colorectal neoplastic lessions were more likely to be above age of 62 years and to be male. The adenoma and carcinoma groups had significantly higher mean age than control group (P: 0.023 and P: 0.039; respectively). The carcinoma group had significantly lower hemoglobine (Hb), total cholesterol, low density lipoprotein-cholesterol (LDL-C) levels than the adenoma (respectively: P: 0.026, P: 0.005, P: 0.008) and control groups (respectively P: 0.022, P: 0.002, P: 0.007). The carcinoma group had higher median HOMA-IR value than the control group (P: 0.034). There were no significant differences in other baseline characteristics between groups. The carcinoma group had significantly higher rate of IR than the adenoma and control groups (respectively P = 0.008 and P: 0.005). Results of USG measurements by two experts have well concordance (Mc Nemar test, chi-square value: 0.112, P = 0.25). there was lower prevalance of patients with NAFLD in the carcinoma group compared to the adenoma and control groups (respectively; P: 0,012, P: 0,023) (Table 1).
Table 1.
Basal Characteristics of the Study Population
| PARAMETERS | CONTROL | ADENOM | CARSINOM |
|---|---|---|---|
| N | 83 | 26 | 18 |
| Age (year) | 55.7 ± 12.1 | 62.2 ± 11.6 | 62.0 ± 14.5a |
| Female (n, %) | 50 (60.2%) | 11 (42.3%) | 7 (38.9%) |
| Male (n, %) | 33 (39.8%) | 15 (57.7%) | 11 (61.1%)a |
| BMI (kg/m2) | 30.1 ± 5.0 | 27.5 ± 4.0 | 28.6 ± 5.6 |
| DM (n, %) | 23 (27.7%) | 4 (15.4%) | 6 (33.3%) |
| HT (n, %) | 22 (26.5%) | 6 (23.1%) | 2 (11.1%) |
| MS (n, %) | 32 (39%) | 8 (31%) | 7 (39%) |
| Increased WC (n, %) | 38 (45.8%) | 11 (42.3%) | 6 (33.3%) |
| SBP (mmHg) | 127.3 ± 17.7 | 130.3 ± 13.3 | 130.3 ± 18.7 |
| DBP (mmHg) | 77.3 ± 15.9 | 74.4 ± 8.7 | 74.7 ± 7.9 |
| Glu (mg/dl) | 105.9 ± 26.2 | 108.4 ± 41.3 | 110.7 ± 40.8 |
| ALT (IU/ml) | 24.9 ± 8.6 | 22.8 ± 8.1 | 26.4 ± 2.7 |
| TG (mg/dl) | 156.7 ± 55.5 | 160.4 ± 81.5 | 119.8 ± 60.1 |
| Cholesterol (mg/dl) | 199.3 ± 36.7 | 202.3 ± 35.5 | 166.9 ± 34.2a,b |
| HDL-C (mg/dl) | 45.5 ± 9.6 | 44.4 ± 8.8 | 43.5 ± 14.0 |
| LDL-C (mg/dl) | 124.1 ± 29.3 | 128.1 ± 34.8 | 99.6 ± 29.2a,b |
| HbA1c (%) | 6.2 ± 1.3 | 6.0 ± 0.7 | 6.7 ± 1.6 |
| Hb (g/dl) | 13.2 ± 2.1 | 13.5 ± 2.1 | 11.7 ± 2.8a,b |
| HOMA-IR* | 1.8 (0.38-15.1) | 2.2 (0.01-17.8) | 3.7 (0.23-14.3)a,b |
| Presence of IR (n, %) | 34 (41%) | 17 (65%)a | 14 (77.8%)a,b |
| Presence of NAFLD (n, %) | 49 (59%) | 13 (50%) | 3 (16.7%)a,b |
Abbreviations: BMI: body mass index, CVD: cardiovascular disease, DBP: diastolic blood pressure, DM: diabetes mellitus, HPL: hyperlipidemia, HT: hypertension, MS: Metabolic syndrome, NS: not significant SBP: systolic blood pressure, ALT: Alanin amino transferase, Glu: Glucose, Hb: Hemoglobin, HDL-C: High density lipoprotein-cholesterol, LDL-C: Low density lipoprotein-cholesterol, NS: not significant, TG: triglyceride. Results were expressed as mean ± SD or number of patients.
Results were expressed as median (minimum-maximum).
Compared to control P < 0.005.
Compared to adenoma groups P < 0.005.
Prevalence of colorectal neoplasm in relation to NAFLD and IR presence
Figure 1 shows prevalance of adenoma and carcinoma in patients with NAFLD and seperately in patients with IR.
Figure 1.
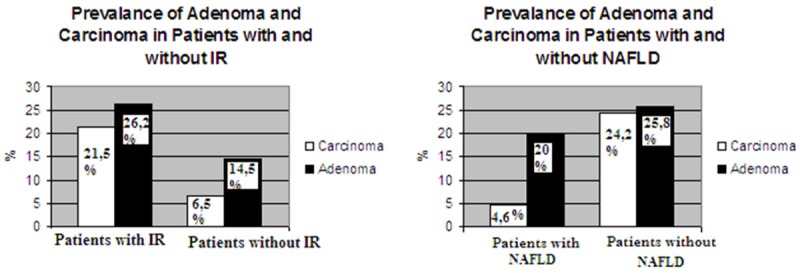
The prevalence of Colorectal Neoplasm in patients with NAFLD and IR.
The prevalences of adenomas and carcinomas were found significantly higher in patients with IR than without IR (respectively; P: 0.008, P: 0.005) and prevalence of carcinoma was found significantly lower in patients with NAFLD than without NAFLD (P: 0.001) (Figure 1).
Association between NAFLD and IR in patients with adenoma and carcinoma
We also made subgroup analysis in adenoma and carcinoma patients with IR for investigating presence of NAFLD. While 21.4% of carcinoma patients with IR had NAFLD, 78.6% of carcinoma patients with IR did not have NAFLD and 29% of adenoma patients had NAFLD, although 71% of adenoma patients with IR did not have NAFLD. Seperately we investigated rate of IR in adenoma and carcinoma patients with NAFLD. While, all of carcinoma patients with NAFLD had IR, 43% of adenoma patients with NAFLD had IR (Figure 2).
Figure 2.
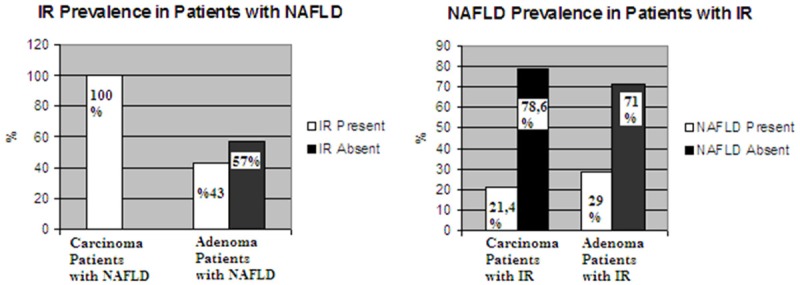
Association between IR and NAFLD in patients with carcinoma and adenoma.
Risk estimation for adenoma and carcinoma according to presence of NAFLD and IR
Risk of adenoma and carcinoma in relation to NAFLD and IR status were shown in Table 2. In multivariate analysis; presence of IR was associated with significantly high risk for both adenoma and CRC (respectively; OR: 2.338, 95% CI: 1.080-4.993, P: 0.003 and OR: 5.023, 95% CI: 1.789-9.789, P: 0.001) (Table 2). Absence of NAFLD was significantly associated with high risk for carcinoma (OR: 7,380, 95% CI: 3.069-7.961, P: 0.010). Absence of NAFLD in the presence of IR was significantly associated with high risk for CRC (OR: 5.218, 95% CI: 1.538-7.448, P: 0.017).
Table 2.
Risk of Adenoma and Carcinoma in relation to NAFLD and IR presence
| ADENOM | CARCINOM | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|||||||||||
| Univariate | Multivariate | Univariate | Multivariate | ||||||||
|
|
|||||||||||
| Total number of subject | Number of subject | OR (95% CI) | P value | OR (95% CI) | P value | Number of subject | OR (95% CI) | P value | OR (95% CI) | P value | |
| IR | |||||||||||
| Absent | 62 | 9 | 1 | 1 | 4 | 1 | 1 | ||||
| Present | 65 | 17 | 2.732 (1.217-5.219) | 0.008 | 2.338 (1.080-4.993) | 0.003 | 14 | 5.130 (1.984-9.976) | 0.008 | 5.023 (1.789-9.789) | 0.001 |
| NAFLD | |||||||||||
| Present | 65 | 13 | 1 | 1 | 3 | 1 | 1 | ||||
| Absent | 62 | 16 | 1.947 (0.815-2.805) | >0.05 | 1.586 (0.605-2.156) | >0.05 | 15 | 7. 918 (3,223-8.10) | 0.003 | 7.380 (3.069-7.961) | 0.010 |
| In subjects with IR | |||||||||||
| Presence of NAFLD | 30 | 5 | 1 | 1 | 3 | 1 | 1 | ||||
| Absence of NAFLD | 35 | 12 | 4.066 (1.655-5.960) | 0.028 | 2.692 (0.532-4.849) | >0.05 | 10 | 5.654 (1.756-7.660) | 0.015 | 5.218 (1.538-7.448) | 0.017 |
Covariates in adjusted models included age, sex, metabolic syndrome, diabetes mellitus, obesity. Abbrevations: CRN: Colorectal Neoplasia, IR: Insuline resistance, NAFLD: Non-alcholic liver disease.
Discussion
In our study, we found significantly higher rates of IR in the adenoma and carcinoma groups compared to the control group. We found also, presence of IR was associated with more than two fold increase in the risk of adenoma, and five fold increase in the risk of carcinoma.
Some enviromental factors are associated with the high risk for CRC including dietary, physical inactivity. Giovannucci proposed that the effects of dietary and lifestyle risk factors on CRC may be mediated through hyperinsulinemia [4]. In vitro, in vivo, and epidemiologic data support this “hyperinsulinemia-colorectal cancer” hypothesis. In animal models, it has been shown that exogenous insulin injection stimulated the growth of CRC precursors [17,18]. In clinical studies, high circulating insulin levels have been found independently associated with increased risk for CRC [19,20]. In addition, type 2 DM has been found to be associated with a 30%-40% increase in the risk of CRC [21]. Patients with CRC have been shown to have higher rate of IR than the control group in previous studies similar to our results [8-10]. Excess insulin has been shown to affect development of other cancers, including breast, liver, and pancreas, as well as CRC [22-24].
Numerous mechanisms related with IR can be responsible in colorectal carcinogenesis. Insulin is a known growth factor and powerful mitogenic agent. Insulin stimulates cell proliferation through two pathways [25]. The minor pathway involves direct binding of insulin to either insulin or insulin-like growth factor-1 (IGF-1) receptors; the major pathway is via inhibition of IGF-binding proteins (IGFBP) and the resultant increase in IGF-1 availability to the IGF-1 receptor [25]. CRCs have a 10- to 50-fold increase in IGF1, when compared with the adjacent normal colonic mucosa, and overexpression of IGF1-R has been described in human CRC. The IGF system is a potent growth regulator closely linked with carcinogenesis [26]. For example, hypersecretion of IGF-1 induced by growth hormone is responsible for the increased risk of CRC among patients with acromegaly. Because CRC has been strongly suggested to be mediated through the insulin pathway, Karimi K et al. have hypothesized that polymorphisms in the genes involving the insulin pathway are associated with the risk of CRC susceptibility. Although they have not found signifcant difference in genotype and allele frequencies between the cases with CRC and controls for the IGF-I, IGFBP-3, insuline cell surface receptor, and insulin receptor substrate-2 (IRS-2) gene variants, they have presented an evidence for the interaction between the rs2289046 variant of IRS2 gene and BMI in the risk of CRC [27].
In the course of malignant progression of adenomas to carcinoma, insulin also plays a mitogenic role through ras oncogene. Ras mutations are important in colon carcinogenesis and may involve the growth of adenomas into cancers [4].
As a feature of IR, hyperglycemia, may also affect the CRC risk through an inhibition of colonic motility. Hyperglycemia also increases advanced glycation end-product (AGE) formation [28]. The production of intracellular AGE precursors damages target cells by modifying proteins and altering their function. It has been reported that plasma proteins modified by AGE precursors bind to AGE receptors on endothelial cells inducing receptor-mediated production of ROS. Also, AGE receptor ligation, by activating nuclear factor-kappa-B (NFκB), can induce adverse changes in gene expression [29]. NFκB, which is strongly associated with abdominal obesity and IR. This transcription factor is involved in cytokine signaling and in cell survival.
It is also possible that metabolic abnormalities secondary to IR (i.e., triglycerides and glucose) may act as a main source of energy for cancer cell growth [30,31].
As well as contribution of IR to carcinogenesis, the carcinogenesis may further enhance IR via chronic inflammatory pathways. IR is also stimulate with low grade chronic inflammation. Tumor necrosis factor-α (TNF-α) which is a cytokine involved in chronic inflammatory states, including cancer, obesity, and diabetes, it blocks insulin signaling by preventing serine phosphorylation of IRS-1 [32]. Overproduction of TNF-α supports and even amplifies the inflammatory process leading to IR [33]. It has been documented that the expression of interleukin (IL)-6 in adipose tissue and its serum concentrations positively correlate with obesity, IR, and type 2 DM, even in cancer patients with IR [34]. Activation of STAT signaling, via IL-6, is known to induce cancer cell proliferation, survival, and invasion, while suppressing host antitumour immunity [35,36].
In this study, the carcinoma group had significantly lower rate of NAFLD than the adenoma and control groups. The risk of CRC was significantly higher in patients without NAFLD compared to patients with NAFLD. As a main finding in this study, we found the absence of NAFLD in the presence of IR was associated with the high risk for carcinoma.
In the literature, there are some conflicting results about the relationship between NAFLD and colorectal neoplams [8-10]. Hwang et al. presented the first evidence that there was an association between NAFLD and an increased rate of colorectal adenomatous polyps by including results of colonoscopy, abdominal ultrasonography, and liver tests [8]. In a cross-sectional study, Wong et al. showed that patients with NAFLD had a higher prevalence of colorectal adenomas and advanced neoplasms. They also showed that among patients with biopsy-proven NAFLD, patients with NASH had a higher prevalence of adenomas and advanced neoplasms than those with simple steatosis [9]. In a large retrospective cohort study of Korean women, Lee et al. found that the risk of adenomatous polyps was twice and the risk of CRC was three fold in patients with NAFLD compaired to patients without NAFLD [10]. Finally, Stadlmayr et al. showed that the patients with NAFLD had significantly more colorectal adenomas and early CRCs compared to those without NAFLD [37]. In contrast, Touzin et al. in a retrospective observational study, included subjects who underwent screening colonoscopy and were evaluated for hepatosteatosis sonographically and histopathologically. Prevelance of adenoma did not show significant diferences between subjects with and without NASH [38].
Most of these studies have concluded that NAFLD could be a risk factor for CRC. But underlying mechanisms of this association has not been identified yet. IR, decreased adiponectin levels, leptin and chronic inflammatory states have been suspected as common mechanisms in carcinogenesis and NAFLD pathogenesis [8-10]. But, It is unclear whether NAFLD leads to colorectal neoplastic lessions by means of these mechanisms, or this association is an incidental coexistance because of the same risk factors related with IR.
Although IR is common in individuals with NAFLD, It is not known that NAFLD is a cause of IR or a consequence of it.
NAFLD is defined as an accumulation of lipids in the form of triglycerides in hepatocytes. The term is used generally when the pathology of metabolic liver disease is not known, or when specifically referring to the fuller spectrum. Both free fatty acids (FFA) and lipoprotein particles provide lipids to the liver. Although some lipoprotein particles are taken up by receptor-mediated endocytosis in others, the TGs may be broken down by hepatic lipase (HL), to produce FFA, which cross the hepatocyte membrane by a combination of facilitated transport and diffusion [39]. The sources of these FFA include lipolysis of stored TGs in adipocytes and dietary fat. The total uptake of FFA by hepatocytes depends on both the concentration of FFA in plasma and the capacity of the cells for FFA uptake [40]. Influencing factors of capacity of the cells for FFA uptake are not resolved in NAFLD.
IR contribute the increase in serum-FFA secondary to lipolysis. But it is not obvious that these FFA were uptaken by hepatocytes or they were consumed by cancer cells as an energy source.
In the course of carcinogenesis multiple metabolic changes occur. Cancer cells need high energy uptake to grow up. This energy is sustained by impairing peripheral insulin-mediated glucose uptake, increasing hepatic glucose production and increasing levels of FFAs through IR [41]. Increase in FFAs involves abnormalities in host fat metabolism including increased lipolysis with increased FFA syntase (FAS) activity, glycerol turnover, and decreased lipogenesis with lowered lipoprotein lipase activity. Normally FAS suplies storage of excess energy intake in the liver, but in cancer patients it appears to play a pivotal role for the growth and survival of many transformed cells [42]. Lipolysis and utilization of hepatic glucose cause decrease in hepatic lipid deposition. Some cytokines such as TNF-alfa and IL-6 have been shown to have role in these metabolic changes in cancer patients. In advanced carcinoma, this cathabolic mechanism leads to cancer-related cachexia and, IR, TNF-α also has been observed in cachectic cancer patients [41]. IL-1 is an cytokine which is released by tumor enviroment and has also been shown to cause decrease in hepatic lipase activity reducing hepatic FFA uptake [43,44]. According to these results it can be suggested that in carcinogenesis while IR is increasing, fat deposition in the liver is decreased.
In previous studies, which showed increased rates of patients with NAFLD in patients with colorectal neoplastic lessions, the patients had higher rates of MS components different from IR which are related with NAFLD such as increased triglyceride levels, obesity, and high LDL-C levels [8,9]. On the other hand in our study, although the carcinoma group had higher rate of IR, they had lower LDL-C and total cholesterol levels. In accordance with our results, in Framingham study, it has been shown that there was an association between low serum cholesterol levels and the colon cancer risk [45]. In a metaanalysis, investigators have found that total cholesterol was inversely associated with all-cancer incidence including CRC [46]. In addition, there were also no significant differences in terms of BMI and presence of increased WC between groups in our study. Keneko et al. have found also no significant differences between CRC and control groups in WC and BMI measurements. They have suggested that the association between these indices and CRC varies depending on gender, ethnic groups and reports [47]. We found similar rates of patients with MS in all three groups and the adenoma group had similar risk factors for NAFLD with the control group. These different results can be explained by the influence of NAFLD risk factors which were also MS components other than IR.
Although some researcher suggests that, the components of MS may appear to have an additive effect on colon neoplasia development acting through different pathophysiological pathways [48], A recent study conducted on animals, has shown that hyperinsulinemia, but not other components of MS, seems to have a more direct role in CRC risk [49].
We also evaluated the subgroup analysis among patients with NAFLD and patients with IR. Although all carcinoma patients with NAFLD had IR, 21.4% of carcinoma patients with IR had NAFLD. These results showed that the presence of NAFLD in carcinoma patients only depends on the presence of IR. Our results suggest that the presence of NAFLD in carcinoma patients can accompany IR, but the presence of IR appears not to be associated with the development of NAFLD in these patients.
Our study has several limitations; our sample size was relatively small. We investigated hepatosteatosis by means of USG. USG with a sensitivity of 60-70% is used frequently inclinical practice [50]. Although magnetic resonance imaging may have a little higher sensitivity of detecting hepatosteatosis, cost effectiveness of USG precludes its use routinely [50]. Liver biopsy, due to its invasive nature, might be appropriate in cases with only elevated transaminases levels. The sensitivity rate of USG may be increased with two experts. In our study results of two experts were similar. Lastly, we could not analyse effect of cancer stage, because of limited numbers of patients with CRC.
According to our results, the presence of NAFLD may be only a coexistence because of the presence of MS components. Although the risk of CRC is positively associated with increased IR, it is negatively associated with NAFLD. Accordingly, we may suggest that because of the activation of catabolic mechanisms and high energy uptake of tumor cells in carcinogenesis, while IR is increasing in carcinoma patients, hepatic fat deposition is decreasing. Further studies should be conducted to clarify the causality between IR and CRC.
Acknowledgements
This study was funded by authors ownself.
Disclosure of conflict of interest
None.
References
- 1.US Cancer Statistics Working Group. United States Cancer Statistics: 1999-2005 Incidence and Mortality Website Report. Published. 2009 [Google Scholar]
- 2.Komninou D, Ayonote A, Richie JP Jr, Rigas B. Insulin resistance and its contribution to colon carcinogenesis. Exp Biol Med (Maywood) 2003;228:396–405. doi: 10.1177/153537020322800410. [DOI] [PubMed] [Google Scholar]
- 3.McKeown-Eyssen G. Epidemiology of colorectal cancer revisited: are serum triglycerides and/or plasma glucose associated with risk? Cancer Epidemiol Biomarkers Prev. 1994;3:687–95. [PubMed] [Google Scholar]
- 4.Giovannucci E, Ascherio A, Rimm EB, Colditz GA, Stampfer MJ, Willett WC. Physical activity, obesity, and risk for colon cancer and adenoma in men. Ann Intern Med. 1995;122:327–34. doi: 10.7326/0003-4819-122-5-199503010-00002. [DOI] [PubMed] [Google Scholar]
- 5.Lu K, Song XL, Han SL Wang CH, Zhong N, Qi LF. Potential Study Perspectives on Mechanisms and Correlations Between Adiposity and Malignancy. Asian Pac J Cancer Prev. 2014;15:1057–60. doi: 10.7314/apjcp.2014.15.2.1057. [DOI] [PubMed] [Google Scholar]
- 6.La Brecque DR, Abbas Z, Anania F, Ferenci P, Khan AG, Goh KL, Hamid SS, Isakov V, Lizarzabal M, Peñaranda MM, Ramos JFR, Sarin S, Stimac D, Thomson ABR, Umar M, Krabshuis J, LeMair A. World Gastroenterology Organisation Global Guidelines Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. J Clin Gastroenterol. 2014;48:467–73. doi: 10.1097/MCG.0000000000000116. [DOI] [PubMed] [Google Scholar]
- 7.Forootan M, Tabatabaeefar M, Yahyaei M, Maghsoodi N. Metabolic Syndrome and Colorectal Cancer: A Cross-Sectional Survey. Asian Pac J Cancer Prev. 2012;13:4999–5002. doi: 10.7314/apjcp.2012.13.10.4999. [DOI] [PubMed] [Google Scholar]
- 8.Hwang ST, Cho YK, Park JH, Kim HJ, Park DI, Sohn CI, Jin W. Relationship of non alcoholic fatty liver disease to colorectal adenomatous polyps. J Gastroenterol Hepatol. 2010;25:562–7. doi: 10.1111/j.1440-1746.2009.06117.x. [DOI] [PubMed] [Google Scholar]
- 9.Wong VW, Wong GL, Tsang SW, Fan T, Chu WC, Woo J, Chan AW, Choi PC, Chim AM, Lau JY, Chan FK, Sung JJ, Chan HL. High prevalence of colorectal neoplasm in patients with non-alcoholic steatohepatitis. Gut. 2011;60:829–36. doi: 10.1136/gut.2011.237974. [DOI] [PubMed] [Google Scholar]
- 10.Lee YI, Lim YS, Park HS. Colorectal neoplasms in relation to non alcoholic fatty liver disease in Korean women: A retrospective cohort study. J Gastroenterol Hepatol. 2012;27:91–5. doi: 10.1111/j.1440-1746.2011.06816.x. [DOI] [PubMed] [Google Scholar]
- 11.Mancia G, Fagard R, Narkiewicz K, Redán J, Zanchetti A, Böhm M, Zannad F. Practice guidelines for the management of arterial hypertension of the European Society of Hypertension (ESH) and the European Society of Cardiology (ESC): ESH/ESC Task Force for the Management of Arterial Hypertension. J Hypertens. 2013;31:1925–38. doi: 10.1097/HJH.0b013e328364ca4c. [DOI] [PubMed] [Google Scholar]
- 12.WHO/IASO/IOTF. The Asia-Pacific Perspective: Redefining Obesity and Its Treatment. Sydney: Health Communications Australia Pty Ltd; 2000. [Google Scholar]
- 13.American Diabetes Association (ADA) Clinical practice recommendations. Diabetes Care. 2007;30(Suppl 1):S42–7. [Google Scholar]
- 14.Grundy SM, Cleeman JI, Daniels SR, Donato KA, Eckel RH, Franklin BA, Gordon DJ, Krauss RM, Savage PJ, Smith SC Jr, Spertus JA, Costa F American Heart Association; National Heart, Lung, and Blood Institute. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation. 2005;112:2735–52. doi: 10.1161/CIRCULATIONAHA.105.169404. [DOI] [PubMed] [Google Scholar]
- 15.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: Insulin resistance and b-cell function from fasting plasma glucose and insulin concentration in man. Diabetologia. 1985;28:412–9. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 16.Kojima S, Watanabe N, Numata M, Ogawa T, Matsuzaki S. Increase in the prevalence of fatty liver in Japan over the past 12 years: analysis of clinical background. J Gastroenterol. 2003;38:954–61. doi: 10.1007/s00535-003-1178-8. [DOI] [PubMed] [Google Scholar]
- 17.Tran TT, Medline A, Bruce WR. Insulin promotion of colon tumors in rats. Cancer Epidemiol Biomarkers Prev. 1996;5:1013–5. [PubMed] [Google Scholar]
- 18.Corpet DE, Jacquinet C, Peiffer G, Tache S. Insulin injections promote the growth of aberrant crypt foci in the colon of rats. Nutr Cancer. 1997;27:316–20. doi: 10.1080/01635589709514543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schoen RE, Tangen CM, Kuller LH, Burke GL, Cushman M, Tracy RP, Dobs A, Savage PJ. Increased blood glucose, body size, and incident colorectal cancer. J Natl Cancer Inst. 1999;91:1147–54. doi: 10.1093/jnci/91.13.1147. [DOI] [PubMed] [Google Scholar]
- 20.Kaaks R, Toniolo P, Akhmedkhanov A. Serum C-peptide, insulin-like growth factor (IGF)-I, IGF-binding proteins ,and colorectal cancer risk in women. J Natl Cancer Inst. 2000;92:1592–600. doi: 10.1093/jnci/92.19.1592. [DOI] [PubMed] [Google Scholar]
- 21.Yang YX, Hennessy S, Lewis JD. Noninsulin dependent diabetes mellitus and colorectal cancer risk. Gastroenterology. 2003;124:A547. [Google Scholar]
- 22.Hsing AW, Gao YT, Chua S, Deng J, Stanczyk FZ. Insulin resistance and prostate cancer risk. J Natl Cancer Inst. 2003;95:67–71. doi: 10.1093/jnci/95.1.67. [DOI] [PubMed] [Google Scholar]
- 23.Rose DP, Komninou D, Stephenson GD. Obesity, adipocytokines, and insulin resistance in breast cancer. Obes Rev. 2004;5:153–165. doi: 10.1111/j.1467-789X.2004.00142.x. [DOI] [PubMed] [Google Scholar]
- 24.Stolzenberg-Solomon RZ, Graubard BI, Chari S, Limburg P, Taylor PR, Virtamo J, Albanes D. Insulin, glucose, insulin resistance, and pancreatic cancer in male smokers. JAMA. 2005;294:2872–8. doi: 10.1001/jama.294.22.2872. [DOI] [PubMed] [Google Scholar]
- 25.Sandhu MS, Dunger DB, Giovannucci EL. Insulin, insulin-like growth factor-I (IGF-I), IGF binding proteins, their biologic interactions, and colorectal cancer. J Natl Cancer Inst. 2002;94:972–80. doi: 10.1093/jnci/94.13.972. [DOI] [PubMed] [Google Scholar]
- 26.Remacle-Bonnet MM, Culouscou JM, Garrouste FL, Rabenandrasana C, Marvaldi JL, Pommier GJ. Expression of type 1, but not type 2 insulin-like growth factor (IGF) receptor on both undifferentiated and differentiated HT29 human colon carcinoma cell lines. J Clin Endocr Metab. 1992;75:609–16. doi: 10.1210/jcem.75.2.1322432. [DOI] [PubMed] [Google Scholar]
- 27.Karimi K, Mahmoudi T, Karimi N, Dolatmoradi H, Arkani M, Farahani H, Vahedi M, Parsimehr E, Dabiri R, Nobakht H, Asadi A, Zali MR. Is there an Association between Variants in Candidate Insulin Pathway Genes IGF-I, IGFBP-3, INSR, and IRS2 and Risk of Colorectal Cancer in the Iranian Population? Asian Pac J Cancer Prev. 2013;14:5011–6. doi: 10.7314/apjcp.2013.14.9.5011. [DOI] [PubMed] [Google Scholar]
- 28.Hammes HP, Martin S, Federlin K, Geisen K, Brownlee M. “Aminoguanidine treatment inhibits the development of experimental diabetic retinopathy. ” Proc Natl Acad Sci U S A. 1991;24:11555–8. doi: 10.1073/pnas.88.24.11555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Giardino I, Edelstein D, Brownlee M. Non-enzymatic glycosylation in vitro and in bovine endothelial cells alters basic fibroblast growth factor activity. A model for intracellular glycosylation in diabetes. J Clin Invest. 1994;1:110–7. doi: 10.1172/JCI117296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moller DE, Flier JS. Insulin resistance-mechanisms, syndromes, and implications. N Engl J Med. 1991;325:938–48. doi: 10.1056/NEJM199109263251307. [DOI] [PubMed] [Google Scholar]
- 31.Sims MA, Hasler WL, Chey WD, Kim MS, Owyang C. Hyperglycemia inhibits mechanoreceptor-mediated gastrocolonic responses, and colonic peristaltic reflexes in healthy humans. Gastroenterology. 1995;108:350–9. doi: 10.1016/0016-5085(95)90060-8. [DOI] [PubMed] [Google Scholar]
- 32.Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-α- and obesity-induced insulin resistance. Science. 1996;271:665–8. doi: 10.1126/science.271.5249.665. [DOI] [PubMed] [Google Scholar]
- 33.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-α: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 34.Vozarova B, Weyer C, Hanson K, Tataranni PA, Bogardus C, Pratley RE. Circulating interleukin-6 in relation to adiposity, insulin action, and insulin secretion. Obes Res. 2001;9:414–7. doi: 10.1038/oby.2001.54. [DOI] [PubMed] [Google Scholar]
- 35.Vakkila J, Lotze MT. Inflammation and necrosis promote tumour growth. Nat Rev Immunol. 2004;4:641–648. doi: 10.1038/nri1415. [DOI] [PubMed] [Google Scholar]
- 36.Zheng L, Dai H, Zhouet M. Fen 1 mutations result in autoimmunity, chronic inflammation and cancers. Nat Med. 2007;13:812–9. doi: 10.1038/nm1599. [DOI] [PubMed] [Google Scholar]
- 37.Stadlmayr A, Aigner E, Stegeretal B. Nonalcoholic fatty liver disease: an independent risk factor for colorectal neoplasia. J Intern Med. 2011;270:41–49. doi: 10.1111/j.1365-2796.2011.02377.x. [DOI] [PubMed] [Google Scholar]
- 38.Touzin NT, Bush KN, Williams CD, Harrison SA. Prevalence of colonic adenomas in patients with nonalcoholic fatty liver disease. Therap Adv Gastroenterol. 2011;4:169–76. doi: 10.1177/1756283X11402118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lambert MS, Avella MA, Berhane Y, Shervill E, Botham KM. The fatty acid composition of chylomicron remnants influences their binding and internalization by isolated hepatocytes. Eur J Biochem. 2001;268:3983–92. doi: 10.1046/j.1432-1327.2001.02311.x. [DOI] [PubMed] [Google Scholar]
- 40.Bradbury MW. Lipid Metabolism and Liver Inflammation. I. Hepatic fatty acid uptake: possible role in steatosis. Am J Physiol Gastrointest Liver Physiol. 2006;290:G194–G198. doi: 10.1152/ajpgi.00413.2005. [DOI] [PubMed] [Google Scholar]
- 41.Mc Namara MJ, Alexander HR, Norton JA. Cytokines and their role in the pathophysiology of cancer cachexia. JPEN J Parenter Enteral Nutr. 1992;16(Suppl):50S–55S. doi: 10.1177/014860719201600603. [DOI] [PubMed] [Google Scholar]
- 42.Kuhajda FP. Fatty acid synthase and cancer: new application of an old pathway. Cancer Res. 2006;66:5977–80. doi: 10.1158/0008-5472.CAN-05-4673. [DOI] [PubMed] [Google Scholar]
- 43.Feingold KR, Memon RA, Moser AH, Shigenaga JK, Grunfeld C. Endotoxin and interleukin-1 decrease hepatic lipase mRNA levels. Atherosclerosis. 1999;142:379–387. doi: 10.1016/s0021-9150(98)00265-2. [DOI] [PubMed] [Google Scholar]
- 44.Apte RN, Dotan S, Elkabets M, White MR, Reich E, Carmi Y, Song X, Dvozkin T, Krelin Y, Voronov E. The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumor-host interactions. Cancer Metastasis Rev. 2006;25:387–408. doi: 10.1007/s10555-006-9004-4. [DOI] [PubMed] [Google Scholar]
- 45.Kreger BE, Anderson KM, Schatzkin A, Splansky GL. Serum cholesterol level, body mass index, and the risk of colon cancer. The Framing-ham Study. Cancer. 1992;70:1038–43. doi: 10.1002/1097-0142(19920901)70:5<1038::aid-cncr2820700505>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 46.Kitahara CM, de González AB, Freedman ND, Huxley R, Mok Y, Jee SH, Samet JM. Total cholesterol and cancer risk in a large prospective study in Korea. J. Clin. Oncol. 2011;29:1592–8. doi: 10.1200/JCO.2010.31.5200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Keneko R, Nakazaki N, Tagawa T, Ohishi C, Kusayanagi S, Kim M, Baba T, Ogawa M, Sato Y. A New Index of Abdominal Obesity which Effectively Predicts Risk of Colon Tumor Development in Female Japanese. Asian Pac J Cancer Prev. 2014;15:1005–10. doi: 10.7314/apjcp.2014.15.2.1005. [DOI] [PubMed] [Google Scholar]
- 48.Pais R, Silaghi H, Silaghi AC, Rusu ML, Dumitrascu DL. Metabolic syndrome and risk of subsequent colorectal cancer. World J Gastroenterol. 2009;15:5141–8. doi: 10.3748/wjg.15.5141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tran TT, Naigamwalla D, Oprescu AI, Lam L, McKeown-Eyssen G, Bruce WR, Giacca A. Hyperinsulinemia, but not other factors associated with insulin resistance, acutely enhances colorectal epithelial proliferation in vivo. Endocrinology. 2006;147:1830–7. doi: 10.1210/en.2005-1012. [DOI] [PubMed] [Google Scholar]
- 50.Scatarige JC, Scott WW, Donovan PJ, Siegelman SS, Sanders RC. Fatty infiltration of the ultrasonographic and computed tomographic correlation. J Ultrasound Med. 1984;3:9–14. doi: 10.7863/jum.1984.3.1.9. [DOI] [PubMed] [Google Scholar]