

Abstract
Objective: The present study aimed to examine the effect of mycophenolate mofetil (MMF), a new immunosuppressive agent, on hypertrophy and apoptosis of podocyte, and investigate the underlying mechanisms. Methods: Cultured rat podocyte were exposed to 5.6 mmol/L normal glucose or 25 mmol/L high glucose with mycophenolic acid (MPA) or Valsartan for 72 h. For animal studies, streptozotocin-induced diabetic rats were untreated or treated with MMF or Valsartan for 16 weeks. After 16 weeks of treatment, the weight of kidney and body, 24 hours urinary protein excretion and serum glucose was detected. Histomorphology of renal tissue was observed by optical microscope and electron microscope. Apoptosis of podocytes were determined by transferase-mediated dUTP nick-end labeling (TUNEL) test. The protein expressions of p21cip1, p27kip1, bax and bcl-2 were examined by Western blot. Results: p27kip1, p21cip1 protein expression in podocytes exposed to high glucose for 72 h and in 16 weeks diabetic glomeruli significantly increased (P<0.01). The expressions of bax, cleaved caspase-3 increased while the expression of bcl-2 decreased in diabetic glomeruli as well as in high glucose. But they were all ameliorated in the groups treated with either MMF or Valsartan. Conclusion: MMF can inhibit abnormal hypertrophy and apoptosis of podocytes in the early stage of diabetes, partly by regulating the expression of cell cycle related protein p27kip1, p21cip1 and apoptosis related genes, such as bax, bcl-2 and cleaved caspase-3. These suggest that the protective effects of MMF on renal function maybe partly through inhibiting abnormal renal cell growth by regulating cell cycle or apoptosis related genes.
Keywords: Mycophenolate mofetil, diabetic nephropathy, podocyte, hypertrophy, apoptosis
Introduction
Renal hypertrophy in the early stage and glomerulosclerosis in the end stage are the characteristics of diabetic nephropathy (DN). Both hypertrophy and apoptosis of renal cells are thought to be involved in the pathogenesis of renal enlargement. Our prior studies have suggested that the apoptosis rate, the expressions of bax, caspase-3 activity and cleavage increased while the expression of bcl-2 decreased in DN group, but they were all ameliorated in the groups treated with mycophenolate mofetil (MMF) [1]. Therefore, in this study we examined the effect of MMF on the hypertrophy and apoptosis of podocyte in vivo and in vitro, and investigated the potential mechanism by examining the expression profile of the cell cycle and apoptosis associated genes.
Materials and methods
Podocyte culture
Conditionally immortalized mouse podocytes were provided by Institute of Basic Medical Science, Chinese Academy of Medical Science (No. 3111C0001CCC000230) and were cultured as previously described [2,3]. Briefly, frozen podocytes were first grown under permissive conditions at 33°C in RPMI-1640 media containing 10% fetal bovine serum (FBS, Hyclone company, America), 50 U/ml interferon-γ (INF-γ) and 100 U/ml of penicillin/streptomycin in collagen-coated flasks, and INF-γ was tapered down to 10 U/ml in successive passages. Then, subcultivation under non-permissive conditions without INF-γ was started at 37°C with media changed on alternate days. Differentiation of podocytes grown for 10 days at 37°C was confirmed by the podocyte marker synaptopodin (polyclonal rabbit antibody, Abcam, England) and validated by western blotting.
After confirming differentiation of podocytes and serum restriction for 24 h, medium was changed to RPMI medium containing normal glucose (5.6 mmol/L glucose, NG), high glucose (25 mmol/L glucose, HG), high glucose + MPA 2.5×10-7 mol/L (Sigma, U.S.), (HG + M), high glucose + Valsartan 10-7 mol/L (Novartis, Beijing, China), (HG + V). After 72 h, podocytes were harvested for either RNA or protein.
Measurement of cellular hypertrophy
Hypertrophy was determined by measurement of cellular protein/cell counts and by flow cytometry. After treatment either with MMF or Valsartan, cells were harvested and washed twice with phosphate-buffered saline (PBS). A small aliquot of cells was counted in a Neubauer chamber after resuspension in PBS. The remaining cells were lysed in Complete Lysis M reagent (Roche Diagnostics) and total protein content was measured by a modified Lowry method. Total protein content was expressed as μg protein per 105 cells and normalized to the corresponding control. Each experiment was independently performed five times. Finally, cell size was measured by FACS analysis (FACS Calibur, Becton Dickenson) as forward cell scatter (FSC). All experiments were performed at least in triplicates.
Animals
32 male healthy Wistar rats, aged 6-7 weeks and weighed 180-240 g, were from the Experimental Animal Center of Shandong University Medical College, Shandong, China. 8 were injected with diluent (NC, n=8) and 24 with 55 mg/kg streptozotocin (Sigma, U.S.) intraperitoneally. Serum glucose levels were measured 72 hours after to confirm the development of diabetes if the serum glucose was higher than 16.7 mmol/L. Diabetic rats were then randomly subdivided into third groups: One group treated with dissolvent (DM), another treated with 15 mg/kg/d of MMF (Roche, Shanghai, China), (DM + M), the third treated with Valsartan (Novartis, Beijing, China), (DM + V), by intragastric administration each day and lasted for 16 weeks. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The animal use protocol has been reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of Yuhuangding Hospital.
After 16 weeks of treatment, the serum glucose, blood pressure and body weight were measured. Kidney weights were measured at the time of sacrifice. The 24 hours urinary protein excretion was determined by enzyme linked immunosorbent assay. One kidney of each rat was collected for histopathologic analysis.
Western blot analysis
Sieved glomeruli and podocytes harvested from plates were homogenized with 500 μl tissue lysate on ice. Then the homogenate was centrifuged at 12000 rpm for 10 min at 4°C. The supernatant was collected and quantified with a bicinchoninic acid (BCA) protein quantitative kit (Pierce, UK). 20 μg of each protein sample was separated by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane (PVDF) (Invitrgen, U.S.). After blocked with defatted milk powder, the PVDF membrane was incubated with P21cip, P27kip, Bcl-2, Bax, β-actin (Santa Cruz, U.S.) or Caspase-3 (R&D, U.S.) at 4°C overnight. After washed, the PVDF membrane was then incubated with a secondary goat anti-rabbit antibody. The expression of targeted protein was detected with enhanced chemiluminescence test and auto scanned by a chemiluminescence detector.
Immunofluorescence study
Immunofluorescence staining for nephrin was performed with frozen kidney sections. Four-micrometer sections were fixed in acetone, blocked with 10% goat serum, and incubated overnight with polyclonal goat anti-rat nephrin antibody (Santa Cruz). Then, sections were stained with fluorescein isothiocyanate-conjugated anti-goat IgG for 30 minutes at room temperature. After washing with PBS, sections were observed through a confocal laser fluorescence microscope (LSM-510; Carl Zeiss, Jena, Germany).
Statistical analysis
Data were presented as mean ± standard error of the mean (SEM) and analyzed by SPSS13.0 software. Comparison between two groups was carried out using t-test. Statistical significance was defined as 0.05 or less.
Results
Celluar hypertrophy
The ratio of proterin/cell numbers as a well-established hypertrophy index revealed a significant increase in podocytes exposed to high glucose (0.828±0.23 vs 0.678±0.16, P<0.05) for 24 hours, and continued to increase with the prolongation of time (Figure 1A). A 1.9-fold increase in the ratio of proterin/cell numbers was observed in podocytes exposed to high glucose for 72 hours, and the increment in the ratio of protein/cell numbers by high glucose was inhibited with MPA or Valsartan in podocytes (P<0.05, Figure 1B). Celluar hypertrophy was also confirmed by FACScan flow cytometer.
Figure 1.
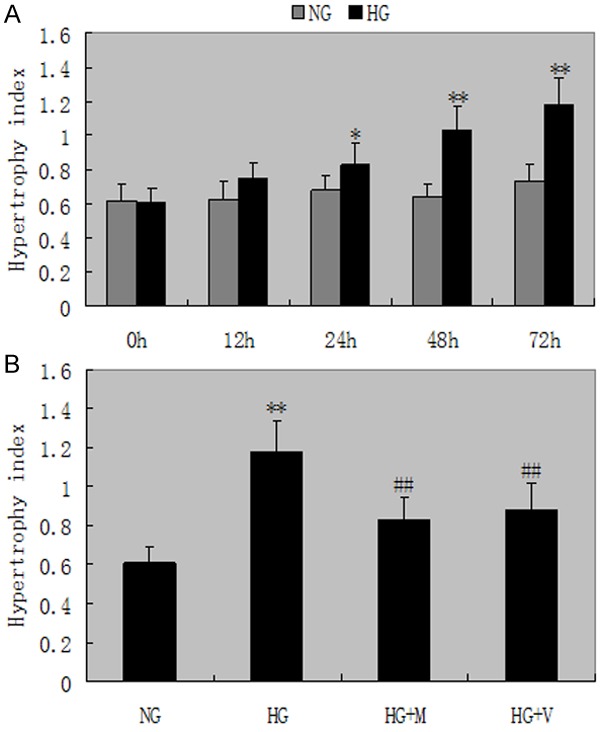
Cellular hypertrophy. A. The ratio of proterin/cell numbers revealed a significant increase in podocytes exposed to high glucose for 24 hours, and continued to increase with the prolongation of time. NG: normal glucose; HG: high glucose; M: mycophenolic acid; V: Valsartan. *P<0.05 and **P<0.01 vs with the NG group. B. The increment in the ratio of protein/cell numbers by high glucose was inhibited with MPA or Valsartan in podocytes for 72 h. #P<0.05 and ##P<0.01 vs HG group. Date are mean ± SEM.
Podocytes p27kip1, p21cip1 protein expression
The p27kip1, p21cip1 protein expression were significantly higher in high glucose than in normal glucose podocytes at 24 h. There were 1.8-fold and 1.6-fold increase in p27kip1, p21cip1 protein expression in high glucose compared with normal glucose cells for 72 h assessed by densitometry (Figure 2). Figure 3 shows that MPA or Valsartan ameliorated this high glucose-induced increase p27kip1, p21cip1 protein expression. Moreover, MPA had more significantly inhibitory effect on the protein expressions of p27kip1 better than valsartan after 72 h (P<0.05).
Figure 2.
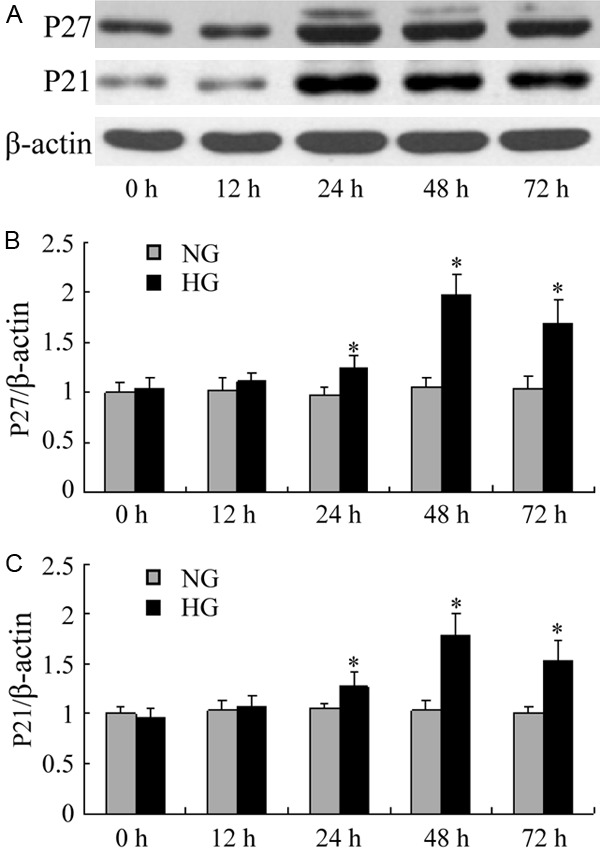
Western blot of p27kip1, p21cip1 in podocytes. A-C. The protein expressions of p27kip1, p21cip1 revealed a significant increase in podocytes exposed to high glucose for 24 hours, and continued to increase with the prolongation of time. *P<0.05 vs with the NG group. Date are mean ± SEM.
Figure 3.
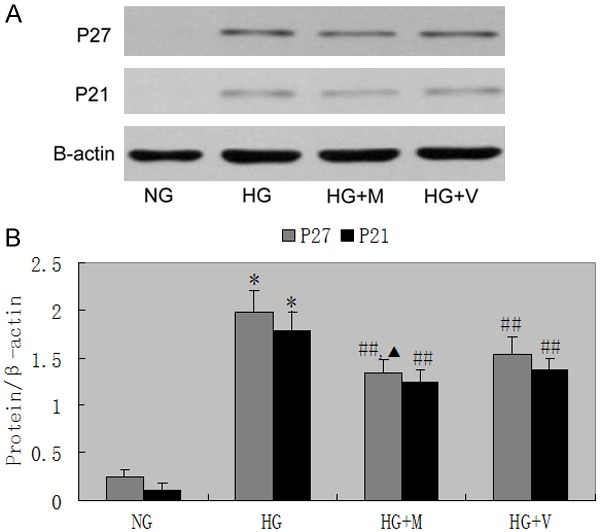
Western blot of p27kip1, p21cip1 in podocytes. Administration of MPA or valsartan significant ameliorated the increase in the protein expressions of p27kip1, p21cip1. ##P<0.01 vs HG group for 72 h. Moreover, MPA had more significantly inhibitory effect on the protein expressions of P27kip1 better than valsartan after 72 h, ▲P<0.05. Date are mean ± SEM.
Apoptosis of podocytes
Exposure to HG for 48 hours resulted in a remarkable increase in apoptotic podocytes (4.97±0.58 vs 2.77±0.31, P<0.05), and these increments in apoptosis were markedly ameliorated by the administration of MPA after 72 hours. Moreover, MPA had more significantly inhibitory effect on apoptosis indexes of podocytes better than valsartan after 72 h (P<0.05, Figure 4).
Figure 4.
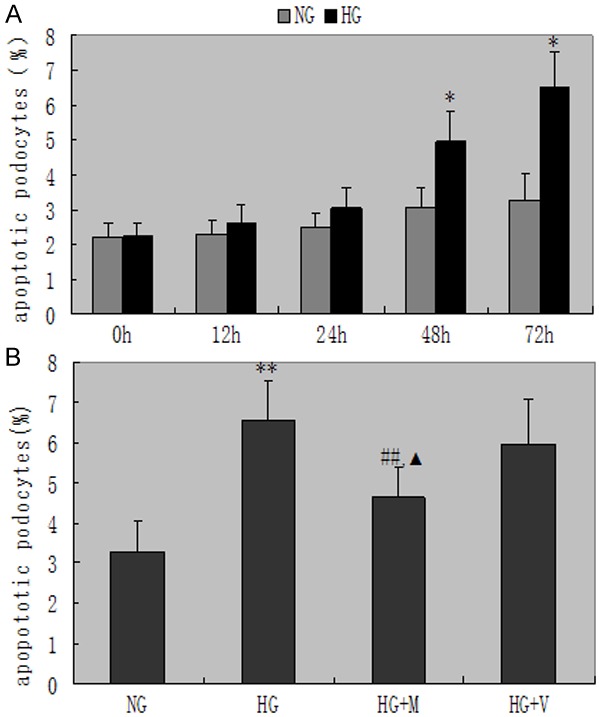
Apoptosis indexes of podocytes in each group. A. The apoptosis indexes of podocytes indicates a significant increase exposed to high glucose for 48 hours, and continued to increase with the prolongation of time, *P<0.05 vs with the NG group. B. Apoptosis indexes of podocytes indicate significant difference as compared with HG and HG + M, ##P<0.01. ▲P<0.05 vs HG + V and HG + M. Date are mean ± SEM.
Bax, bcl-2 and active fragments of caspase-3 protein expression in less and more podocyte
We secondly determined the changes of bax, bcl-2 and active fragments of caspase-3 protein expression in hypertrophied podocyte. Compared with incubation in NG, exposure to high glucose for 48 h significantly induced bax and cleaved caspase-3 protein expressions and reduced bcl-2 protein expressions (bax/bcl-2: 1.53±0.21 vs 1.07±0.12, P<0.01; caspase-3/β-actin: 2.06±0.27 vs 1.11±0.14, P<0.01) (Figure 5). These changes induced by HG was significantly abrogated by MPA or Valsartan. Moreover, MPA had more significantly inhibitory effect than valsartan on the radio of bax/bcl-2 and cleaved caspase-3 protein expressions in HG after 72 h (P<0.05, Figure 6).
Figure 5.
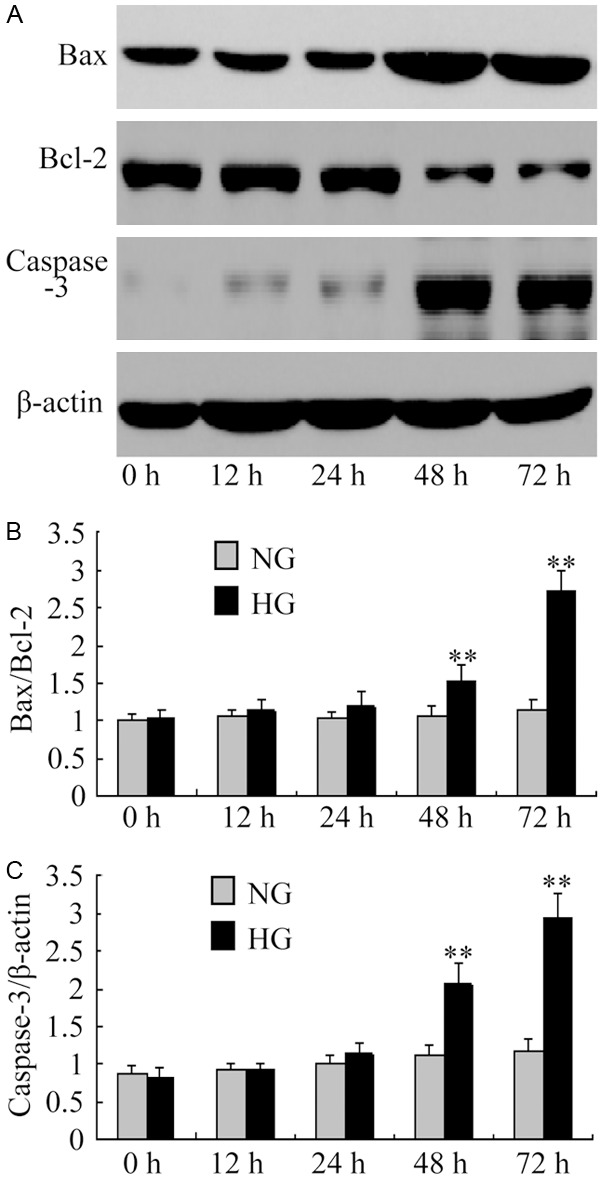
The expressions of Bax/Bcl-2 and caspase-3 detected by Western blot. A-C. The protein expressions of Bax/Bcl-2 and caspase-3 revealed a significant increase in podocytes exposed to high glucose for 48 hours, and continued to increase with the prolongation of time. **P<0.01 vs with the NG group. Date are mean ± SEM.
Figure 6.
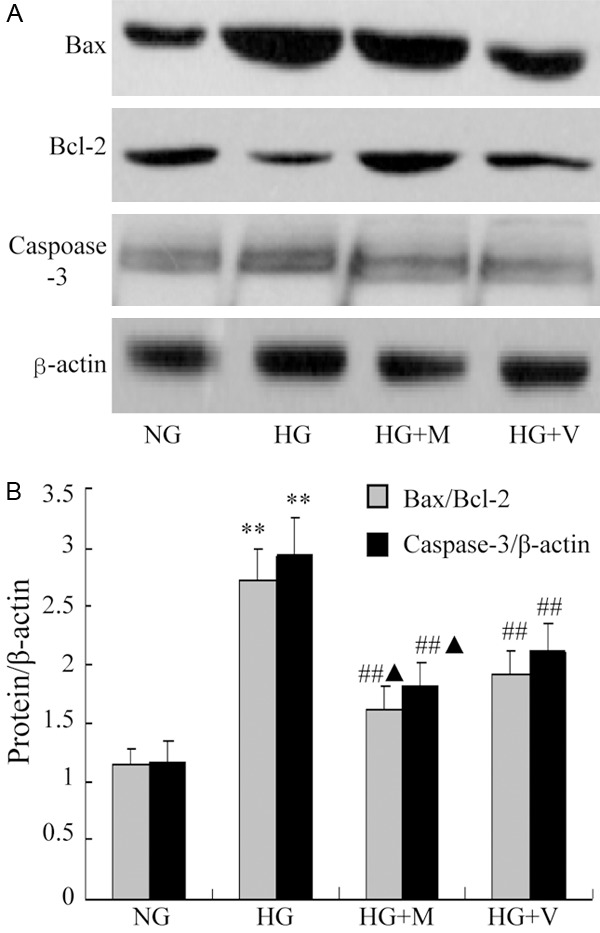
The expressions of Bax/Bcl-2 and caspase-3 detected by Western blot in cultural podocytes, **P<0.01 indicate significant difference as compared with the NG group; ##P<0.01 indicate significant difference as compared with the HG group and HG + M or HG + V, ▲P<0.05 vs HG + V and HG + M. Date are mean ± SEM.
Animal data
After 16 weeks of treatment, systolic blood pressure, glucose and 24 hour urinary protein excretion were measured. We found that the ratio of kidney weight to body weight and 24 hour urinary protein excretion in DN group increased sharply but they were significantly reduced by MMF or Valsartan (P<0.01). The serum glucose in DN group was also higher than that in NC group, but it couldn’t be reduced by MMF or Valsartan (Table 1).
Table 1.
Clinical parameters measured at 16 weeks after treatment with MMF or Valsartan
| Groups (n=8) | NC | DN | DN + M | DN + V |
|---|---|---|---|---|
| Kw/Bw (‰) | 4.57±0.24 | 8.49±0.43** | 5.86±0.25*,# | 6.25±0.37**,# |
| Blood glucose (mg/dl) | 108.1±2.3 | 328.7±4.54** | 323.4±3.36** | 335.9±4.78** |
| Serum creatinine (mg/dL) | 0.96±0.02 | 0.97±0.05 | 0.93±0.04 | 0.99±0.03 |
| urinary protein (mg/24 h) | 16.52±0.66 | 55.37±2.33** | 23.84±1.83*,# | 29.10±1.27**,# |
MMF: mycophenolate mofetil; NC: normal control; DN: dissolvent (DM); DM + M: 15 mg/kg/d of MMF; DM + V: Valsartan. Data are shown as mean ± SEM.
P<0.05 vs NC group;
P<0.01 vs NC group.
P<0.01 indicates the significant difference as compared with DN group.
Renal morphological changes of rats
Under the optical microscope and electron microscope, we found that in DN group, the foot process of podocytes fused and flattened, glomerular basement membrane exposed and some podocytes ablated, the glomerular basement membrane thickened and the mesangial matrix increased. However, these changes were alleviated after 16 w of treatment, which was in accord with our previous studies [1].
Glomerular p27kip1, p21cip1 protein expression
Figure 7 shows a representative Western blot of equal amounts of protein from the lysates of sieved control, diabetes mellitus, diabetes mellitus + MMF, and diabetes mellitus + Valsartan. Glomerular p27kip1, p21cip1 protein expression were also increased in diabetes rats at 4 weeks (P<0.05). Administration of Valsartan or MMF in diabetic inhibited the increase in p27kip1, p21cip1 protein expression (Figure 8).
Figure 7.
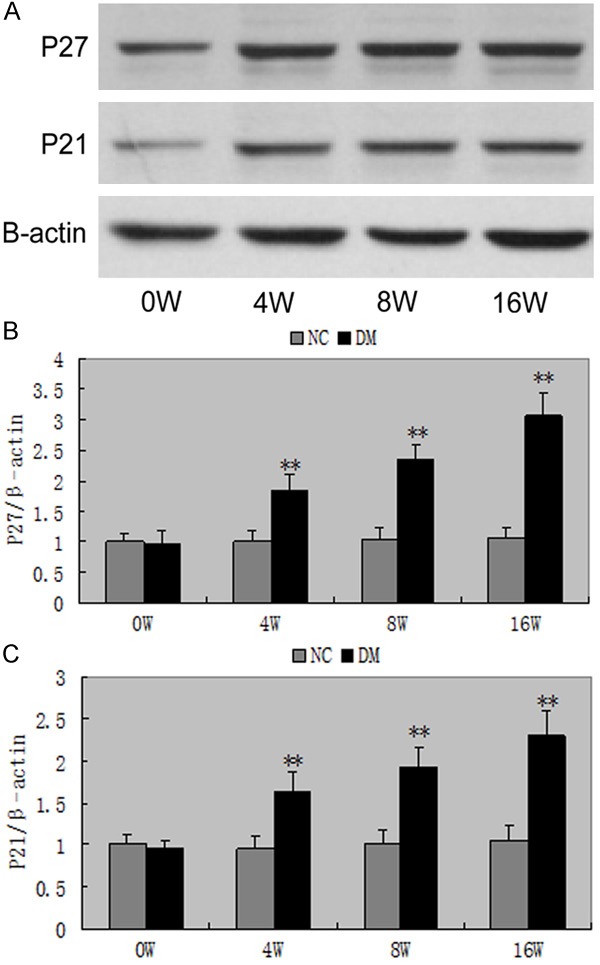
Western blot of p27kip1, p21cip1 in DM. A-C. The protein expressions of p27kip1, p21cip1 revealed a significant increase in rats exposed to high glucose for 4 weeks, and continued to increase. **P<0.01 vs with the NC group. Date are mean ± SEM.
Figure 8.
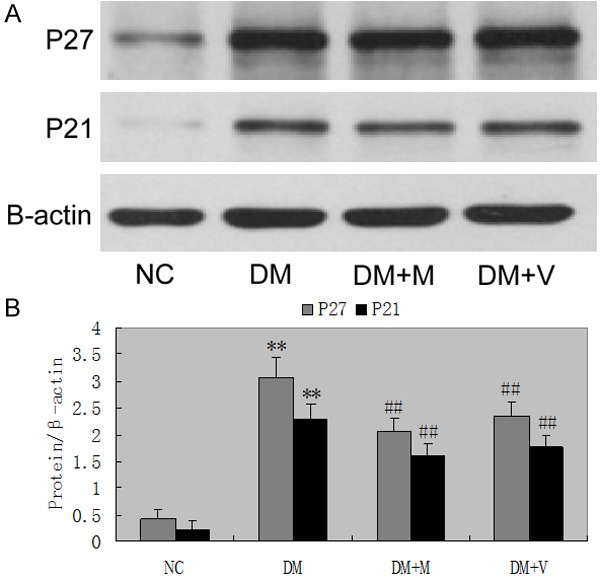
The protein expressions of p27kip1, p21cip1 in rats. Administration of MMF or valsartan significant ameliorated the increase in the protein expressions of p27kip1, p21cip1. ##P<0.01 vs DM group for 16 weeks. Date are mean ± SEM.
Nephrin expression
Immunofluorescence staining also revealed the fact that untreated diabetic rats had a decreased nephrin expression as well as altered the expression pattern which mainly locates in the central area of glomeruli rather than in peripheral area. MMF or valsartan treatment can stop nephrin expression decrease along the glomerular capillary area. These results suggest that MMF or valsartan have a supportive role in maintaining nephrin and thus may be the glomerular filtration barriers (Figure 9).
Figure 9.
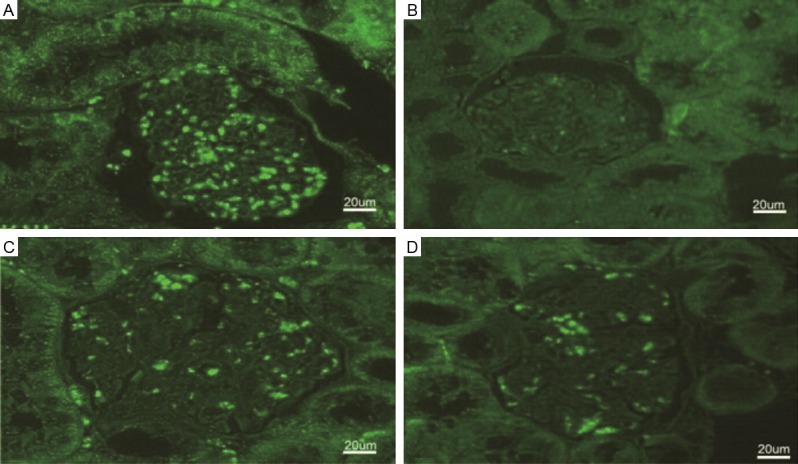
Immunofluorescent staining of nephrin in kidneys (original magnification ×300). A. Non-diabetic control rat; B. Untreated diabetic control rat; C. MMF treated diabetic rat; D. Valsartan treated diabetic rat.
Apoptosis of podocytes
Then number of apoptotic podocytes was significantly increased in DM (2.63±0.31) compared to NC (0.84±0.26) (P<0.01) at 8 weeks, and continued to decrease with the prolongation of time, (3.72±0.54) compared to NC (1.17±0.54) (P<0.01) at 16 weeks. It could be inhibited by MMF and Valsartan [1] (P<0.05).
Protein expressions of of bax, bcl-2 and cleaved caspase-3
Bax and cleaved caspase-3 protein expressions were significantly increased, while bcl-2 protein expressions were markedly decreased in the DM compared to NC from 8 weeks (bax/bcl-2: 2.27±0.20 vs 1.02±0.07, P<0.05; caspase-3/β-actin: 1.69±0.28 vs 1.04±0.15, P<0.05) (Figure 10). Administration of MMF or valsartan significantly ameliorated the increases in the radio of bax/bcl-2 (P<0.05) and cleaved caspase-3 protein expressions in DM glomeruli (P<0.05), which was in accord with our previous studie [1].
Figure 10.
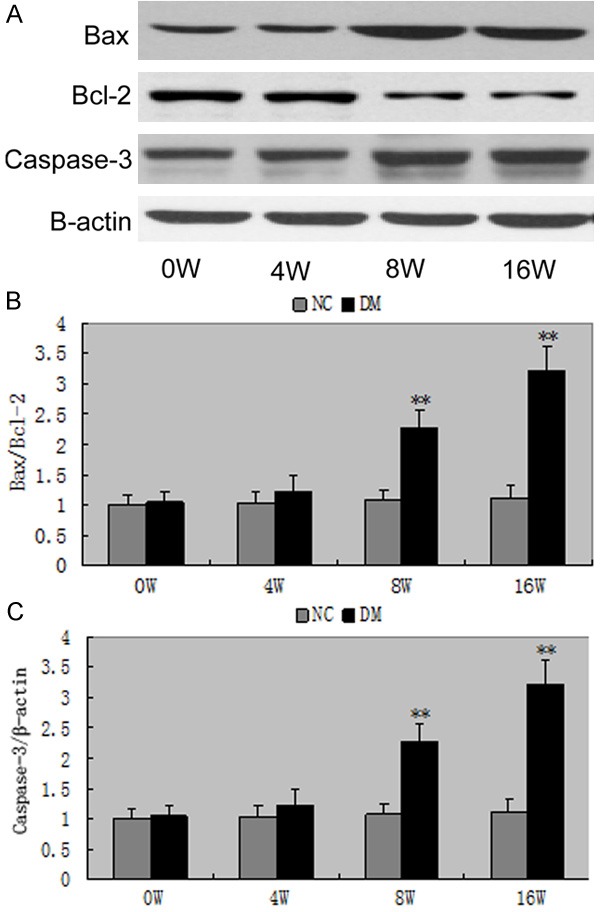
The expressions of Bax/Bcl-2 and caspase-3 in rats. A-C. The protein expressions of Bax/Bcl-2 and caspase-3 revealed a significant increase exposed to high glucose for 4 weeks and continued to increase. **P<0.01 vs with the NG group. Date are mean ± SEM.
Discussion
Glomerular hypertrophy is a hallmark of diabetic nephropathy. Beyond the paradigm of extracellular matrix expansion resulting in complex renal structural and functional changes, podocyte injury has emerged to play a pivotal role in diabetic nephropathy [4]. Podocyte depletion could be a major mechanism driving glomerulosclerosis and progressive deterioration of renal function [5,6]. Podocytes are terminally differentiated cells and die rather than proliferate in diabetic nephropathy [7]. Therefore, there is a baseline expression of p27Kip1 in normal podocytes; however, under diabetic conditions p27Kip1 is upregulated and leads to further cell cycle arrest which drives the cells towards hypertrophy. This is of particular importance in diabetes since the p27Kip1-/- mice are partially protected from diabetic nephropathy [8].
In this study, we demonstrated that p27kip1 and p21cip1 protein expression increased, as assessed by Western blot in diabetic glomeruli and in podocytes under high glucose conditions, in accordance with previous studies [9,10]. The immunohistochemical staining also revealed that glomerular p27kip1 and p21cip1 protein expression was increased in diabetic rats, which was in accord with the results of the Western blot. Taken together, it seems that glomerular hypertrophy in diabetes is a consequence of the hypertrophy of podocytes as well as mesangial cells.
Recent evidence underscores the role of the cyclin-dependent kinase inhibitors (CKI) p27Kip1 and p21Cip1 as important mediators of early structural and functional renal abnormalities in diabetic nephropathy, including podocyte injury with subsequent proteinuria and renal hypertrophy [8,11]. The diabetic milieu per se, hemodynamic changes, and local growth factor such as transforming growth factor-β (TGF-β) and angiotensin II are considered mediators in the pathogenesis of cellular hypertrophy in diabetes [12,13].
One of the important upstream factors inducing p27Kip1 is angiotensin II. Angiotensin exerts a hypertrophic effect on mesangial cells and renal tubular cells [14,15]. In podocytes, angiotensin II is known to mediate rearrangement of the actin cytoskeleton [16], depolarization and increase in cytosolic calcium activity [17], and apoptosis [18]. To understand the underlying molecular mechanism of angiotensin II induced hypertrophy, a number of investigation have been performed upon the effect of angiotensin II on cell cycle regulatory proteins and CKIs. Xu et al. studied podocytes in glucose-stimulated podocytes and in diabetic glomeruli and found an increase in p27Kip1 mRNA and protein expression in podocytes exposed to high glucose [19]. This increase in p27Kip1 expression was associated with podocyte hypertrophy. Interestingly, high glucose-induced p27Kip1 expression and hypertrophy of podocytes were attenuated by an angiotensin II receptor blocker indicating that some of the effects may be mediated by high glucose-induced angiotensin II [19]. Angiotensin-converting enzyme inhibitors or angiotensin II receptor blocker treatment, which became the standard therapy for diabetic nephropathy, may prevent or even reverse renal hypertrophy in diabetes [20-22]. In this study, we also demonstrate that Valsartan inhibits increased p27kip1 and p21cip1 expression both in vivo and in vitro. Taken together, the observation that either ACE inhibitor or ARB inhibits p27Kip1 and p21Cip1 expression suggests that the mechanism is related to an angiotensin II blockade rather than direct antioxidant effects.
In addition to hypertrophy, apoptosis has also been implicated in the pathogenesis of diabetic nephropathy [23-25]. The number of podocytes is decreased in the glomeruli of patients with type 1 and type 2 diabetes [26,27], and a good deal of evidence has shown that apoptosis is also implicated in the process of podocyte loss under diabetic conditions [28]. Changes in the interaction of podocytes with the underlying glomerular basement membrane and apoptosis of podocytes contribute to this depletion. Our previous study found hyperglycaemia induced podocytes apoptosis accompanied with increased expression of cleaved caspase-3, bax protein and decreased bcl-2 protein expression [1]. In this study, we found podocyte numbers were significantly decreased for 48 h in high glucose or in 8-week diabetic glomeruli compared to normal control; furthermore, podocyte numbers were significantly lower in 16-week DN relative to 8-week DN. Double immunofluorescence staining for nephrin revealed that apoptosis within 16-week DN was significantly increased mainly in podocytes. However, the hypertrophy of podocyte was found in 24 h high glucose or 4-week diabetic glomeruli, with a concomitant increase in p27Kip1 and p21cip1 expression. Taken together, we assume that apoptosis occurs principally after 8 weeks of DN induction especially in more hypertrophied diabetic glomeruli, resulting in fewer podocytes in DN. A recent study by Menini et al. [29] also suggested that glomerular cell apoptosis was not an early feature in the course of experimental diabetic glomerulopathy, since it is preceded by glomerular hypertrophy, supporting our assumption. It has been previously shown that hyperglycaemia directly induces hypertrophy and later apoptosis in cultured podocytes [19,24,30]. These findings suggest that apoptosis is more operative in more hypertrophied diabetic glomeruli.
Podocyte loss is closely correlated with the onset and magnitude of glomerular sclerosis [4] and a better understanding of podocyte pathophysiology in diabetic nephropathy is necessary to develop innovative therapeutic strategies. MMF is an immunosuppressant and prodrug of mycophenolic acid, known for its anti-inflammatory and antiproliferative properties and used extensively in allograft rejection in recent years [31]. MMF prevent renal disease progression by suppressing lymphocytes, macrophages infiltration and adhesion molecules expression [32]. MMF can reduce renal injury in anti-Thy1. 1-nephritis rats, and has the anti-oxidant effect by blocking fibrosis in streptozotocin (STZ)-induced diabetic rats [33,34]. The present study demonstrated that MMF could significantly inhibit hypertrophy and apoptosis of podocyte. To further elucidate the underlying mechanism of reduced viability of podocyte induced by MMF, we investigated the cell cycle arrest and apoptosis of podocyte treated by MMF and found that treatment of podocyte with MMF for 72 h caused a significant increase of p27Kip1 and p21Cip1 expression. These results demonstrate that the inhibitory effect of MMF on podocyte hypertrophy is mediated by significantly up-regulated p27Kip1 and p21Cip1 expression. Moreover, we found that MMF was able to inhibit podocyte apoptosis via increasing cleaved caspase-3 and bax expression, decreasing bcl-2 expression. Taken together, the coordinate inhibition of cell cycle arrest and apoptosis contributes to the effect of MMF on podocyte, consistent with the results observed in other cell lines.
In summary, the present study revealed that MMF could inhibit the hypertrophy and apoptosis of podocytes, probably via the regulation of the expression of cell cycle-related genes and bax/bcl-2. These data provide the support for the potential application of MMF in the treatment of diabetic nephropathy.
Acknowledgements
Promotive research fund for excellent young and middle-aged scientists of Shandong Province (No. 2008BS02027).
Disclosure of conflict of interest
None.
References
- 1.Lv W, Zhang Y, Guan G, Li P, Wang J, Qi D. Mycophenolate mofetil and valsartan inhibit podocyte apoptosis in streptozotocin-induced diabetic rats. Pharmacology. 2013;92:227–234. doi: 10.1159/000354600. [DOI] [PubMed] [Google Scholar]
- 2.Mundel P, Reiser J, Zúñiga Mejía Borja A, Pavenstädt H, Davidson GR, Kriz W, Zeller R. Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Exp Cell Res. 1997;236:248–258. doi: 10.1006/excr.1997.3739. [DOI] [PubMed] [Google Scholar]
- 3.Shankland SJ, Pippin JW, Reiser J, Mundel P. Podocytes in culture: past, present, and future. Kidney Int. 2007;72:26–36. doi: 10.1038/sj.ki.5002291. [DOI] [PubMed] [Google Scholar]
- 4.Wolf G, Chen S, Ziyadeh FN. From the periphery of the glomerular capillary wall toward the center of disease: podocyte injury comes of age in diabetic nephropathy. Diabetes. 2005;54:1626–1634. doi: 10.2337/diabetes.54.6.1626. [DOI] [PubMed] [Google Scholar]
- 5.Wharram BL, Goyal M, Wiggins JE, Sanden SK, Hussain S, Filipiak WE, Saunders TL, Dysko RC, Kohno K, Holzman LB, Wiggins RC. Podocyte depletion causes glomerulosc-lerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol. 2005;16:2941–2952. doi: 10.1681/ASN.2005010055. [DOI] [PubMed] [Google Scholar]
- 6.Macconi D, Bonomelli M, Benigni A, Plati T, Sangalli F, Longaretti L, Conti S, Kawachi H, Hill P, Remuzzi G, Remuzzi A. Pathophysiological implications of reduced podocyte number in a rat model of progressive glomerular injury. Am J Pathol. 2006;168:42–54. doi: 10.2353/ajpath.2006.050398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marshall CB, Shankland SJ. Cell cycle regulatory proteins in podocyte health and disease. Nephron Exp Nephrol. 2007;106:e51–e59. doi: 10.1159/000101793. [DOI] [PubMed] [Google Scholar]
- 8.Wolf G, Schanze A, Stahl RA, Shankland SJ, Amann K. p27 (Kip1) knockout mice are protected from diabetic nephropathy: evidence for p27 (Kip1) haplotype insufficiency. Kidney Int. 2005;68:1583–1589. doi: 10.1111/j.1523-1755.2005.00570.x. [DOI] [PubMed] [Google Scholar]
- 9.Wolf G, Wenzel U, Ziyadeh FN, Stahl RA. ACE inhibitor treatment reduces glomerular p16INK4 and p27Kip1 expression in diabetic BBdp rats. Diabeologia. 1999;42:1425–1432. doi: 10.1007/s001250051314. [DOI] [PubMed] [Google Scholar]
- 10.Hoshi S, Shu Y, Yoshida F, Inagaki T, Sonoda J, Watanabe T, Nomoto K, Nagata M. Podocyte injury promotes progressive nephropathy in Zuker diabetic fatty rats. Lab Invest. 2002;82:25–34. doi: 10.1038/labinvest.3780392. [DOI] [PubMed] [Google Scholar]
- 11.Rüster C, Bondeva T, Franke S, Förster M, Wolf G. Advanced glycation end-products induce cell cycle arrest and hypertrophy in podocytes. Nephrol Dial Transplant. 2008;23:2179–2191. doi: 10.1093/ndt/gfn085. [DOI] [PubMed] [Google Scholar]
- 12.Leehey DJ, Singh AK, Alavi N, Singh R. Role of angiotensin II in diabetic nephropathy. Kidney Int. 2000;58:S93–S98. doi: 10.1046/j.1523-1755.2000.07715.x. [DOI] [PubMed] [Google Scholar]
- 13.Hoffman BB, Sharma K, Ziyadeh FN. Potential role of TGF-β in diabetic nephropathy. Miner Electrolyte Metab. 1998;24:190–196. doi: 10.1159/000057369. [DOI] [PubMed] [Google Scholar]
- 14.Wolf G, Schroeder R, Ziyadeh FN, Thaiss F, Zahner G, Stahl RA. High glucose stimulates expression of p27Kip1 in cultured mouse mesangial cells: relationship to hypertrophy. Am J Physiol. 1997;273:348–356. doi: 10.1152/ajprenal.1997.273.3.F348. [DOI] [PubMed] [Google Scholar]
- 15.Wolf G, Wenzel U, Hannken T, Stahl RA. Angiotensin II induces p27Kip1 expression in renal tubules in vivo: role of oxygen species. J Mol Med. 2001;79:382–389. doi: 10.1007/s001090100241. [DOI] [PubMed] [Google Scholar]
- 16.Benigni A, Gagliardini E, Remuzzi G. Changes in glomerular perm-selectivity induced by angiotensin II imply podocyte dysfuncion and slit diaphragm protein rearrangement. Semin Nephol. 2004;24:131–140. doi: 10.1016/j.semnephrol.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 17.Gloy J, Henger A, Fischer KG, Nitschke R, Bleich M, Mundel P, Schollmeyer P, Greger R, Pavenstädt H. Angiotensin II modulates celluar functions of podocytes. Kidney Int. 1998;67:S168–S170. doi: 10.1046/j.1523-1755.1998.06736.x. [DOI] [PubMed] [Google Scholar]
- 18.Durvasula RV, Petermann AT, Hiromura K, Blonski M, Pippin J, Mundel P, Pichler R, Griffin S, Couser WG, Shankland SJ. Activation of a local tissue angiotensin system in podocytes by mechanical strain. Kidney Int. 2004;65:30–39. doi: 10.1111/j.1523-1755.2004.00362.x. [DOI] [PubMed] [Google Scholar]
- 19.Xu ZG, Yoo TH, Ryu DR, Cheon Park H, Ha SK, Han DS, Adler SG, Natarajan R, Kang SW. Angiotensin II receptor blocker inhibits p27Kip1 expression in glucose-stimulated podocytes and in diabetic glomeruli. Kidney Int. 2005;67:944–952. doi: 10.1111/j.1523-1755.2005.00158.x. [DOI] [PubMed] [Google Scholar]
- 20.Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The collaborative study group. N Engl J Med. 1993;329:1456–1465. doi: 10.1056/NEJM199311113292004. [DOI] [PubMed] [Google Scholar]
- 21.Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, Remuzzi G, Snapinn SM, Zhang Z, Shahinfar S RENAAL Study Investigators. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl Med. 2001;345:861–869. doi: 10.1056/NEJMoa011161. [DOI] [PubMed] [Google Scholar]
- 22.Gross ML, El-Shakmak A, Szábó A, Koch A, Kuhlmann A, Münter K, Ritz E, Amann K. ACE-inhibitions but not endothelin recepor blockers prevent podocyte loss in early diabetic nephropathy. Diabetologia. 2003;46:856–868. doi: 10.1007/s00125-003-1106-8. [DOI] [PubMed] [Google Scholar]
- 23.Dalla Vestra M, Saller A, Mauer M, Fioretto P. Role of mesangial expansion in the pathogenesis of diabetic nephropathy. J Nephrol. 2001;14:S51–S57. [PubMed] [Google Scholar]
- 24.Susztak K, Raff AC, Schiffer M, Böttinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55:225–233. [PubMed] [Google Scholar]
- 25.Verzola D, Gandolfo MT, Ferrario F, Rastaldi MP, Villaggio B, Gianiorio F, Giannoni M, Rimoldi L, Lauria F, Miji M, Deferrari G, Garibotto G. Apoptosis in the kidneys of patients with type II diabetic nephropathy. Kidney Int. 2007;72:1262–1272. doi: 10.1038/sj.ki.5002531. [DOI] [PubMed] [Google Scholar]
- 26.Steffes MW, Schmidt D, McCrery R, Basgen JM International Diabetic Nephropathy Study Group. Glomerular cell number in normal subjects and in type 1 diabetic patients. Kidney Int. 2001;59:2104–2113. doi: 10.1046/j.1523-1755.2001.00725.x. [DOI] [PubMed] [Google Scholar]
- 27.Pagtalunan ME, Miller PL, Jumping-Eagle S, Nelson RG, Myers BD, Rennke HG, Coplon NS, Sun L, Meyer TW. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest. 1997;99:342–348. doi: 10.1172/JCI119163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shankland SJ. The podocyte’s response to injury: role in proteinuria and glomerulosclerosis. Kidney Int. 2006;69:2131–2147. doi: 10.1038/sj.ki.5000410. [DOI] [PubMed] [Google Scholar]
- 29.Menini S, Iacobini C, Oddi G, Ricci C, Simonelli P, Fallucca S, Grattarola M, Pugliese F, Pesce C, Pugliese G. Increased glomerular cell (podocyte) apoptosis in rats with streptozotocin-induced diabetes mellitus: role in the development of diabetic glomerular disease. Diabetologia. 2007;50:2591–2599. doi: 10.1007/s00125-007-0821-y. [DOI] [PubMed] [Google Scholar]
- 30.Kim NH. Podocyte hypertrophy in diabetic nephropathy. Nephrology. 2005;10:S14–S16. doi: 10.1111/j.1440-1797.2005.00450.x. [DOI] [PubMed] [Google Scholar]
- 31.Utimura R, Fujihara CK, Mattar AL, Malheiros DM, Noronha IL, Zatz R. Mycophenolate mofetil prevents the development of glomerular injury in experimental diabetes. Kidney Int. 2003;63:209–216. doi: 10.1046/j.1523-1755.2003.00736.x. [DOI] [PubMed] [Google Scholar]
- 32.Mycophenolate mofetil in renal transplantation: 3-year results from the placebo-controlled trial. European Mycophenolate Mofetil Cooperative Study Group. Transplantation. 1999;68:391–396. [PubMed] [Google Scholar]
- 33.Romero F, Rodríguez-Iturbe B, Parra G, González L, Herrera-Acosta J, Tapia E. Mycophenolate mofetil prevents the progressive renal failure induced by 5/6 renal ablation in rats. Kidney Int. 1999;55:945–955. doi: 10.1046/j.1523-1755.1999.055003945.x. [DOI] [PubMed] [Google Scholar]
- 34.Wu YG, Lin H, Qi XM, Wu GZ, Qian H, Zhao M, Shen JJ, Lin ST. Prevention of early renal injury by mycophenolate mofetil and its mechanism in experimental diabetes. Int Immunopharmacol. 2006;6:445–453. doi: 10.1016/j.intimp.2005.09.006. [DOI] [PubMed] [Google Scholar]